
Tracking Co-Occurrence of N501Y, P681R, and Other Key Mutations in SARS-CoV-2 Spike for Surveillance

 , and
, and
Abstract
:1. Introduction
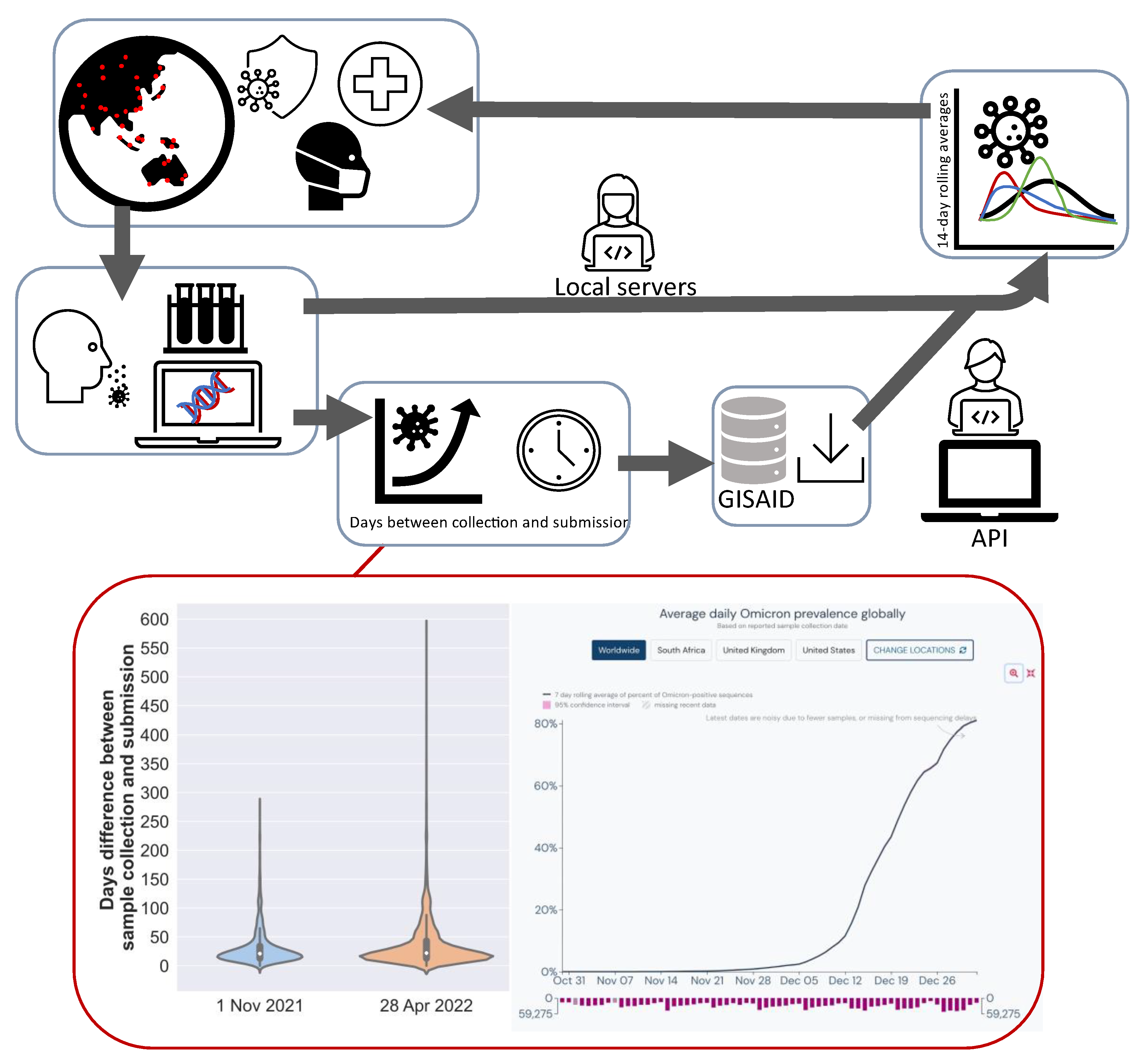
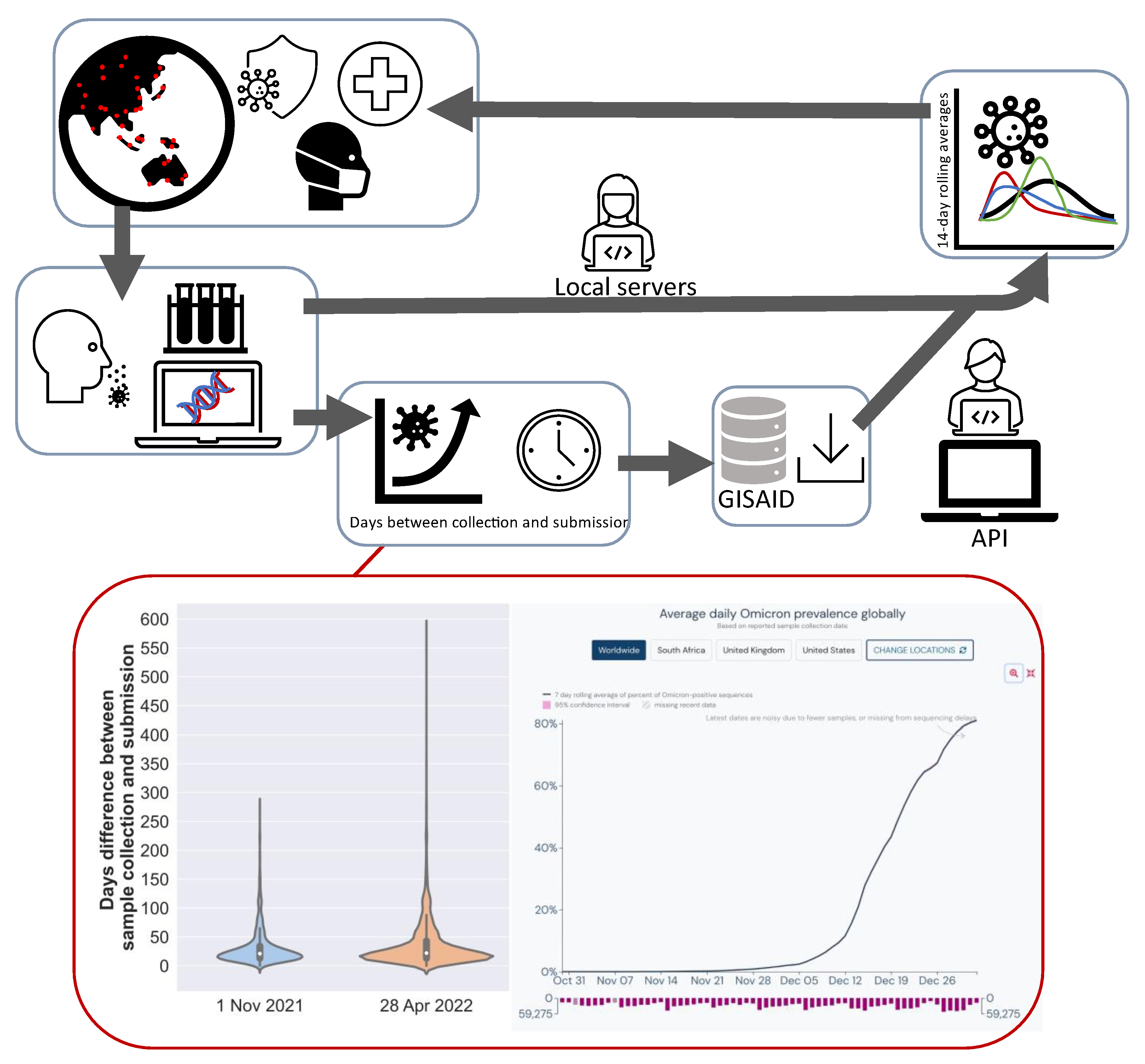
2. Methods
2.1. Study Rationale
2.2. Bioinformatics Analysis
2.3. Biomolecular Modelling
3. Results
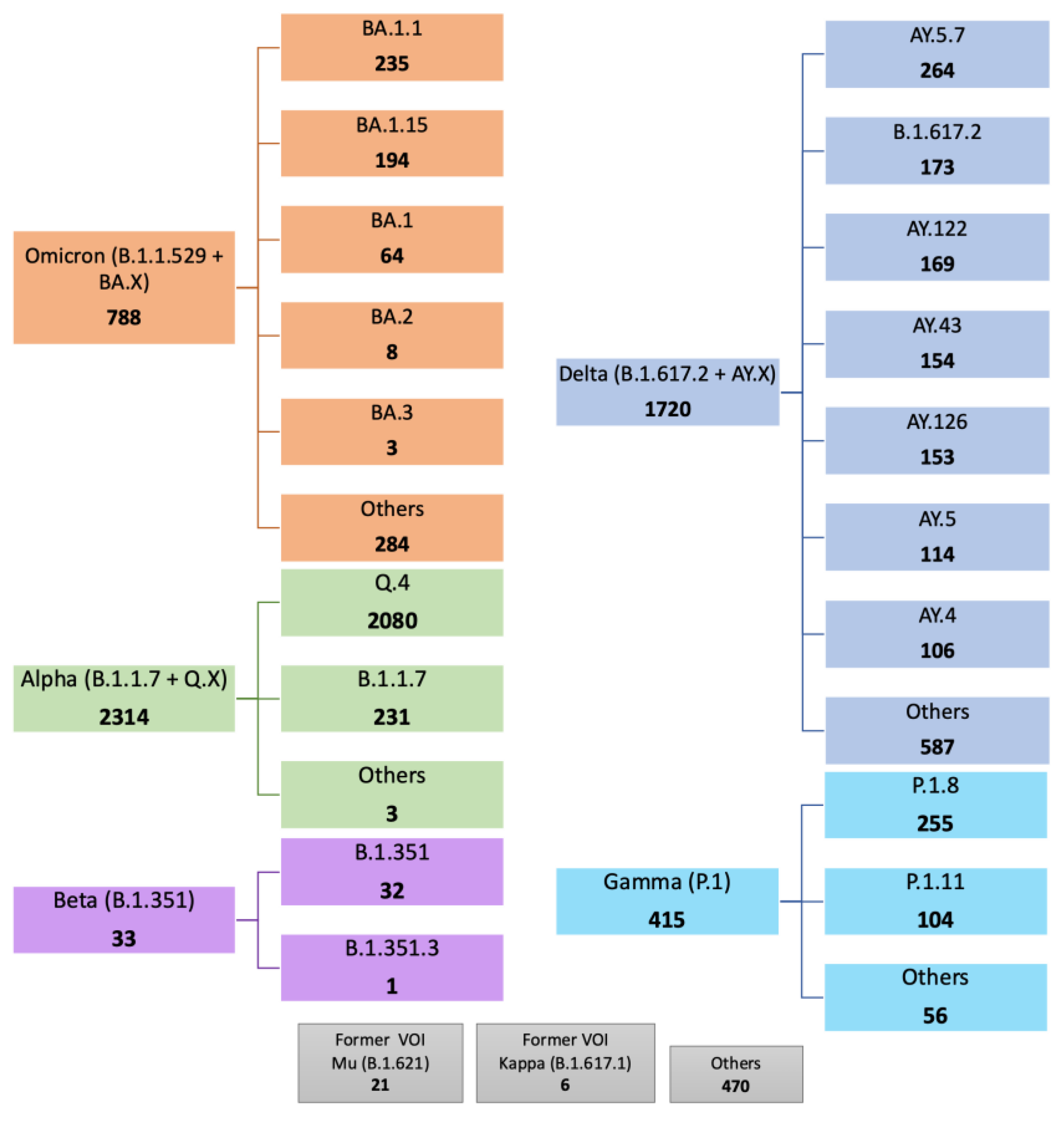
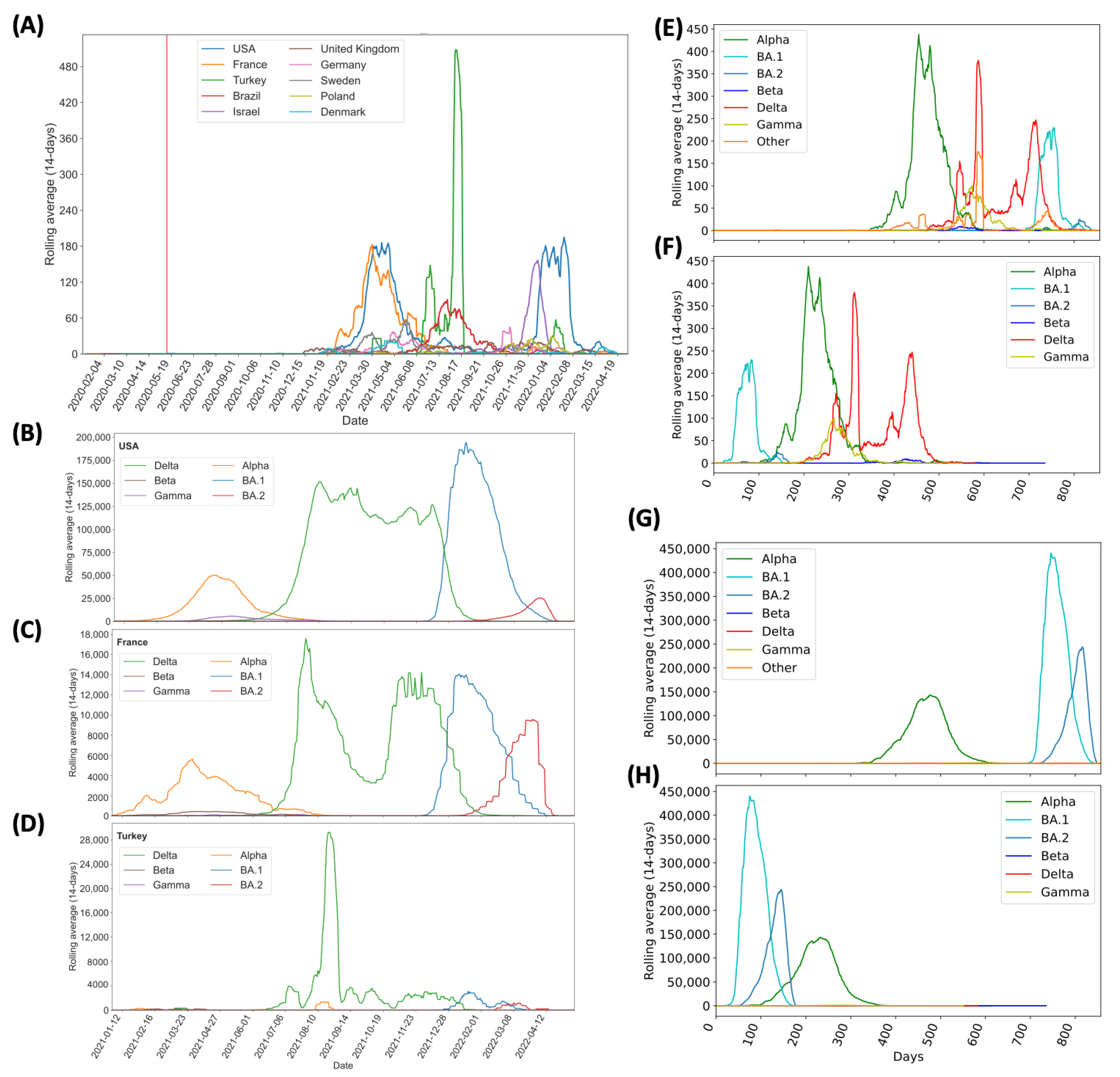
3.1. Predominance of Alpha Q.4 acquiring P681R and Delta Acquiring N501Y
3.2. Frequency and Distribution of D614G, N501Y and P681R Mutations
3.3. In Silico Modelling of the Spike Trimer
4. Discussion
4.1. Y501-R681 Almost Always in the G614 Background
4.2. Alpha Acquiring P681R Ahead of Other VOC Could Be Due to P/H681R
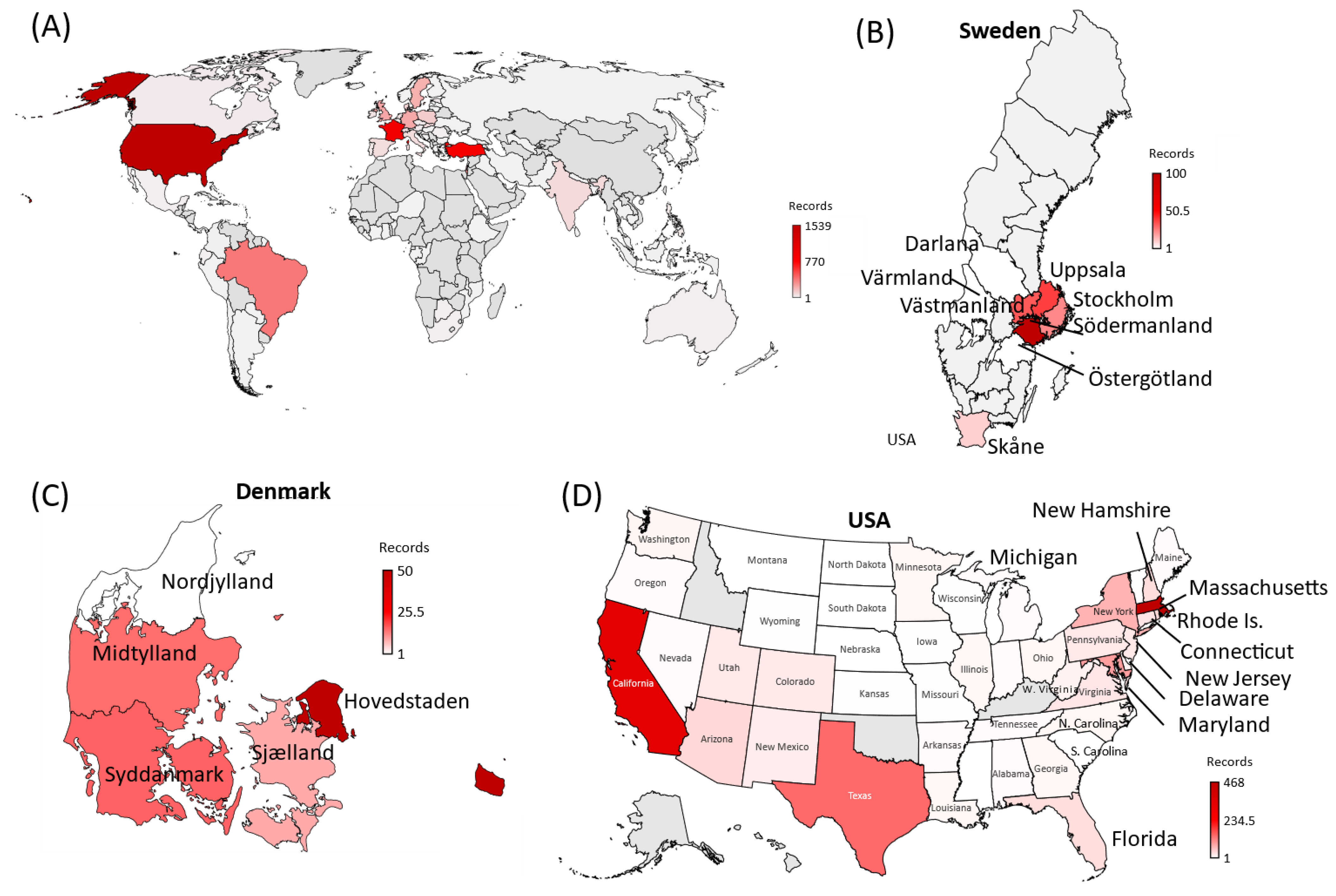
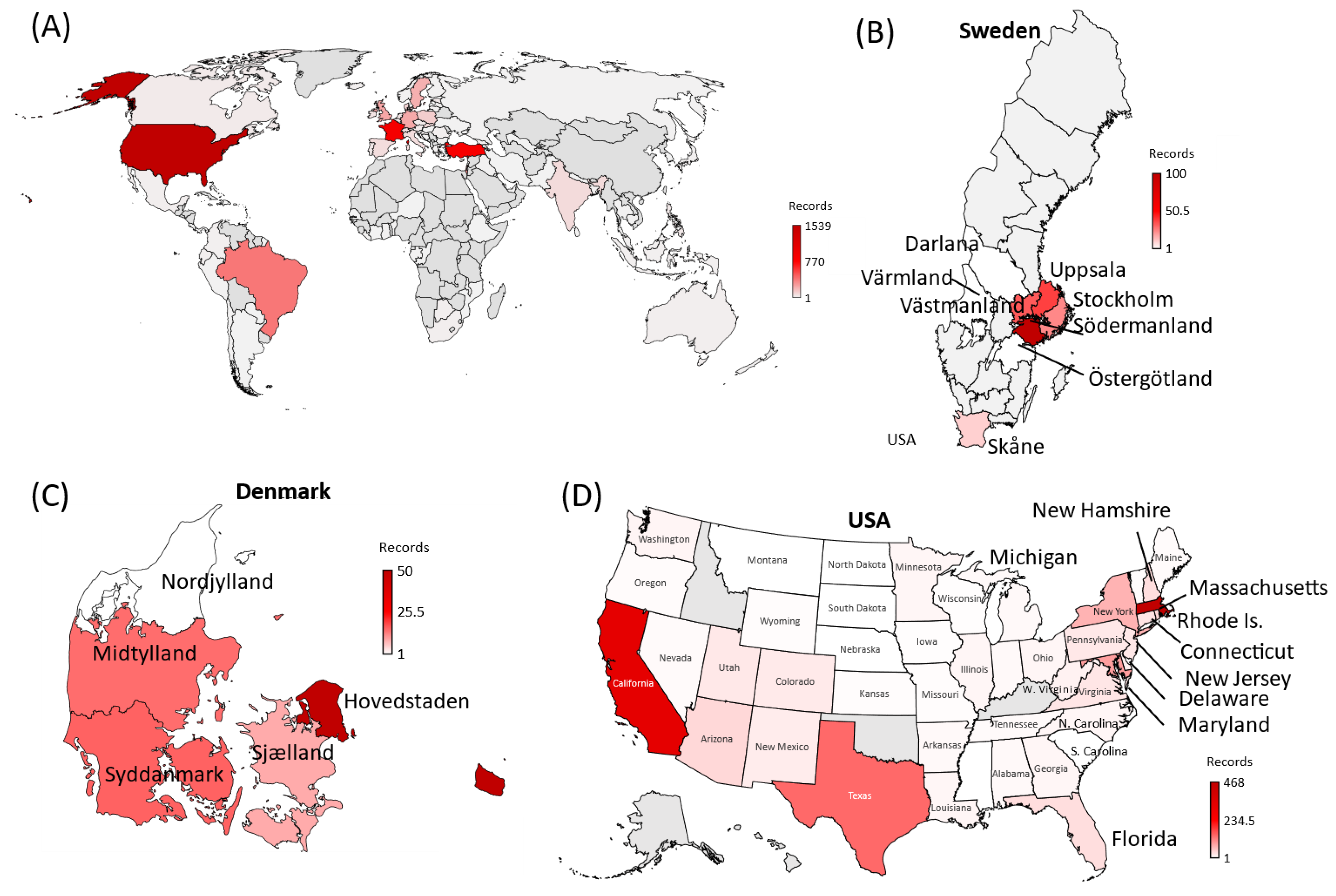
4.3. Co-Occurrences Have Been Reported Predominantly in Eight Countries
4.4. Y501-R681 Does Not Outcompete Other Variants
4.5. Implications for the Omicron VOC
4.6. SARS-CoV-2 Evolution and Selective Pressures on Quasispecies
4.7. Error Catastrophe, Muller’s Ratchet, and Implications for SARS-CoV-2
5. Conclusions and Limitations
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.C.; Tay, A.P.; Wilson, L.O.W.; Reti, D.; Hosking, C.; McAuley, A.J.; Pharo, E.; Todd, S.; Stevens, V.; Neave, M.J.; et al. Supporting Pandemic Response Using Genomics and Bioinformatics: A Case Study on the Emergent SARS-CoV-2 Outbreak. Transbound. Emerg. Dis. 2020, 67, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Wilke, C.O.; Wang, J.L.; Ofria, C.; Lenski, R.E.; Adami, C. Evolution of Digital Organisms at High Mutation Rates Leads to Survival of the Flattest. Nature 2001, 412, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Petrone, M.E.; Holmes, E.C. We Shouldn’t Worry When a Virus Mutates during Disease Outbreaks. Nat. Microbiol. 2020, 5, 529–530. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. The Coronavirus Is Mutating—Does It Matter? Nature 2020, 585, 174–177. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- McAuley, A.J.; Kuiper, M.J.; Durr, P.A.; Bruce, M.P.; Barr, J.; Todd, S.; Au, G.G.; Blasdell, K.; Tachedjian, M.; Lowther, S.; et al. Experimental and in silico Evidence Suggests Vaccines Are Unlikely to Be Affected by D614G Mutation in SARS-CoV-2 Spike Protein. npj Vaccines 2020, 5, 96. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y Spike Substitution Enhances SARS-CoV-2 Infection and Transmission. Nature 2021, 602, 294–299. [Google Scholar] [CrossRef]
- Karim, S.S.A.; de Oliveira, T. New SARS-CoV-2 Variants—Clinical, Public Health, and Vaccine Implications. N. Engl. J. Med. 2021, 384, 1866–1868. [Google Scholar] [CrossRef]
- Riddell, S.; Goldie, S.; McAuley, A.J.; Kuiper, M.J.; Durr, P.A.; Blasdell, K.R.; Tachedjian, M.; Druce, J.D.; Smith, T.R.F.; Broderick, K.E.; et al. Live Virus Neutralisation of the 501Y.V1 and 501Y.V2 SARS-CoV-2 Variants Following INO-4800 Vaccination of Ferrets. Front. Immunol. 2021, 12, 694857. [Google Scholar] [CrossRef]
- Kuiper, M.J.; Wilson, L.O.W.; Mangalaganesh, S.; Lee, C.; Reti, D.; Vasan, S.S. “But Mouse, You Are Not Alone”: On Some Severe Acute Respiratory Syndrome Coronavirus 2 Variants Infecting Mice. ILAR J. 2021, 62, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. The Mutation That Helps Delta Spread like Wildfire. Nature 2021, 596, 472–473. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta Spike P681R Mutation Enhances SARS-CoV-2 Fitness over Alpha Variant. Cell Rep. 2022, 39, 110829. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P.; Sheppard, C.M.; Brown, J.C.; Goonawardane, N.; Zhou, J.; Whiteley, M.; Consortium, P.V.; de Silva, T.I.; Barclay, W.S. The SARS-CoV-2 Variants Associated with Infections in India, B.1.617, Show Enhanced Spike Cleavage by Furin. bioRxiv 2021. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef]
- Bauer, D.C.; Metke-Jimenez, A.; Maurer-Stroh, S.; Tiruvayipati, S.; Wilson, L.O.W.; Jain, Y.; Perrin, A.; Ebrill, K.; Hansen, D.P.; Vasan, S.S. Interoperable Medical Data: The Missing Link for Understanding COVID-19. Transbound. Emerg. Dis. 2021, 68, 1753–1760. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.Q. Higher Infectivity of the SARS-CoV-2 New Variants Is Associated with K417N/T, E484K, and N501Y Mutants: An Insight from Structural Data. J. Cell. Physiol. 2021, 236, 7045–7057. [Google Scholar] [CrossRef]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced Fusogenicity and Pathogenicity of SARS-CoV-2 Delta P681R Mutation. Nature 2021, 602, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Remacle, A.G.; Shiryaev, S.A.; Oh, E.-S.; Cieplak, P.; Srinivasan, A.; Wei, G.; Liddington, R.C.; Ratnikov, B.I.; Parent, A.; Desjardins, R.; et al. Substrate Cleavage Analysis of Furin and Related Proprotein Convertases. J. Biol. Chem. 2008, 283, 20897–20906. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, M.; Damalanka, V.C.; Tartell, M.A.; Chung, D.H.; Lourenco, A.L.; Pwee, D.; Bridwell, A.E.M.; Hoffmann, M.; Voss, J.; Karmakar, P.; et al. A Novel Class of TMPRSS2 Inhibitors Potently Block SARS-CoV-2 and MERS-CoV Viral Entry and Protect Human Epithelial Lung Cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2108728118. [Google Scholar] [CrossRef]
- Statista Demographics I Statista. f. Available online: https://www.statista.com/markets/411/topic/466/demographics/ (accessed on 22 December 2021).
- Lythgoe, K.A.; Hall, M.; Ferretti, L.; de Cesare, M.; MacIntyre-Cockett, G.; Trebes, A.; Andersson, M.; Otecko, N.; Wise, E.L.; Moore, N.; et al. SARS-CoV-2 within-Host Diversity and Transmission. Science 2021, 372, 6539. [Google Scholar] [CrossRef]
- Latif, A.A.; Mullen, J.L.; Alkuzweny, M.; Tsueng, G.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. United Kingdom Variant Report. Available online: https://outbreak.info/location-reports?loc=GBR&selected=Delta&selected=Alpha (accessed on 22 December 2021).
- Ritchie, H.; Mathieu, E.; Rodés-Guirao, L.; Appel, C.; Giattino, C.; Ortiz-Ospina, E.; Hasell, J.; Macdonald, B.; Beltekian, D.; Roser, M. Coronavirus Pandemic (COVID-19). Available online: https://ourworldindata.org/coronavirus (accessed on 22 December 2021).
- Timeline of UK Government Coronavirus Lockdowns. Available online: https://www.instituteforgovernment.org.uk/charts/uk-government-coronavirus-lockdowns (accessed on 22 December 2021).
- Ballotpedia Travel Restrictions Issued by States in Response to the Coronavirus (COVID-19) Pandemic, 2020–2021. Available online: https://ballotpedia.org/Travel_restrictions_issued_by_states_in_response_to_the_coronavirus_(COVID-19)_pandemic,_2020-2021 (accessed on 22 December 2021).
- Thi Nhu Thao, T.; Labroussaa, F.; Ebert, N.; V’kovski, P.; Stalder, H.; Portmann, J.; Kelly, J.; Steiner, S.; Holwerda, M.; Kratzel, A.; et al. Rapid Reconstruction of SARS-CoV-2 Using a Synthetic Genomics Platform. Nature 2020, 582, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Muruato, A.; Lokugamage, K.G.; Narayanan, K.; Zhang, X.; Zou, J.; Liu, J.; Schindewolf, C.; Bopp, N.E.; Aguilar, P.V.; et al. An Infectious CDNA Clone of SARS-CoV-2. Cell Host Microbe 2020, 27, 841–848.e3. [Google Scholar] [CrossRef]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H.; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446.e14. [Google Scholar] [CrossRef]
- Xie, X.; Lokugamage, K.G.; Zhang, X.; Vu, M.N.; Muruato, A.E.; Menachery, V.D.; Shi, P.-Y. Engineering SARS-CoV-2 Using a Reverse Genetic System. Nat. Protoc. 2021, 16, 1761–1784. [Google Scholar] [CrossRef]
- Ferguson, N.; Ghani, A.; Cori, A.; Hogan, A.; Hinsley, W.; Volz, E. Report 49—Growth, Population Distribution and Immune Escape of Omicron in England; Faculty of Medicine, Imperial College London: London, UK, 2021. [Google Scholar]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.; et al. SARS-CoV-2 Omicron Has Extensive but Incomplete Escape of Pfizer BNT162b2 Elicited Neutralization and Requires ACE2 for Infection. medRxiv 2021. [Google Scholar] [CrossRef]
- Pulliam, J.R.C.; van Schalkwyk, C.; Govender, N.; von Gottberg, A.; Cohen, C.; Groome, M.J.; Dushoff, J.; Mlisana, K.; Moultrie, H. Increased Risk of SARS-CoV-2 Reinfection Associated with Emergence of Omicron in South Africa. medRxiv 2022, 376, eabn4947. [Google Scholar] [CrossRef]
- Heguy, A.; (New York University Langone Health). Personal Communication, 2021.
- Corey, L.; Beyrer, C.; Cohen, M.S.; Michael, N.L.; Bedford, T.; Rolland, M. SARS-CoV-2 Variants in Patients with Immunosuppression. N. Engl. J. Med. 2021, 385, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Grépin, K.A.; Ho, T.-L.; Liu, Z.; Marion, S.; Piper, J.; Worsnop, C.Z.; Lee, K. Evidence of the Effectiveness of Travel-Related Measures during the Early Phase of the COVID-19 Pandemic: A Rapid Systematic Review. BMJ Glob. Health 2021, 6, e004537. [Google Scholar] [CrossRef] [PubMed]
- Liebig, J.; Najeebullah, K.; Jurdak, R.; El Shoghri, A.; Paini, D. Should International Borders Re-Open? The Impact of Travel Restrictions on COVID-19 Importation Risk. BMC Public Health 2021, 21, 1573. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.R.; Jaffe, G.; Doane, M.; Glaser, A.; Gremillion, T.M.; Ho, M.D. Responsible Governance of Gene Editing in Agriculture and the Environment. Nat. Biotechnol. 2021, 39, 1055–1057. [Google Scholar] [CrossRef]
- Abbud, A.; Castilho, E.A. A Call for a More Comprehensive SARS-CoV-2 Sequence Database for Brazil. Lancet Reg. Health Am. 2022, 5, 100095. [Google Scholar] [CrossRef]
- Domingo, E.; Perales, C. Viral Quasispecies. PLoS Genet. 2019, 15, e1008271. [Google Scholar] [CrossRef]
- Drew, T.W. The Emergence and Evolution of Swine Viral Diseases: To What Extend Have Husbandry Systems and Global Trade Contributed to Their Distribution and Diversity? Rev. Sci. Tech. l’OIE 2011, 30, 95–106. [Google Scholar] [CrossRef]
- Sun, F.; Wang, X.; Tan, S.; Dan, Y.; Lu, Y.; Zhang, J.; Xu, J.; Tan, Z.; Xiang, X.; Zhou, Y.; et al. SARS-CoV-2 Quasispecies Provides an Advantage Mutation Pool for the Epidemic Variants. Microbiol. Spectr. 2021, 9, e00261-21. [Google Scholar] [CrossRef]
- Lin, L.; Li, Q.; Wang, Y.; Shi, Y. Syncytia Formation during SARS-CoV-2 Lung Infection: A Disastrous Unity to Eliminate Lymphocytes. Cell Death Differ. 2021, 28, 2019–2021. [Google Scholar] [CrossRef]
- Tejero, H.; Marín, A.; Montero, F. The Relationship between the Error Catastrophe, Survival of the Flattest, and Natural Selection. BMC Evol. Biol. 2011, 11, 2. [Google Scholar] [CrossRef] [Green Version]
- de Alcañíz, J.G.G.; López-Rodas, V.; Costas, E. Sword of Damocles or Choosing Well. Population Genetics Sheds Light into the Future of the COVID-19 Pandemic and SARS-CoV-2 New Mutant Strains. medRxiv 2021. [Google Scholar] [CrossRef]
- Brüssow, H. COVID-19: Emergence and Mutational Diversification of SARS-CoV-2. Microb. Biotechnol. 2021, 14, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Haddad, D.; John, S.E.; Mohammad, A.; Hammad, M.M.; Hebbar, P.; Channanath, A.; Nizam, R.; Al-Qabandi, S.; Al Madhoun, A.; Alshukry, A.; et al. SARS-CoV-2: Possible Recombination and Emergence of Potentially More Virulent Strains. PLoS ONE 2021, 16, e0251368. [Google Scholar] [CrossRef] [PubMed]
- Pollett, S.; Conte, M.A.; Sanborn, M.; Jarman, R.G.; Lidl, G.M.; Modjarrad, K.; Berry, I.M. A Comparative Recombination Analysis of Human Coronaviruses and Implications for the SARS-CoV-2 Pandemic. Sci. Rep. 2021, 11, 17365. [Google Scholar] [CrossRef] [PubMed]

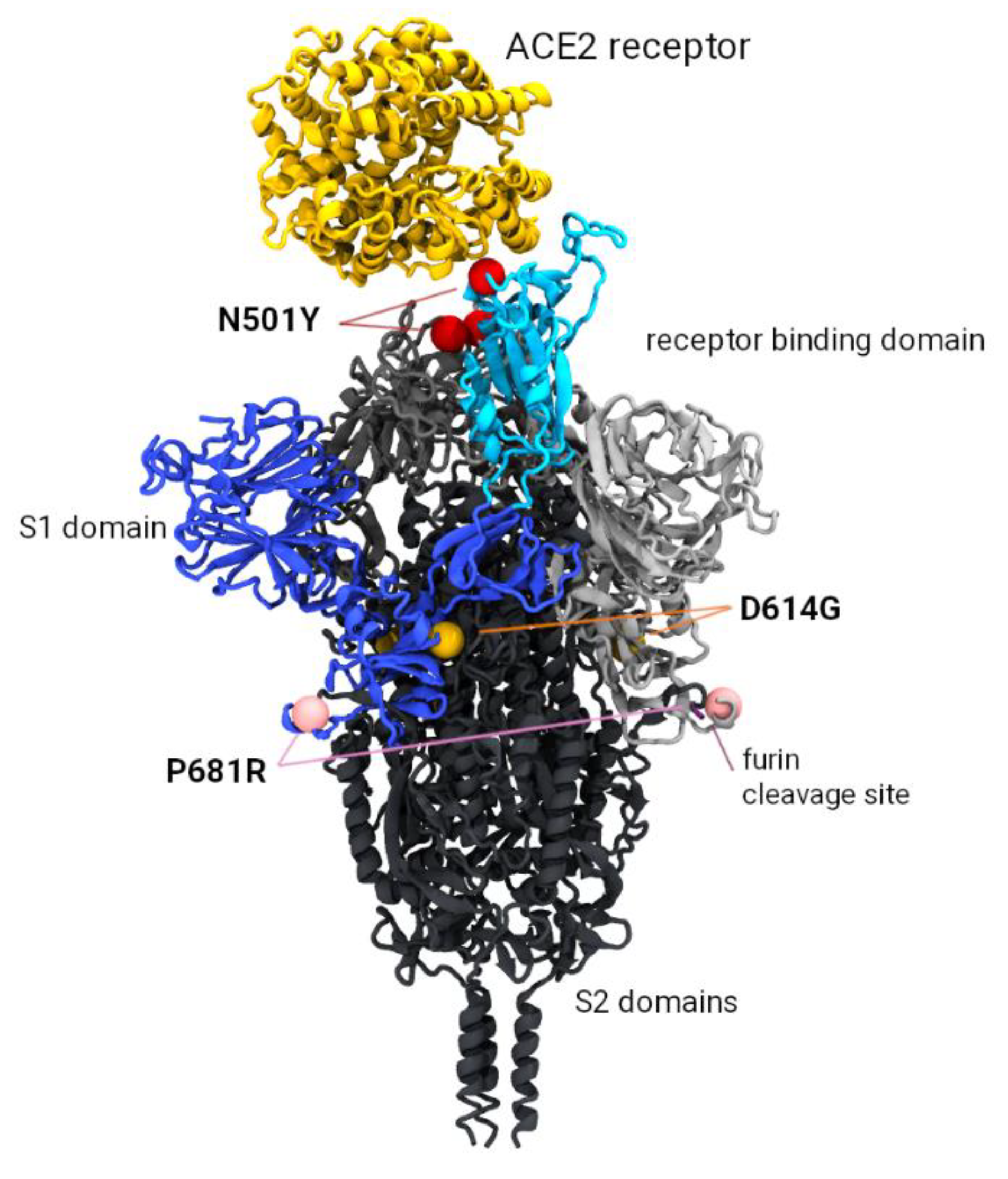

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country | Sequencing-to-Positivity Ratio | Observed Number of MOCI Co-Occurrences from GISAID | Predicted True Number of MOCI Co-Occurrences Based on the Ratio | Difference between the Predicted and Observed Number of MOCI Co-Occurrences |
|---|---|---|---|---|
| USA | 1:25 | 1539 | 38,707 | 37,168 |
| France | 1:86 | 881 | 75,908 | 75,027 |
| Turkey | 1:165 | 854 | 141,105 | 140,251 |
| Brazil | 1:219 | 389 | 85,139 | 84,750 |
| Israel | 1:54 | 348 | 18,678 | 18,330 |
| United Kingdom | 1:8 | 241 | 1925 | 1684 |
| Germany | 1:42 | 231 | 9777 | 9546 |
| Sweden | 1:13 | 210 | 2768 | 2558 |
| Poland | 1:73 | 117 | 8574 | 8457 |
| Denmark | 1:6 | 95 | 594 | 499 |
| All remaining countries with the MOCI co-occurrences (n = 81) | 1:48 | 862 | 41,376 | 40,514 |
| Total | 1:48 | 5747 | 275,856 | 271,877 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.; Mangalaganesh, S.; Wilson, L.O.W.; Kuiper, M.J.; Drew, T.W.; Vasan, S.S. Tracking Co-Occurrence of N501Y, P681R, and Other Key Mutations in SARS-CoV-2 Spike for Surveillance. Zoonotic Dis. 2022, 2, 147-162. https://doi.org/10.3390/zoonoticdis2030014
Lee C, Mangalaganesh S, Wilson LOW, Kuiper MJ, Drew TW, Vasan SS. Tracking Co-Occurrence of N501Y, P681R, and Other Key Mutations in SARS-CoV-2 Spike for Surveillance. Zoonotic Diseases. 2022; 2(3):147-162. https://doi.org/10.3390/zoonoticdis2030014
Chicago/Turabian StyleLee, Carol, Shruthi Mangalaganesh, Laurence O. W. Wilson, Michael J. Kuiper, Trevor W. Drew, and Seshadri S. Vasan. 2022. "Tracking Co-Occurrence of N501Y, P681R, and Other Key Mutations in SARS-CoV-2 Spike for Surveillance" Zoonotic Diseases 2, no. 3: 147-162. https://doi.org/10.3390/zoonoticdis2030014
APA StyleLee, C., Mangalaganesh, S., Wilson, L. O. W., Kuiper, M. J., Drew, T. W., & Vasan, S. S. (2022). Tracking Co-Occurrence of N501Y, P681R, and Other Key Mutations in SARS-CoV-2 Spike for Surveillance. Zoonotic Diseases, 2(3), 147-162. https://doi.org/10.3390/zoonoticdis2030014