

The Role of Substance P and NK1 Receptors in Mild to Severe Traumatic Brain Injury: From CTE to ICP


Abstract
:1. Introduction
2. Mild TBI
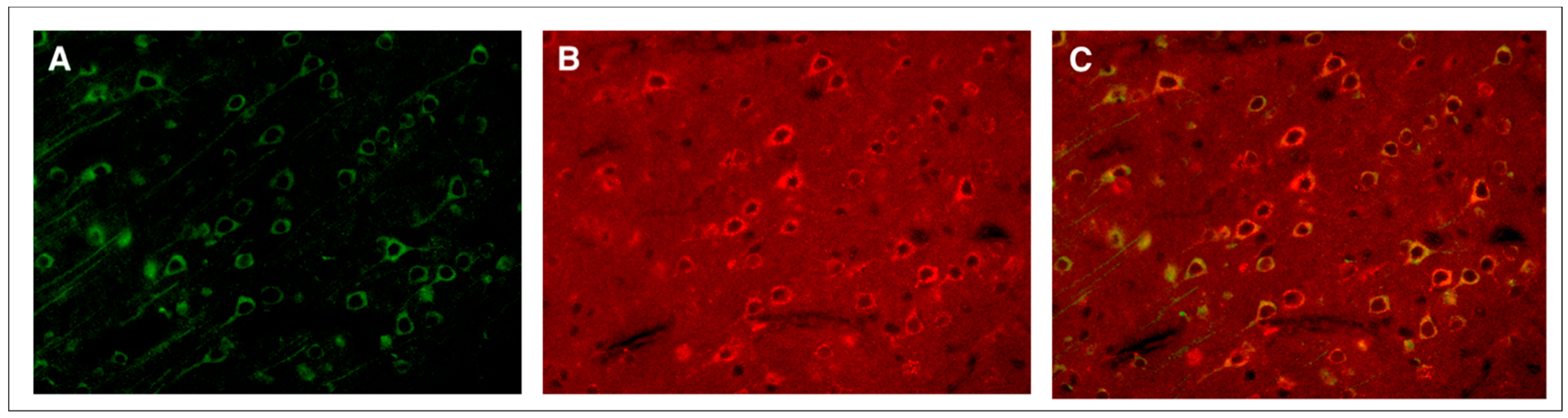
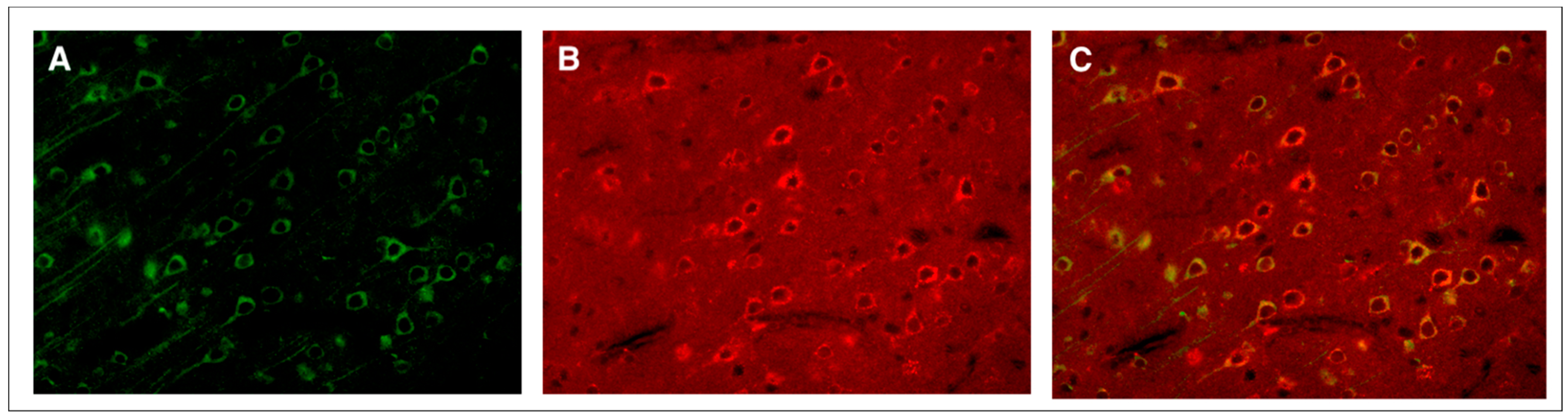
3. Moderate TBI
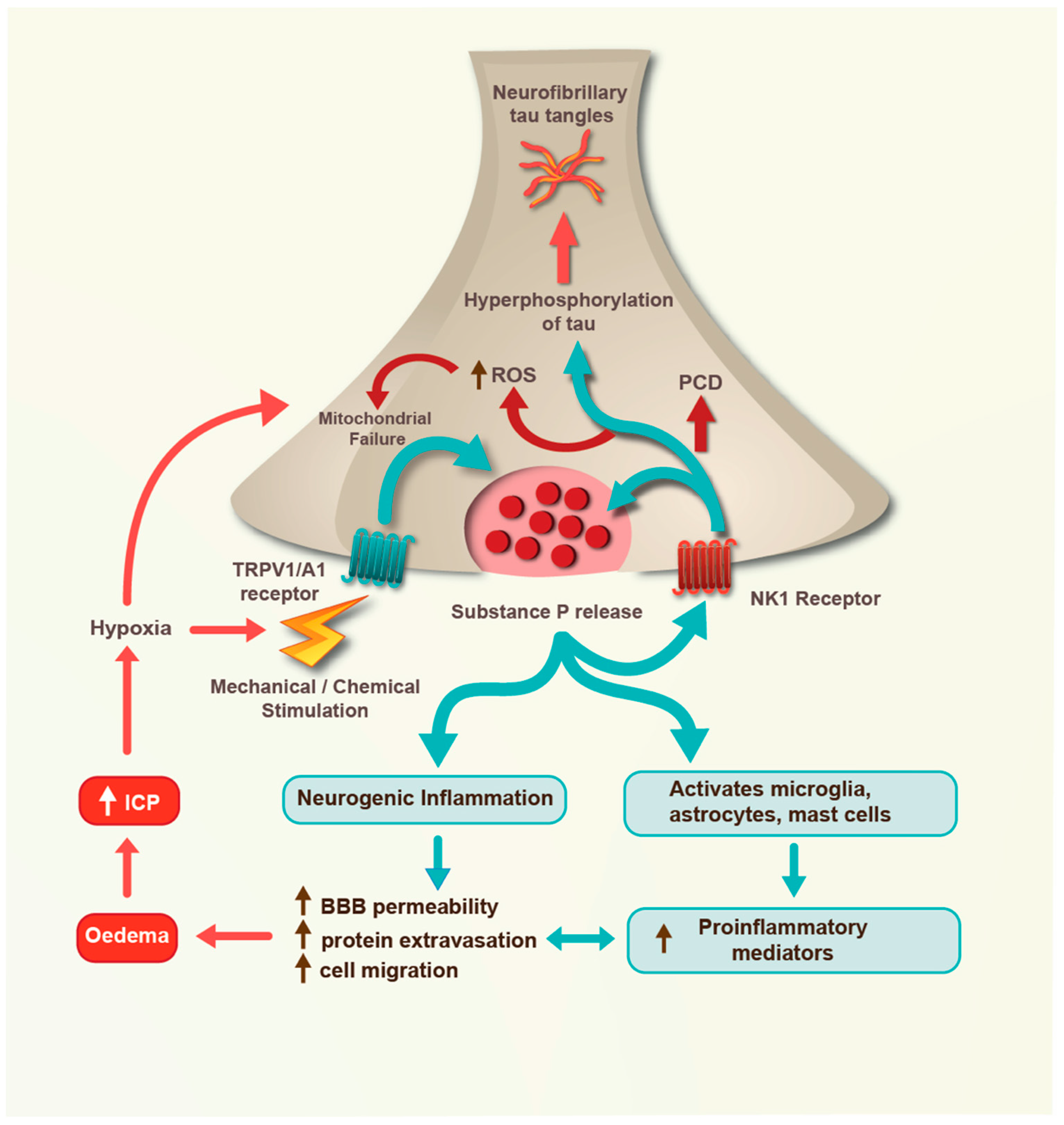
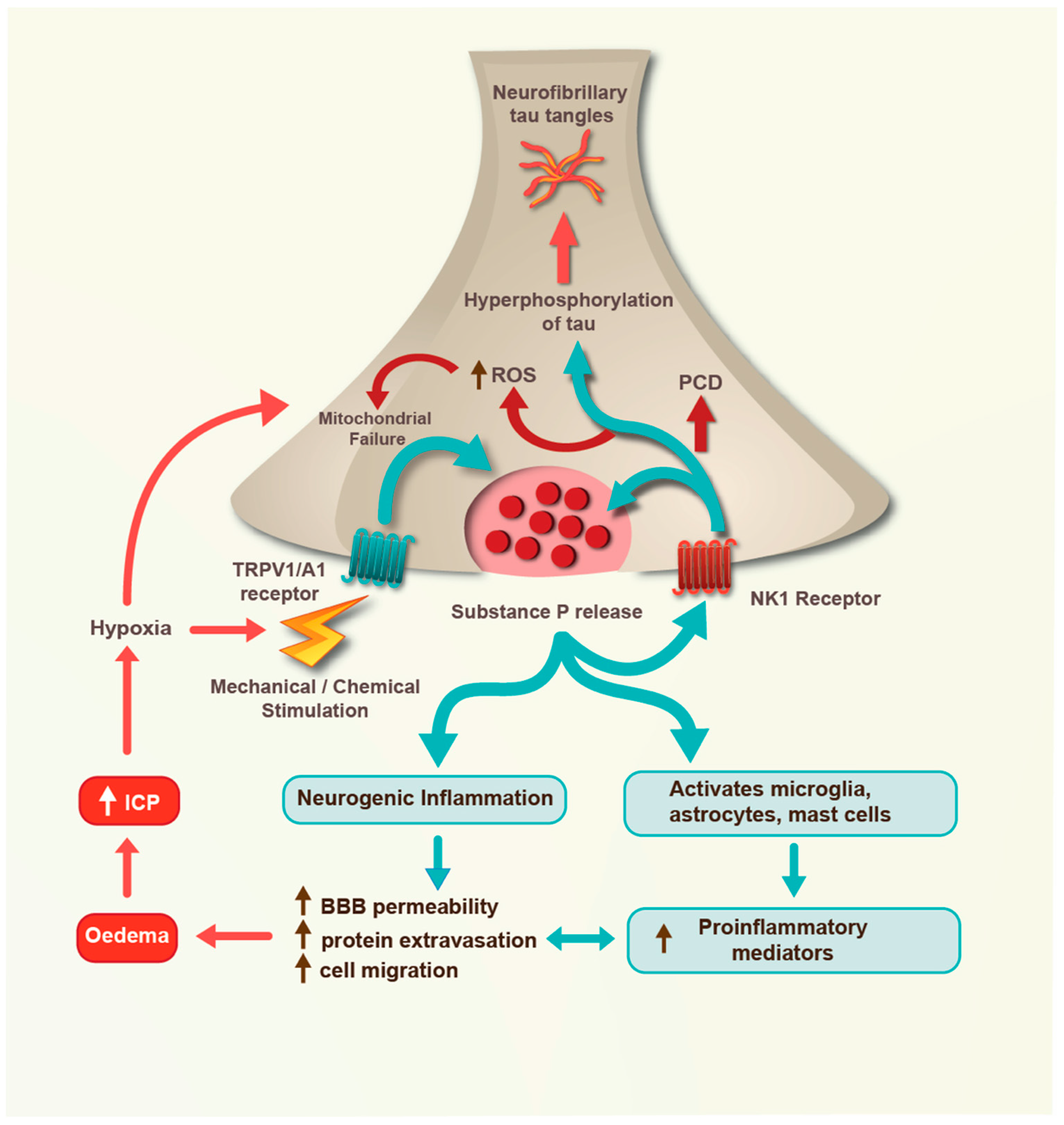
4. Severe TBI
5. Conclusions and Future Directions

{kind=link}
{kind=link}
| Condition | Species | Model | Notable Observations | Citation |
|---|---|---|---|---|
| Mild TBI | Rat | Single weight drop | Increased perivascular and astrocytic SP immunoreactivity | Donkin et al., 2004 [13] |
| Rat | Repeated weight drop | Neuronal tau phosphorylation after injury is inhibited by NK1 antagonist | Corrigan et al., 2021 [18] | |
| Mouse | Blast | NK1 antagonist reduced tau phosphorylation at 28 days after injury and improved functional outcome | Corrigan et al., 2021 [18] | |
| Moderate TBI | Rat | Weight drop | Capsaicin pre-treatment reduces brain oedema and functional deficits | Vink et al., 2003 [27]; Nimmo et al., 2004 [26] |
| Rat | Weight drop | NK1 antagonist increases brain free magnesium after TBI | Vink et al., 2004 [43] | |
| Rat | Weight drop | Increased brain SP immunoreactivity and serum SP after injury; NK1 antagonist reduces post-traumatic BBB permeability and oedema, and improves functional outcome | Donkin et al., 2009 [30] | |
| Rat | Weight drop | NK1 antagonist reduces axonal injury and improves functional outcome even when administered up to 12 h post-trauma | Donkin et al., 2011 [29] | |
| Rat | Weight drop | NK1 antagonist reduces axonal injury and improve functional outcome in female animals | Corrigan et al., 2012 [28] | |
| Rat | Weight drop | NK1 antagonists inhibit microglial proliferation | Carthew et al., 2012 [72] | |
| Mouse | Cortical impact | NK1 antagonist and SP deletion inhibits post-traumatic oxidative stress, apoptosis, neuroinflammation, and cell death; improves functional outcome | Li et al., 2019 [40] | |
| Severe TBI | Human | Clinical | Perivascular SP immunoreactivity is increased after trauma and may be released in response to neuronal stress | Zacest et al., 2010 [31] |
| Sheep | Captive bolt | NK1 antagonist reduces post-traumatic elevated ICP | Gabrielian et al., 2013 [50] | |
| Sheep | Captive bolt | NK1 antagonist reduces post-traumatic elevated ICP and improves brain oxygen | Gabrielian et al., 2017 [51] | |
| Human | Clinical | Elevated serum SP associated with injury severity and mortality in adults | Lorente et al., 2015 [44]; 2017 [45] | |
| Human | Clinical | Elevated post-traumatic serum SP in children | Zhou et al., 2021 [46] | |
| Human | Clinical | Elevated serum SP part of a multi-staged neuropeptide response after TBI | Alves et al., 2022 [47] |
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Centers for Disease Control and Prevention. Leading Causes of Death and Injury. Available online: https://www.cdc.gov/injury/wisqars/leadingcauses.html (accessed on 25 June 2023).
- Goldstein, M. Traumatic brain injury: A silent epidemic. Ann. Neurol. 1990, 27, 327. [Google Scholar] [CrossRef] [PubMed]
- Nowinski, C.J.; Bureau, S.C.; Buckland, M.E.; Curtis, M.A.; Daneshvar, D.H.; Faull, R.L.M.; Grinberg, L.T.; Hill-Yardin, E.L.; Murray, H.C.; Pearce, A.J.; et al. Applying the Bradford Hill criteria for causation to repetitive head impacts and chronic traumatic tncephalopathy. Front. Neurol. 2022, 13, 938163. [Google Scholar] [CrossRef] [PubMed]
- Stocchetti, N.; Carbonara, M.; Citerio, G.; Ercole, A.; Skrifvars, M.B.; Smielewski, P.; Zoerle, T.; Menon, D.K. Severe traumatic brain injury: Targeted management in the intensive care unit. Lancet Neurol. 2017, 16, 452–464. [Google Scholar] [CrossRef]
- Hokfelt, T.; Pernow, B.; Wahren, J. Substance P: A pioneer amongst neuropeptides. J. Intern. Med. 2001, 249, 27–40. [Google Scholar] [CrossRef]
- Coveñas, R.; Rodriguez, F.D.; Muñoz, M. The Neurokinin-1 receptor: A promising antitumor target. Receptors 2022, 1, 72–97. [Google Scholar] [CrossRef]
- Almeida, T.A.; Rojo, J.; Nieto, P.M.; Pinto, F.M.; Hernandez, M.; Martin, J.D.; Candenas, M.L. Tachykinins and tachykinin receptors: Structure and activity relationships. Curr. Med. Chem. 2004, 11, 2045–2081. [Google Scholar] [CrossRef]
- Saria, A. The tachykinin NK1 receptor in the brain: Pharmacology and putative functions. Eur. J. Pharmacol. 1999, 375, 51–60. [Google Scholar] [CrossRef]
- Caberlotto, L.; Hurd, Y.L.; Murdock, P.; Wahlin, J.P.; Melotto, S.; Corsi, M.; Carletti, R. Neurokinin 1 receptor and relative abundance of the short and long isoforms in the human brain. Eur. J. Neurosci. 2003, 17, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Coveñas, R. Neurokinin/Tachykinin receptors. In Encyclopedia of Molecular Pharmacology, 3rd ed.; Offermanns, S., Rosenthal, W., Eds.; Springer: Cham, Switzerland, 2021; pp. 1093–1103. [Google Scholar]
- Iverson, G.L.; Gardner, A.J.; Terry, D.P.; Ponsford, J.L.; Sills, A.K.; Broshek, D.K.; Solomon, G.S. Predictors of clinical recovery from concussion: A systematic review. Br. J. Sports Med. 2017, 51, 941–948. [Google Scholar] [CrossRef]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.S.; Kubilus, C.A.; Stern, R.A. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef]
- Donkin, J.J.; Cernak, I.; Rodgers, K.M.; Vink, R. Mild concussive head injury results in increased brain substance P immunoreactivity. In 7th International Neurotrauma Symposium; Medimond International Proceedings: Bologna, Italy, 2004; pp. 75–78. [Google Scholar]
- Campos, M.M.; Calixto, J.B. Neurokinin mediation of edema and inflammation. Neuropeptides 2000, 34, 314–322. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Stein, T.D.; Kiernan, P.T.; Alvarez, V.E. The neuropathology of chronic traumatic encephalopathy. Brain Pathol. 2015, 25, 350–364. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Stein, T.D.; Huber, B.R.; Crary, J.F.; Bieniek, K.; Dickson, D.; Alvarez, V.E.; Cherry, J.D.; Farrell, K.; Butler, M.; et al. Chronic traumatic encephalopathy (CTE): Criteria for neuropathological diagnosis and relationship to repetitive head impacts. Acta Neuropathol. 2023, 145, 371–394. [Google Scholar] [CrossRef]
- O’Keeffe, E.; Kelly, E.; Liu, Y.; Giordano, C.; Wallace, E.; Hynes, M.; Tiernan, S.; Meagher, A.; Greene, C.; Hughes, S.; et al. Dynamic blood-brain barrier regulation in mild traumatic brain injury. J. Neurotrauma 2020, 37, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, F.; Cernak, I.; McAteer, K.; Hellewell, S.C.; Rosenfeld, J.V.; Turner, R.J.; Vink, R. NK1 antagonists attenuate tau phosphorylation after blast and repeated concussive injury. Sci. Rep. 2021, 11, 8861. [Google Scholar] [CrossRef]
- Hoffmann, T.; Nimmo, A.J.; Sleight, A.; Vankan, P.; Vink, R. Use of NK-1 Receptor Antagonists with Pyridinic Structure, for the Treatment of Brain, Spinal or Nerve Injury. WO2003006016A3, 2003. [Google Scholar]
- Fiebich, B.L.; Schleicher, S.; Butcher, R.D.; Craig, A.; Lieb, K. The neuropeptide substance P activates p38 mitogen-activated protein kinase resulting in IL-6 expression independently from NF-kappa B. J. Immunol. 2000, 165, 5606–5611. [Google Scholar] [CrossRef]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef]
- Sun, J.; Ramnath, R.D.; Tamizhselvi, R.; Bhatia, M. Role of protein kinase C and phosphoinositide 3-kinase-Akt in substance P-induced proinflammatory pathways in mouse macrophages. FASEB J. 2009, 23, 997–1010. [Google Scholar] [CrossRef]
- Mantyh, P.W. Neurobiology of substance P and the NK1 receptor. J. Clin. Psychiatry 2002, 63, 6–10. [Google Scholar]
- Collins-Praino, L.; Gutschmidt, D.; Sharkey, J.; Arulsamy, A.; Corrigan, F. Temporal changes in tau phosphorylation and related kinase and phosphatases following two models of traumatic brain injury. J. Neuroinflamm. Neurodegener. Dis. 2018, 2, 100007. [Google Scholar]
- Murphy, J.E.; Roosterman, D.; Cottrell, G.S.; Padilla, B.E.; Feld, M.; Brand, E.; Cedron, W.J.; Bunnett, N.W.; Steinhoff, M. Protein phosphatase 2A mediates resensitization of the neurokinin 1 receptor. Am. J. Physiol. Cell Physiol. 2011, 301, C780–C791. [Google Scholar] [CrossRef]
- Nimmo, A.J.; Cernak, I.; Heath, D.L.; Hu, X.; Bennett, C.J.; Vink, R. Neurogenic inflammation is associated with development of edema and functional deficits following traumatic brain injury in rats. Neuropeptides 2004, 38, 40–47. [Google Scholar] [CrossRef]
- Vink, R.; Young, A.; Bennett, C.J.; Hu, X.; Connor, C.O.; Cernak, I.; Nimmo, A.J. Neuropeptide release influences brain edema formation after diffuse traumatic brain injury. Acta Neurochir. Suppl. 2003, 86, 257–260. [Google Scholar] [PubMed]
- Corrigan, F.; Leonard, A.; Ghabriel, M.; Van Den Heuvel, C.; Vink, R. A substance P antagonist improves outcome in female Sprague Dawley rats following diffuse traumatic brain injury. CNS Neurosci. Ther. 2012, 18, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Donkin, J.J.; Cernak, I.; Blumberg, P.C.; Vink, R. A substance P antagonist reduces axonal injury and improves neurologic outcome when administered up to 12 hours after traumatic brain injury. J. Neurotrauma 2011, 28, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Donkin, J.J.; Nimmo, A.J.; Cernak, I.; Blumbergs, P.C.; Vink, R. Substance P is associated with the development of brain edema and functional deficits after traumatic brain injury. J. Cereb. Blood Flow. Metab. 2009, 29, 1388–1398. [Google Scholar] [CrossRef]
- Zacest, A.C.; Vink, R.; Manavis, J.; Sarvestani, G.T.; Blumbergs, P.C. Substance P immunoreactivity increases following human traumatic brain injury. Acta Neurochir. Suppl. 2010, 106, 211–216. [Google Scholar]
- Donkin, J.J.; Vink, R. Mechanisms of cerebral edema in traumatic brain injury: Therapeutic developments. Curr. Opin. Neurol. 2010, 23, 293–299. [Google Scholar] [CrossRef]
- Hooper, C.; Pinteaux-Jones, F.; Fry, V.A.; Sevastou, I.G.; Baker, D.; Heales, S.J.; Pocock, J.M. Differential effects of albumin on microglia and macrophages; implications for neurodegeneration following blood-brain barrier damage. J. Neurochem. 2009, 109, 694–705. [Google Scholar] [CrossRef]
- Corrigan, F.; Mander, K.A.; Leonard, A.V.; Vink, R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J. Neuroinflammation 2016, 13, 264. [Google Scholar] [CrossRef] [PubMed]
- Safwat, A.; Helmy, A.; Gupta, A. The role of substance P within traumatic brain injury and implications for therapy. J. Neurotrauma 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Seabrook, G.R.; Shepheard, S.L.; Williamson, D.J.; Tyrer, P.; Rigby, M.; Cascieri, M.A.; Harrison, T.; Hargreaves, R.J.; Hill, R.G. L-733,060, a novel tachykinin NK1 receptor antagonist; effects in [Ca2+]i mobilisation, cardiovascular and dural extravasation assays. Eur. J. Pharmacol. 1996, 317, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Sirianni, A.C.; Jiang, J.; Zeng, J.; Mao, L.L.; Zhou, S.; Sugarbaker, P.; Zhang, X.; Li, W.; Friedlander, R.M.; Wang, X. N-acetyl-l-tryptophan, but not N-acetyl-d-tryptophan, rescues neuronal cell death in models of amyotrophic lateral sclerosis. J. Neurochem. 2015, 134, 956–968. [Google Scholar] [CrossRef]
- Matalinska, J.; Lipinski, P.F.J. Correcting a widespread error: Neuroprotectant N-acetyl-L-tryptophan does not bind to the neurokinin-1 receptor. Mol. Cell Neurosci. 2022, 120, 103728. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, F.; Vink, R.; Turner, R.J. Inflammation in acute CNS injury: A focus on the role of substance P. Br. J. Pharmacol. 2016, 173, 703–715. [Google Scholar] [CrossRef]
- Li, Q.; Wu, X.; Yang, Y.; Zhang, Y.; He, F.; Xu, X.; Zhang, Z.; Tao, L.; Luo, C. Tachykinin NK1 receptor antagonist L-733,060 and substance P deletion exert neuroprotection through inhibiting oxidative stress and cell death after traumatic brain injury in mice. Int. J. Biochem. Cell Biol. 2019, 107, 154–165. [Google Scholar] [CrossRef]
- Habgood, M.D.; Bye, N.; Dziegielewska, K.M.; Ek, C.J.; Lane, M.A.; Potter, A.; Morganti-Kossmann, C.; Saunders, N.R. Changes in blood-brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur. J. Neurosci. 2007, 25, 231–238. [Google Scholar] [CrossRef]
- Castro-Obregón, S.; Del Rio, G.; Chen, S.F.; Swanson, R.A.; Frankowski, H.; Rao, R.V.; Stoka, V.; Vesce, S.; Nicholls, D.G.; Bredesen, D.E. A ligand-receptor pair that triggers a non-apoptotic form of programmed cell death. Cell. Death Differ. 2002, 9, 807–817. [Google Scholar] [CrossRef]
- Vink, R.; Donkin, J.J.; Cruz, M.I.; Nimmo, A.J.; Cernak, I. A substance P antagonist increases brain intracellular free magnesium concentration after diffuse traumatic brain injury in rats. J. Am. Coll. Nutr. 2004, 23, 538S–540S. [Google Scholar] [CrossRef] [PubMed]
- Lorente, L.; Martín, M.M.; Almeida, T.; Hernandez, M.; Ramos, L.; Argueso, M.; Cáceres, J.J.; Solé-Violán, J.; Jiménez, A. Serum substance P levels are associated with severity and mortality in patients with severe traumatic brain injury. Crit. Care 2015, 19, 192. [Google Scholar] [CrossRef]
- Lorente, L.; Martín, M.M.; Pérez-Cejas, A.; González-Rivero, A.F.; Argueso, M.; Ramos, L.; Solé-Violán, J.; Cáceres, J.J.; Jiménez, A.; García-Marin, V. Persistently high serum substance P levels and early mortality in patients with severe traumatic brain injury. World Neurosurg. 2019, 132, e613–e617. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ye, H.; Lu, W. Serum substance P concentration in children with traumatic brain injury: A First Report. World Neurosurg. 2021, 147, e200–e205. [Google Scholar] [CrossRef]
- Alves, J.L.; Mendes, J.; Leitao, R.; Silva, A.P.; Pinto, A.M. A multi-staged neuropeptide response to traumatic brain injury. Eur. J. Trauma Emerg. Surg. 2022, 48, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Gabrielian, L.; Wisllshire, L.W.; Helps, S.C.; van den Heuvel, C.; Mathias, J.L.; Vink, R. Intracranial pressure changes following traumatic brain injury in rats: Lack of significant change in the absence of mass lesions or hypoxia. J. Neurotrauma 2011, 28, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Vink, R.; Bahtia, K.D.; Reilly, P.L. The relationship between intracranial pressure and brain oxygenation following traumatic brain injury in sheep. Acta Neurochir. Suppl. 2008, 102, 189–192. [Google Scholar]
- Gabrielian, L.; Helps, S.C.; Thornton, E.; Turner, R.J.; Leonard, A.V.; Vink, R. Substance P antagonists as a novel intervention for brain edema and raised intracranial pressure. Acta Neurochir. Suppl. 2013, 118, 201–204. [Google Scholar]
- Vink, R.; Gabrielian, L.; Thornton, E. The role of substance P in secondary pathophysiology after traumatic brain injury. Front. Neurol. 2017, 8, 304. [Google Scholar] [CrossRef]
- O’Connor, C.A.; Cernak, I.; Vink, R. The temporal profile of edema formation differs between male and female rats following diffuse traumatic brain injury. Acta Neurochir. Suppl. 2006, 96, 121–124. [Google Scholar]
- Sorby-Adams, A.J.; Leonard, A.V.; Hoving, J.W.; Yassi, N.; Vink, R.; Wells, A.J.; Turner, R.J. NK1-r antagonist treatment comparable to decompressive craniectomy in reducing intracranial pressure following stroke. Front. Neurosci. 2019, 13, 681. [Google Scholar] [CrossRef]
- Nag, S.; Venugopalan, R.; Stewart, D.J. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol. 2007, 114, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Schubert, W.; Frank, P.G.; Razani, B.; Park, D.S.; Chow, C.W.; Lisanti, M.P. Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J. Biol. Chem. 2001, 276, 48619–48622. [Google Scholar] [CrossRef]
- Povlishock, J.T.; Becker, D.P.; Sullivan, H.G.; Miller, J.D. Vascular permeability alterations to horseradish peroxidase in experimental brain injury. Brain Res. 1978, 153, 223–239. [Google Scholar] [CrossRef]
- Barzo, P.; Marmarou, A.; Fatouros, P.; Corwin, F.; Dunbar, J.G. Acute blood-brain barrier changes in experimental closed head injury as measured by MRI and Gd-DTPA. Acta Neurochir. Suppl. 1997, 70, 243–246. [Google Scholar] [PubMed]
- Castejón, O.J. Increased vesicular and vacuolar transendothelial transport in traumatic human brain oedema. A review. Folia Neuropathol. 2013, 51, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Kubale, V.; Abramovic, Z.; Pogacnik, A.; Heding, A.; Sentjurc, M.; Vrecl, M. Evidence for a role of caveolin-1 in neurokinin-1 receptor plasma-membrane localization, efficient signaling, and interaction with beta-arrestin 2. Cell Tissue Res. 2007, 330, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.H.; Segura, J.M.; Martinez, K.L.; Hovius, R.; George, N.; Johnsson, K.; Vogel, H. FRET imaging reveals that functional neurokinin-1 receptors are monomeric and reside in membrane microdomains of live cells. Proc. Natl. Acad. Sci. USA 2006, 103, 2138–2143. [Google Scholar] [CrossRef]
- Monastyrskaya, K.; Hostettler, A.; Buergi, S.; Draeger, A. The NK1 receptor localizes to the plasma membrane microdomains, and its activation is dependent on lipid raft integrity. J. Biol. Chem. 2005, 280, 7135–7146. [Google Scholar] [CrossRef]
- Mineo, C.; Ying, Y.S.; Chapline, C.; Jaken, S.; Anderson, R.G. Targeting of protein kinase Calpha to caveolae. J. Cell Biol. 1998, 141, 601–610. [Google Scholar] [CrossRef]
- Pressura Neuro. Available online: https://pressuraneuro.com (accessed on 25 June 2023).
- Stein, D.G. Embracing failure: What the Phase III progesterone studies can teach about TBI clinical trials. Brain Inj. 2015, 29, 1259–1272. [Google Scholar] [CrossRef]
- Chawla, S.P.; Grunberg, S.M.; Gralla, R.J.; Hesketh, P.J.; Rittenberg, C.; Elmer, M.E.; Schmidt, C.; Taylor, A.; Carides, A.D.; Evans, J.K.; et al. Establishing the dose of the oral NK1 antagonist aprepitant for the prevention of chemotherapy-induced nausea and vomiting. Cancer 2003, 97, 2290–2300. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Montgomery, S.; Ball, W.; Morrison, M.; Snavely, D.; Liu, G.; Hargreaves, R.; Hietala, J.; Lines, C.; Beebe, K.; et al. Lack of efficacy of the substance p (neurokinin1 receptor) antagonist aprepitant in the treatment of major depressive disorder. Biol. Psychiatry 2006, 59, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Rupniak, N.M.J.; Kramer, M.S. NK1 receptor antagonists for depression: Why a validated concept was abandoned. J. Affect. Disord. 2017, 223, 121–125. [Google Scholar] [CrossRef]
- Hargreaves, R. Imaging substance P receptors (NK1) in the living human brain using positron emission tomography. J. Clin. Psychiatry 2002, 63 (Suppl. S11), 18–24. [Google Scholar]
- Bergstrom, M.; Hargreaves, R.J.; Burns, H.D.; Goldberg, M.R.; Sciberras, D.; Reines, S.A.; Petty, K.J.; Ogren, M.; Antoni, G.; Langstrom, B.; et al. Human positron emission tomography studies of brain neurokinin 1 receptor occupancy by aprepitant. Biol. Psychiatry 2004, 55, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Coveñas, R. The Neurokinin-1 receptor antagonist aprepitant: An intelligent bullet against cancer? Cancers 2020, 12, 2692. [Google Scholar] [CrossRef]
- Carthew, H.L.; Ziebell, J.M.; Vink, R. Substance P-induced changes in cell genesis following diffuse traumatic brain injury. Neuroscience 2012, 214, 78–83. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vink, R.; Corrigan, F. The Role of Substance P and NK1 Receptors in Mild to Severe Traumatic Brain Injury: From CTE to ICP. Receptors 2023, 2, 220-231. https://doi.org/10.3390/receptors2040015
Vink R, Corrigan F. The Role of Substance P and NK1 Receptors in Mild to Severe Traumatic Brain Injury: From CTE to ICP. Receptors. 2023; 2(4):220-231. https://doi.org/10.3390/receptors2040015
Chicago/Turabian StyleVink, Robert, and Frances Corrigan. 2023. "The Role of Substance P and NK1 Receptors in Mild to Severe Traumatic Brain Injury: From CTE to ICP" Receptors 2, no. 4: 220-231. https://doi.org/10.3390/receptors2040015
APA StyleVink, R., & Corrigan, F. (2023). The Role of Substance P and NK1 Receptors in Mild to Severe Traumatic Brain Injury: From CTE to ICP. Receptors, 2(4), 220-231. https://doi.org/10.3390/receptors2040015