
Comparison of Agonist Activity between CB1 and CB2 Receptors with Orthosteric Site Mutations
 , ,
, ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
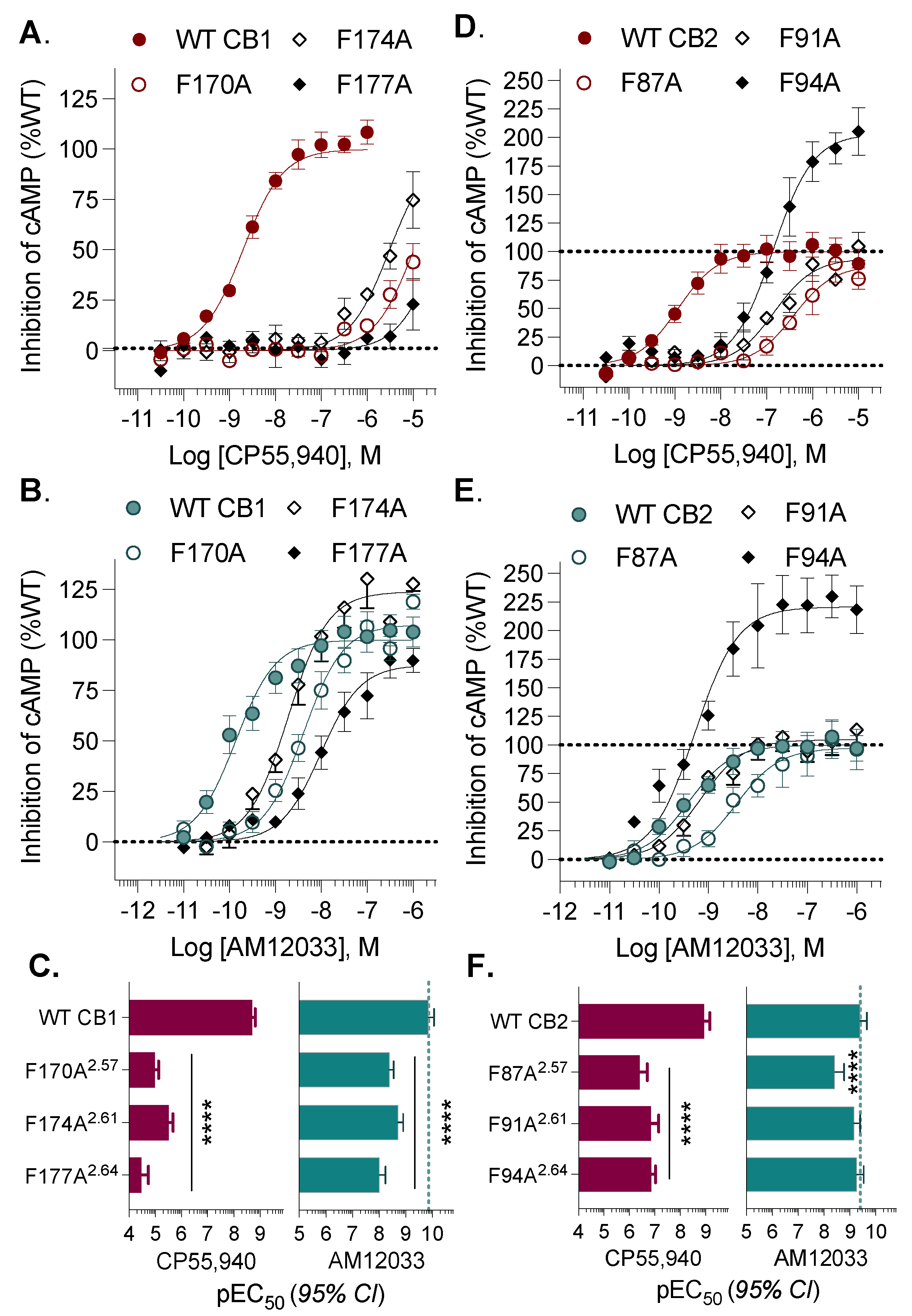
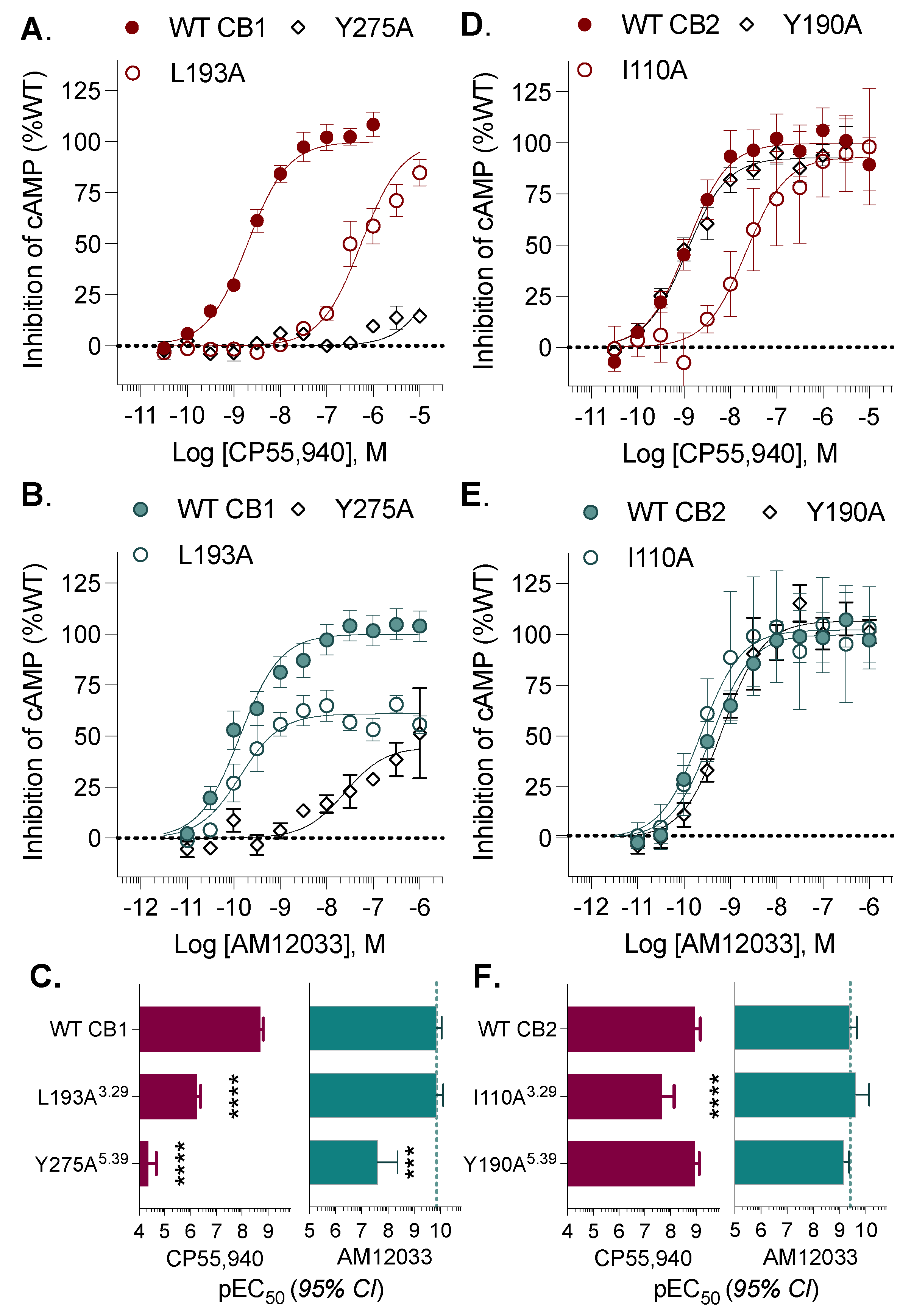
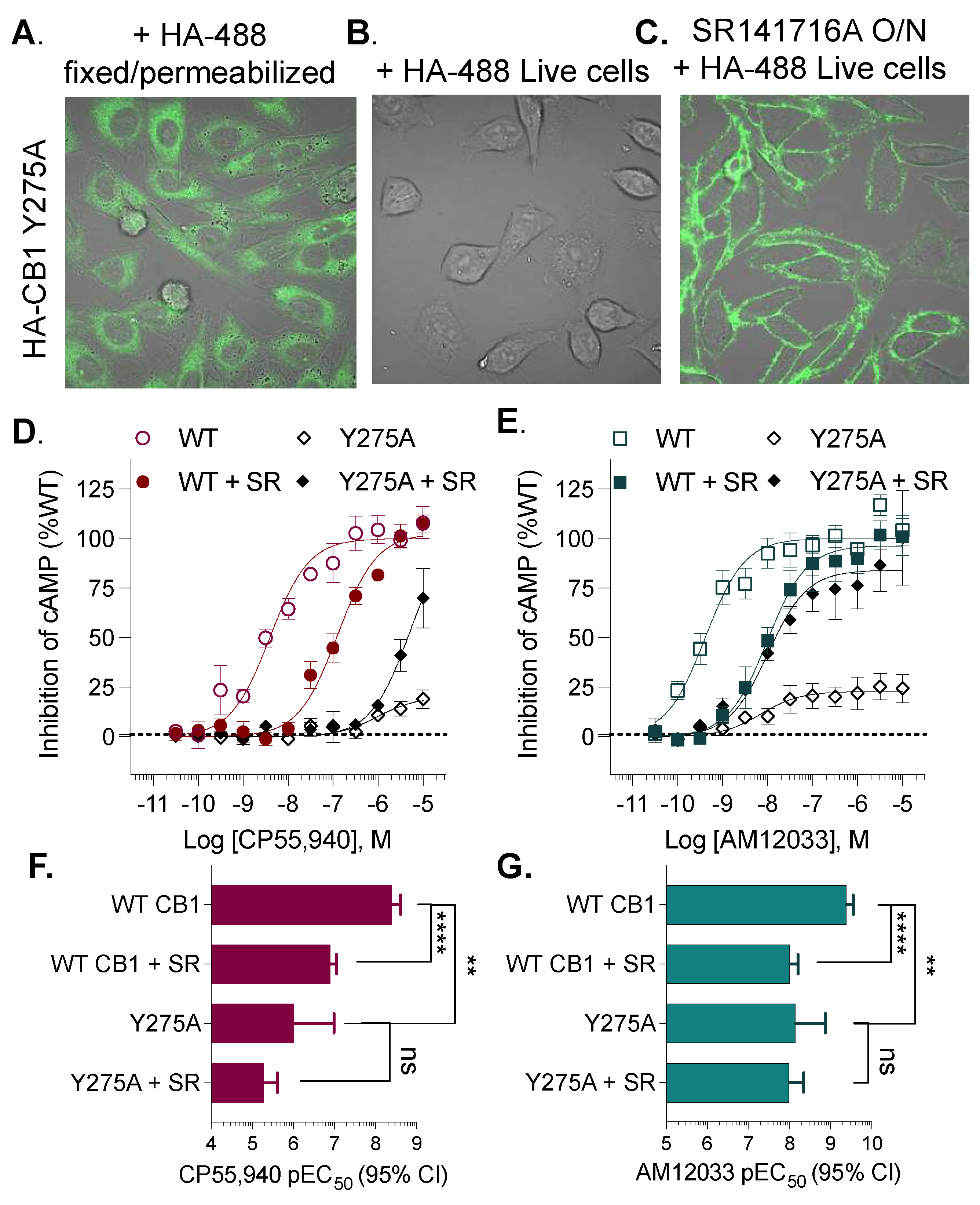
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lowe, H.; Toyang, N.; Steele, B.; Bryant, J.; Ngwa, W. The Endocannabinoid System: A Potential Target for the Treatment of Various Diseases. Int. J. Mol. Sci. 2021, 22, 9472. [Google Scholar] [CrossRef] [PubMed]
- Aizpurua-Olaizola, O.; Elezgarai, I.; Rico-Barrio, I.; Zarandona, I.; Etxebarria, N.; Usobiaga, A. Targeting the endocannabinoid system: Future therapeutic strategies. Drug Discov. Today 2017, 22, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A. Cannabinoid inhibition of adenylate cyclase. Biochemistry of the response in neuroblastoma cell membranes. Mol. Pharmacol. 1985, 27, 429–436. [Google Scholar] [PubMed]
- Priestley, R.; Glass, M.; Kendall, D. Functional selectivity at cannabinoid receptors. Adv. Pharmacol. 2017, 80, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Katona, I.; Freund, T.F. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat. Med. 2008, 14, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Kunos, G.; Osei-Hyiaman, D.; Bátkai, S.; Sharkey, K.A.; Makriyannis, A. Should peripheral CB(1) cannabinoid receptors be selectively targeted for therapeutic gain? Trends Pharmacol. Sci. 2009, 30, 1–7. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Peigneur, S.; Hendrickx, L.A.; Tytgat, J. Targeting Cannabinoid Receptors: Current Status and Prospects of Natural Products. Int. J. Mol. Sci. 2020, 21, 5064. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yan, Q.; Xie, J.; Liu, Z.; Liu, F.; Liu, Y.; Zhou, S.; Pan, S.; Liu, D.; Duan, J.; et al. The intervention of cannabinoid receptor in chronic and acute kidney disease animal models: A systematic review and meta-analysis. Diabetol. Metab. Syndr. 2024, 16, 45. [Google Scholar] [CrossRef] [PubMed]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.J.; Xi, Z.-X. Progress in brain cannabinoid CB2 receptor research: From genes to behavior. Neurosci. Biobehav. Rev. 2019, 98, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Lynn, A.B.; Herkenham, M. Localization of cannabinoid receptors and nonsaturable high-density cannabinoid binding sites in peripheral tissues of the rat: Implications for receptor-mediated immune modulation by cannabinoids. J. Pharmacol. Exp. Ther. 1994, 268, 1612–1623. [Google Scholar] [PubMed]
- Schatz, A.R.; Lee, M.; Condie, R.B.; Pulaski, J.T.; Kaminski, N.E. Cannabinoid receptors CB1 and CB2: A characterization of expression and adenylate cyclase modulation within the immune system. Toxicol. Appl. Pharmacol. 1997, 142, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Gálvez, Y.; Palomo-Garo, C.; Fernández-Ruiz, J.; García, C. Potential of the cannabinoid CB(2) receptor as a pharmacological target against inflammation in Parkinson’s disease. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2016, 64, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Aymerich, M.S.; Aso, E.; Abellanas, M.A.; Tolon, R.M.; Ramos, J.A.; Ferrer, I.; Romero, J.; Fernández-Ruiz, J. Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem. Pharmacol. 2018, 157, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Mboumba Bouassa, R.S.; Sebastiani, G.; Di Marzo, V.; Jenabian, M.A.; Costiniuk, C.T. Cannabinoids and Chronic Liver Diseases. Int. J. Mol. Sci. 2022, 23, 9423. [Google Scholar] [CrossRef] [PubMed]
- LoVerme, J.; Duranti, A.; Tontini, A.; Spadoni, G.; Mor, M.; Rivara, S.; Stella, N.; Xu, C.; Tarzia, G.; Piomelli, D. Synthesis and characterization of a peripherally restricted CB1 cannabinoid antagonist, URB447, that reduces feeding and body-weight gain in mice. Bioorg. Med. Chem. Lett. 2009, 19, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Bi, G.-H.; Galaj, E.; He, Y.; Xi, Z.-X. Cannabidiol inhibits sucrose self-administration by CB1 and CB2 receptor mechanisms in rodents. Addict. Biol. 2020, 25, e12783. [Google Scholar] [CrossRef] [PubMed]
- Rohbeck, E.; Eckel, J.; Romacho, T. Cannabinoid Receptors in Metabolic Regulation and Diabetes. Physiology 2021, 36, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Barutta, F.; Grimaldi, S.; Gambino, R.; Vemuri, K.; Makriyannis, A.; Annaratone, L.; di Marzo, V.; Bruno, G.; Gruden, G. Dual therapy targeting the endocannabinoid system prevents experimental diabetic nephropathy. Nephrol. Dial. Transplant. 2017, 32, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Hidalgo, B.; García-Martín, A.; Muñoz, E.; González-Mariscal, I. Detrimental Effect of Cannabidiol on the Early Onset of Diabetic Nephropathy in Male Mice. Pharmaceuticals 2021, 14, 863. [Google Scholar] [CrossRef] [PubMed]
- Montero, C.; Campillo, N.E.; Goya, P.; Paez, J.A. Homology models of the cannabinoid CB1 and CB2 receptors. A docking analysis study. Eur. J. Med. Chem. 2005, 40, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Ashton, J.C.; Wright, J.L.; McPartland, J.M.; Tyndall, J.D. Cannabinoid CB1 and CB2 receptor ligand specificity and the development of CB2-selective agonists. Curr. Med. Chem. 2008, 15, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Ji, B.; Fang, J.; Wang, J.; Li, J.; Wang, J. Discovery of Potent and Selective CB2 Agonists Utilizing a Function-Based Computational Screening Protocol. ACS Chem. Neurosci. 2023, 14, 3941–3958. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.R.; Tang, J.Q.; Zhang, W.N.; Zhuang, C.L.; Shi, Y. Rational drug design of CB2 receptor ligands: From 2012 to 2021. RSC Adv. 2022, 12, 35242–35259. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB(1). Nature 2017, 547, 468–471. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, D.C.; Perry, E.; MacDougall, L.; Ammerman, Y.; Cooper, T.; Wu, Y.T.; Braley, G.; Gueorguieva, R.; Krystal, J.H. The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: Implications for psychosis. Neuropsychopharmacology 2004, 29, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Brust, C.A.; Nikas, S.P.; Song, F. Activation and signaling mechanism revealed by cannabinoid receptor-Gi complex structures. Cell 2020, 180, 655–665.e18. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chang, H.; Bouma, J.; de Paus, L.V.; Mukhopadhyay, P.; Paloczi, J.; Mustafa, M.; van der Horst, C.; Kumar, S.S.; Wu, L.; et al. Structural basis of selective cannabinoid CB2 receptor activation. Nat. Commun. 2023, 14, 1447. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Wang, C.; Katritch, V.; Han, G.W.; Huang, X.P.; Vardy, E.; McCorvy, J.D.; Jiang, Y.; Chu, M.; Siu, F.Y.; et al. Structural features for functional selectivity at serotonin receptors. Science 2013, 340, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Trzaskowski, B.; Latek, D.; Yuan, S.; Ghoshdastider, U.; Debinski, A.; Filipek, S. Action of molecular switches in GPCRs—Theoretical and experimental studies. Curr. Med. Chem. 2012, 19, 1090–1109. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Kooistra, A.J.; Munk, C.; Heydenreich, F.M.; Veprintsev, D.B.; Bouvier, M.; Babu, M.M.; Gloriam, D.E. GPCR activation mechanisms across classes and macro/microscales. Nat. Struct. Mol. Biol. 2021, 28, 879–888. [Google Scholar] [CrossRef] [PubMed]
- White, K.L.; Eddy, M.T.; Gao, Z.G.; Han, G.W.; Lian, T.; Deary, A.; Patel, N.; Jacobson, K.A.; Katritch, V.; Stevens, R.C. Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling. Structure 2018, 26, 259–269.e5. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Shapovalov, M.V.; Dunbrack, R.L., Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB(1). Cell 2016, 167, 750–762.e14. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hua, T.; Vemuri, K.; Ho, J.-H.; Wu, Y.; Wu, L.; Popov, P.; Benchama, O.; Zvonok, N.; Qu, L. Crystal structure of the human cannabinoid receptor CB2. Cell 2019, 176, 459–467.e13. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors. In Methods in Neurosciences; Elsevier: Amsterdam, The Netherlands, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Chin, C.-n.; Murphy, J.W.; Huffman, J.W.; Kendall, D.A. The third transmembrane helix of the cannabinoid receptor plays a role in the selectivity of aminoalkylindoles for CB2, peripheral cannabinoid receptor. J. Pharmacol. Exp. Ther. 1999, 291, 837–844. [Google Scholar] [PubMed]
- McAllister, S.D.; Tao, Q.; Barnett-Norris, J.; Buehner, K.; Hurst, D.P.; Guarnieri, F.; Reggio, P.H.; Harmon, K.W.N.; Cabral, G.A.; Abood, M.E. A critical role for a tyrosine residue in the cannabinoid receptors for ligand recognition. Biochem. Pharmacol. 2002, 63, 2121–2136. [Google Scholar] [CrossRef] [PubMed]
- Porcu, A.; Melis, M.; Turecek, R.; Ullrich, C.; Mocci, I.; Bettler, B.; Gessa, G.L.; Castelli, M.P. Rimonabant, a potent CB1 cannabinoid receptor antagonist, is a Galpha(i/o) protein inhibitor. Neuropharmacology 2018, 133, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi-Carmona, M.; Barth, F.; Héaulme, M.; Shire, D.; Calandra, B.; Congy, C.; Martinez, S.; Maruani, J.; Néliat, G.; Caput, D.; et al. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994, 350, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.; Brown, S.; Derleth, C.; Mackie, K. Internalization and recycling of the CB1 cannabinoid receptor. J. Neurochem. 1999, 73, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Coutts, A.A.; Anavi-Goffer, S.; Ross, R.A.; MacEwan, D.J.; Mackie, K.; Pertwee, R.G.; Irving, A.J. Agonist-Induced Internalization and Trafficking of Cannabinoid CB1 Receptors in Hippocampal Neurons. J. Neurosci. 2001, 21, 2425–2433. [Google Scholar] [CrossRef] [PubMed]
- Farrens, D.L.; Altenbach, C.; Yang, K.; Hubbell, W.L.; Khorana, H.G. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 1996, 274, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; Shakhnovich, E.I.; et al. Common activation mechanism of class A GPCRs. Elife 2019, 8, e50279. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Mercier, R.W.; Anday, J.K.; Thakur, G.A.; Zvonok, A.M.; Hurst, D.; Reggio, P.H.; Janero, D.R.; Makriyannis, A. Ligand-binding architecture of human CB2 cannabinoid receptor: Evidence for receptor subtype-specific binding motif and modeling GPCR activation. Chem. Biol. 2008, 15, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Picone, R.P.; Khanolkar, A.D.; Xu, W.; Ayotte, L.A.; Thakur, G.A.; Hurst, D.P.; Abood, M.E.; Reggio, P.H.; Fournier, D.J.; Makriyannis, A. (-)-7′-Isothiocyanato-11-hydroxy-1′, 1′-dimethylheptylhexahydrocannabinol (AM841), a high-affinity electrophilic ligand, interacts covalently with a cysteine in helix six and activates the CB1 cannabinoid receptor. Mol. Pharmacol. 2005, 68, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, D.W.; Papanastasiou, M.; Melchior, K.; Zvonok, N.; Mercier, R.W.; Janero, D.R.; Thakur, G.A.; Cha, S.; Wu, B.; Karger, B. Mass spectrometry-based proteomics of human cannabinoid receptor 2: Covalent cysteine 6.47 (257)-ligand interaction affording megagonist receptor activation. J. Proteome Res. 2011, 10, 4789–4798. [Google Scholar] [CrossRef] [PubMed]
- Kapur, A.; Hurst, D.P.; Fleischer, D.; Whitnell, R.; Thakur, G.A.; Makriyannis, A.; Reggio, P.H.; Abood, M.E. Mutation Studies of Ser7.39 and Ser2.60 in the Human CB1 Cannabinoid Receptor: Evidence for a Serine-Induced Bend in CB1Transmembrane Helix 7. Mol. Pharmacol. 2007, 71, 1512–1524. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.-Y.; Bertalovitz, A.C.; Kendall, D.A. Identification of essential cannabinoid-binding domains: Structural insights into early dynamic events in receptor activation. J. Biol. Chem. 2011, 286, 33422–33435. [Google Scholar] [CrossRef]
- Black, J.W.; Leff, P. Operational models of pharmacological agonism. Proc. R. Soc. Lond. B Biol. Sci. 1983, 220, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Shire, D.; Calandra, B.; Bouaboula, M.; Barth, F.; Rinaldi-Carmona, M.; Casellas, P.; Ferrara, P. Cannabinoid receptor interactions with the antagonists SR 141716A and SR 144528. Life Sci. 1999, 65, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Shire, D.; Calandra, B.; Delpech, M.; Dumont, X.; Kaghad, M.; Le Fur, G.; Caput, D.; Ferrara, P. Structural features of the central cannabinoid CB1 receptor involved in the binding of the specific CB1 antagonist SR 141716A. J. Biol. Chem. 1996, 271, 6941–6946. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.H.; Slowey, C.A.; Hurst, D.P.; Reggio, P.H. The difference between the CB(1) and CB(2) cannabinoid receptors at position 5.46 is crucial for the selectivity of WIN55212-2 for CB(2). Mol. Pharmacol. 1999, 56, 834–840. [Google Scholar] [PubMed]
- Wilbanks, A.M.; Laporte, S.A.; Bohn, L.M.; Barak, L.S.; Caron, M.G. Apparent loss-of-function mutant GPCRs revealed as constitutively desensitized receptors. Biochemistry 2002, 41, 11981–11989. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Soto, M.; Verma, R.K.; Willette, B.K.A.; Gonye, E.C.; Moore, A.M.; Moritz, A.E.; Boateng, C.A.; Yano, H.; Free, R.B.; Shi, L.; et al. A structural basis for how ligand binding site changes can allosterically regulate GPCR signaling and engender functional selectivity. Sci. Signal 2020, 13, eaaw5885. [Google Scholar] [CrossRef] [PubMed]
- Hurst, D.P.; Grossfield, A.; Lynch, D.L.; Feller, S.; Romo, T.D.; Gawrisch, K.; Pitman, M.C.; Reggio, P.H. A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J. Biol. Chem. 2010, 285, 17954–17964. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.L.; Reggio, P.H. Cannabinoid CB1 receptor recognition of endocannabinoids via the lipid bilayer: Molecular dynamics simulations of CB1 transmembrane helix 6 and anandamide in a phospholipid bilayer. J. Comput. Aided Mol. Des. 2006, 20, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Fay, J.F.; Dunham, T.D.; Farrens, D.L. Cysteine Residues in the Human Cannabinoid Receptor: Only C257 and C264 Are Required for a Functional Receptor, and Steric Bulk at C386 Impairs Antagonist SR141716A Binding. Biochemistry 2005, 44, 8757–8769. [Google Scholar] [CrossRef] [PubMed]
- Daigle, T.L.; Kwok, M.L.; Mackie, K. Regulation of CB1 cannabinoid receptor internalization by a promiscuous phosphorylation-dependent mechanism. J. Neurochem. 2008, 106, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Atwood, B.K.; Wager-Miller, J.; Haskins, C.; Straiker, A.; Mackie, K. Functional selectivity in CB(2) cannabinoid receptor signaling and regulation: Implications for the therapeutic potential of CB(2) ligands. Mol. Pharmacol. 2012, 81, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Carrier, E.J.; Kearn, C.S.; Barkmeier, A.J.; Breese, N.M.; Yang, W.; Nithipatikom, K.; Pfister, S.L.; Campbell, W.B.; Hillard, C.J. Cultured rat microglial cells synthesize the endocannabinoid 2-arachidonylglycerol, which increases proliferation via a CB2 receptor-dependent mechanism. Mol. Pharmacol. 2004, 65, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Bouaboula, M.; Dussossoy, D.; Casellas, P. Regulation of peripheral cannabinoid receptor CB2 phosphorylation by the inverse agonist SR 144528. Implications for receptor biological responses. J. Biol. Chem. 1999, 274, 20397–20405. [Google Scholar] [CrossRef] [PubMed]
- Irannejad, R.; von Zastrow, M. GPCR signaling along the endocytic pathway. Curr. Opin. Cell Biol. 2014, 27, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, R.; Devi, L.A. Regulation of CB1 cannabinoid receptor trafficking by the adaptor protein AP-3. FASEB J. 2008, 22, 2311–2322. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, G.C.; Deliu, E.; Marcu, J.; Hoffman, N.E.; Console-Bram, L.; Zhao, P.; Madesh, M.; Abood, M.E.; Brailoiu, E. Differential Activation of Intracellular versus Plasmalemmal CB2 Cannabinoid Receptors. Biochemistry 2014, 53, 4990–4999. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, G.C.; Oprea, T.I.; Zhao, P.; Abood, M.E.; Brailoiu, E. Intracellular cannabinoid type 1 (CB1) receptors are activated by anandamide. J. Biol. Chem. 2011, 286, 29166–29174. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CP55,940 | AM12033 | |||||
|---|---|---|---|---|---|---|
| CB1 | EC50, nM | (95% CI) | vs. CB1 | EC50, nM | (95% CI) | vs. CB1 |
| WT CB1 | 1.97 | (1.52–2.54) | 0.137 | (0.0853–0.219) | ||
| F170A2.57 | 10368 | (7440–14779) | **** | 4.07 | (2.82–5.86) | **** |
| F174A2.61 | 3072 | (2126–4427 | **** | 1.90 | (1.24–2.89) | **** |
| F177A2.64 | >10,000 | **** | 9.76 | (5.75–16.3) | **** | |
| L193A3.29 | 549 | (405–744) | **** | 0.139 | (0.0761–0.247 | |
| Y275A5.39 | >10,000 | **** | 25 | (4–130) | *** | |
| C355A6.47 | 0.870 | (0.576–1.315) | ** | 0.100 | (0.0553–0.177) | |
| F379A7.35 | 946 | (760–1173) | **** | 0.042 | (0.0241–0.0718) | ** |
| S383A7.39 | 713 | (471–1075) | **** | 0.685 | (0.322–1.39) | ** |
| CP55,940 | AM12033 | |||||
| CB2 | EC50, nM | (95% CI) | vs. CB2 | EC50, nM | (95% CI) | vs. CB2 |
| WT CB2 | 1.17 | (0.681–1.99) | 0.393 | (0.217–0.705) | ||
| F87A2.57 | 388 | (198–735) | **** | 3.71 | (1.65–8.12) | **** |
| F91A2.61 | 146 | (71–282) | **** | 0.686 | (0.389–1.19 | |
| F94A2.64 | 139 | (93–207) | **** | 0.532 | (0.289–0.944) | |
| I110A3.29 | 22 | (7–64) | **** | 0.233 | (0.073–0.698) | |
| Y190A5.39 | 1.13 | (0.770–1.65) | 0.679 | (0.437–1.04) | ||
| C257A6.47 | 23 | (13–38) | *** | 0.606 | (0.264–1.36) | |
| F281A7.35 | 100 | (34–269) | **** | 0.586 | (0.189–1.76) | |
| S285A7.39 | 424 | (147–1160) | **** | 0.856 | (0.538–1.35) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brust, C.A.; Swanson, M.A.; Tsoutsouvas, C.I.; Dimova, S.T.; Dang, V.Q.; Stahl, E.L.; Ho, J.-H.; Nikas, S.P.; Makriyannis, A.; Bohn, L.M. Comparison of Agonist Activity between CB1 and CB2 Receptors with Orthosteric Site Mutations. Receptors 2024, 3, 380-396. https://doi.org/10.3390/receptors3030018
Brust CA, Swanson MA, Tsoutsouvas CI, Dimova ST, Dang VQ, Stahl EL, Ho J-H, Nikas SP, Makriyannis A, Bohn LM. Comparison of Agonist Activity between CB1 and CB2 Receptors with Orthosteric Site Mutations. Receptors. 2024; 3(3):380-396. https://doi.org/10.3390/receptors3030018
Chicago/Turabian StyleBrust, Christina A., Matthew A. Swanson, Christos Iliopoulos Tsoutsouvas, Snezana T. Dimova, Vuong Q. Dang, Edward L. Stahl, Jo-Hao Ho, Spyros P. Nikas, Alexandros Makriyannis, and Laura M. Bohn. 2024. "Comparison of Agonist Activity between CB1 and CB2 Receptors with Orthosteric Site Mutations" Receptors 3, no. 3: 380-396. https://doi.org/10.3390/receptors3030018
APA StyleBrust, C. A., Swanson, M. A., Tsoutsouvas, C. I., Dimova, S. T., Dang, V. Q., Stahl, E. L., Ho, J.-H., Nikas, S. P., Makriyannis, A., & Bohn, L. M. (2024). Comparison of Agonist Activity between CB1 and CB2 Receptors with Orthosteric Site Mutations. Receptors, 3(3), 380-396. https://doi.org/10.3390/receptors3030018