
Cu-Substituted Hydroxyapatite Powder: Mechanochemical Synthesis Using Different Copper Sources and Thermal Stability

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
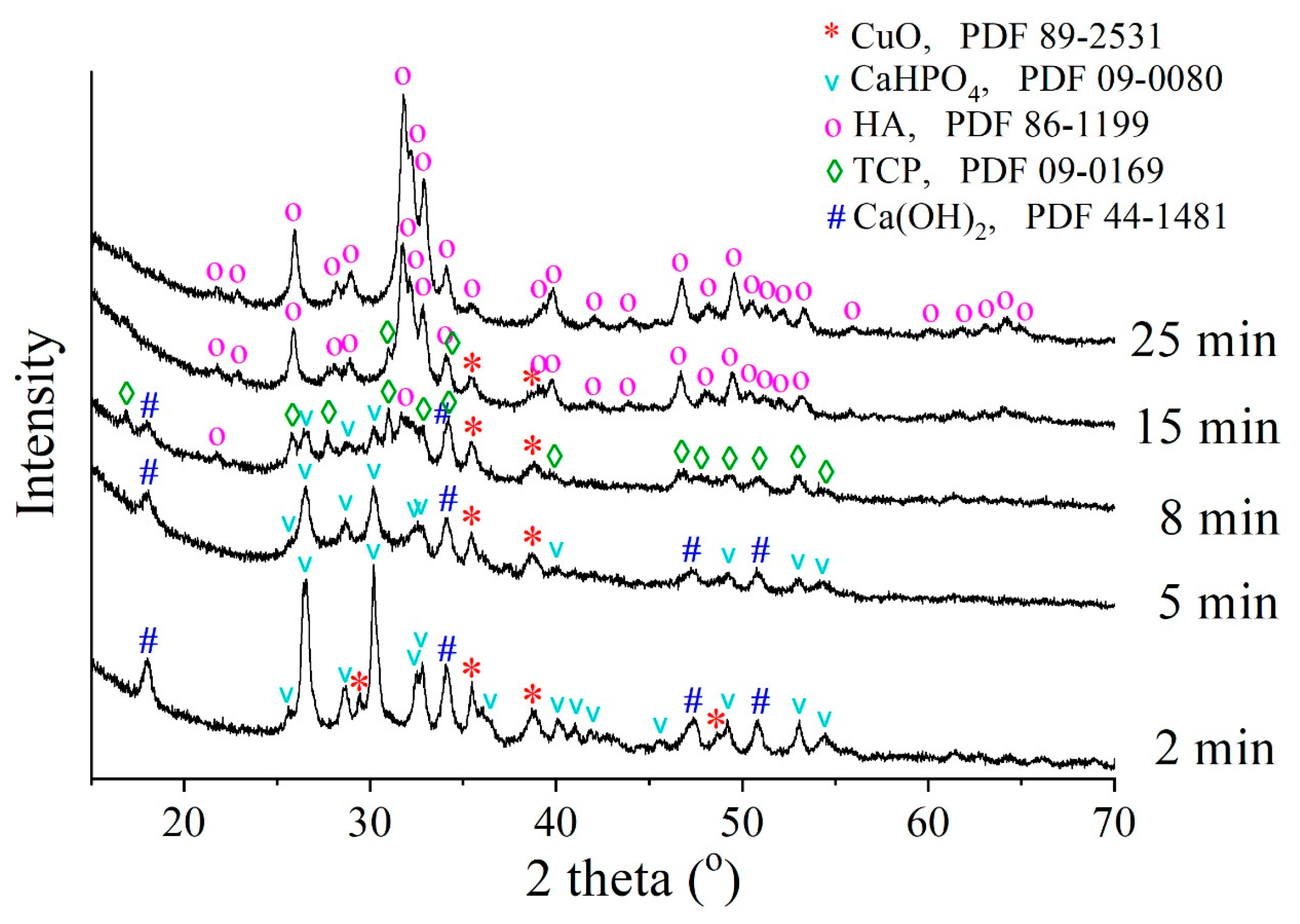
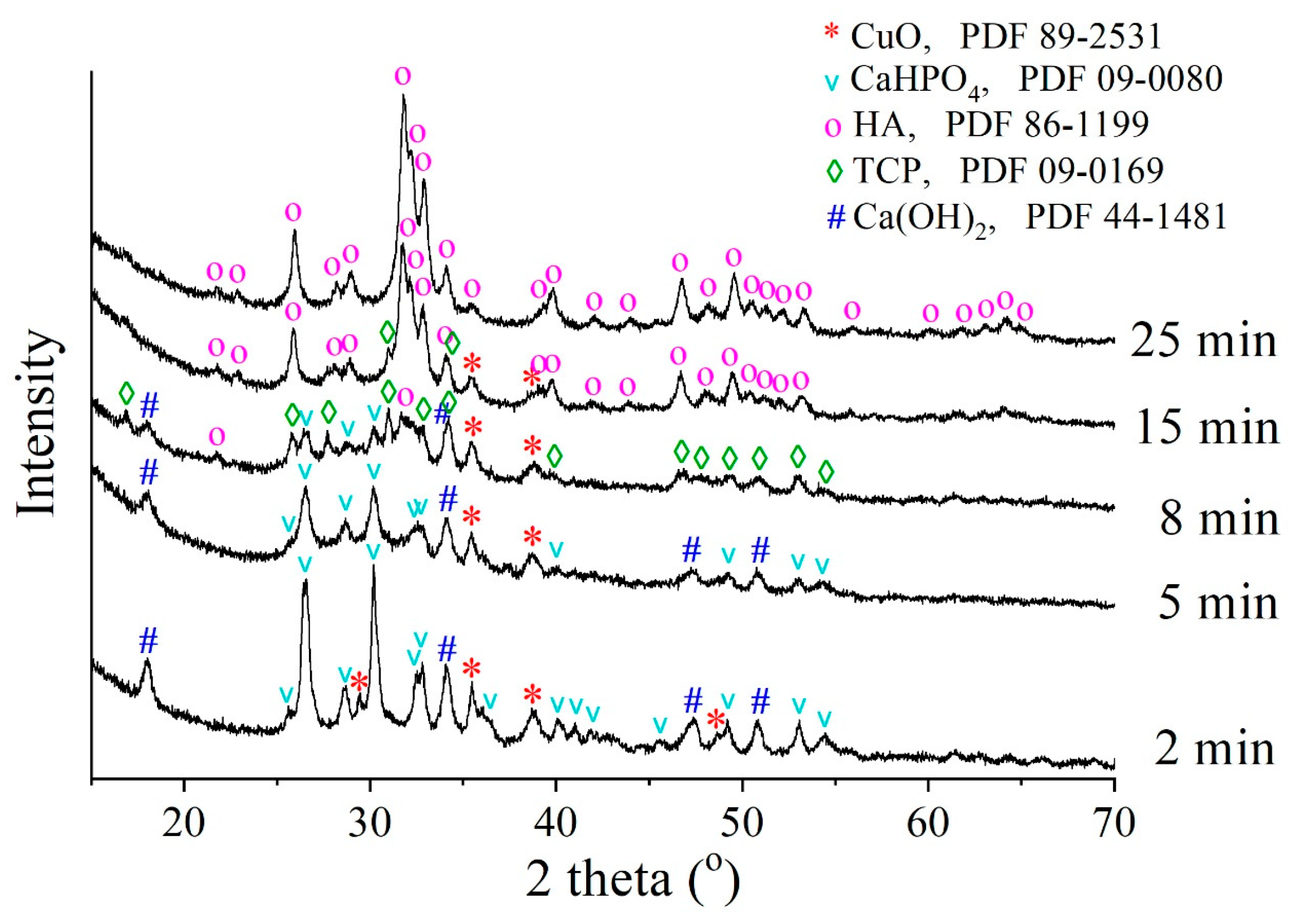
3.1. Optimal Conditions of Mechanochemical Synthesis
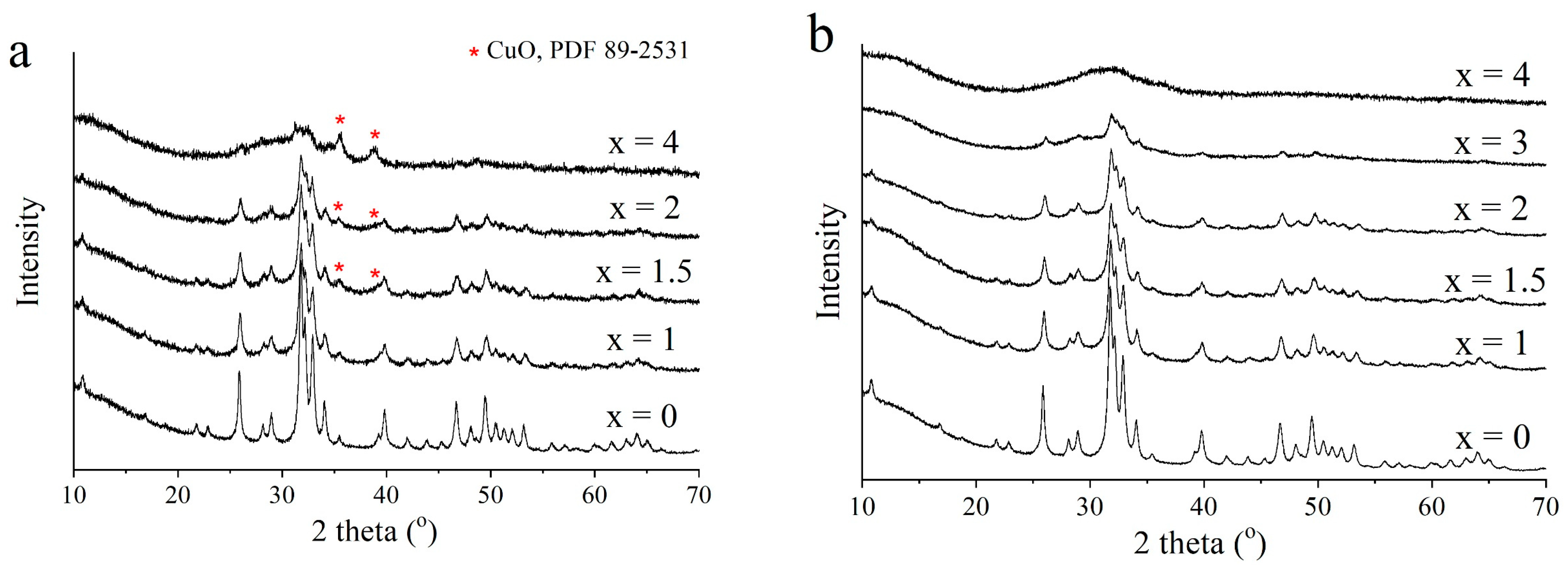
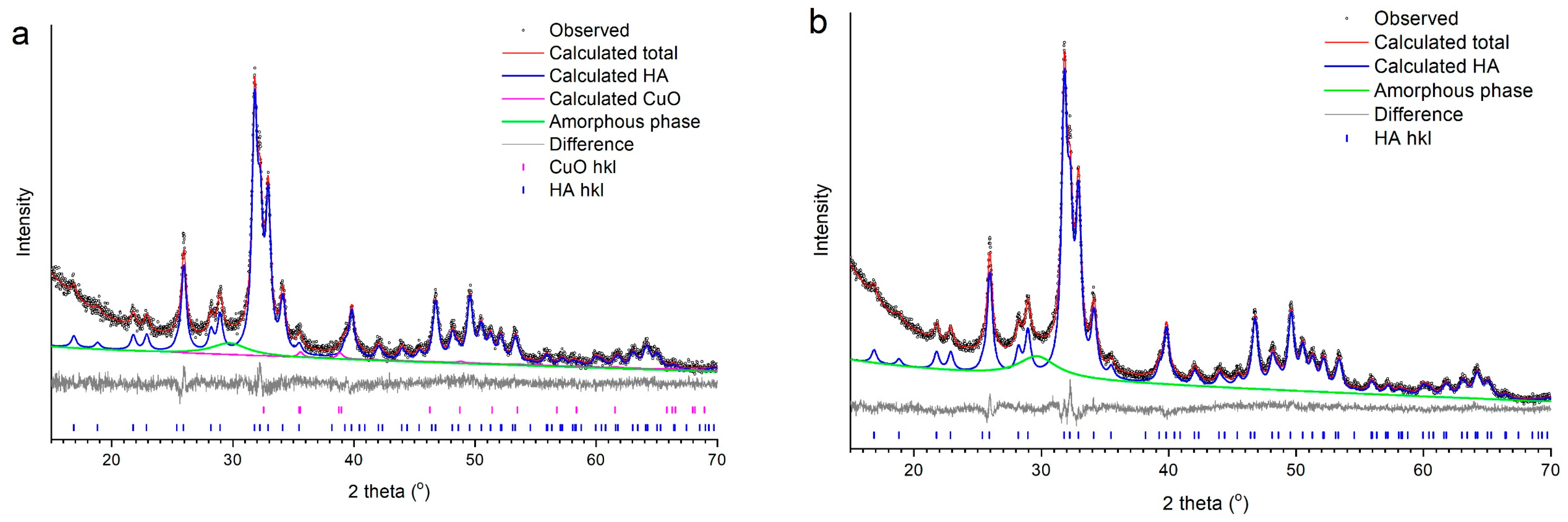
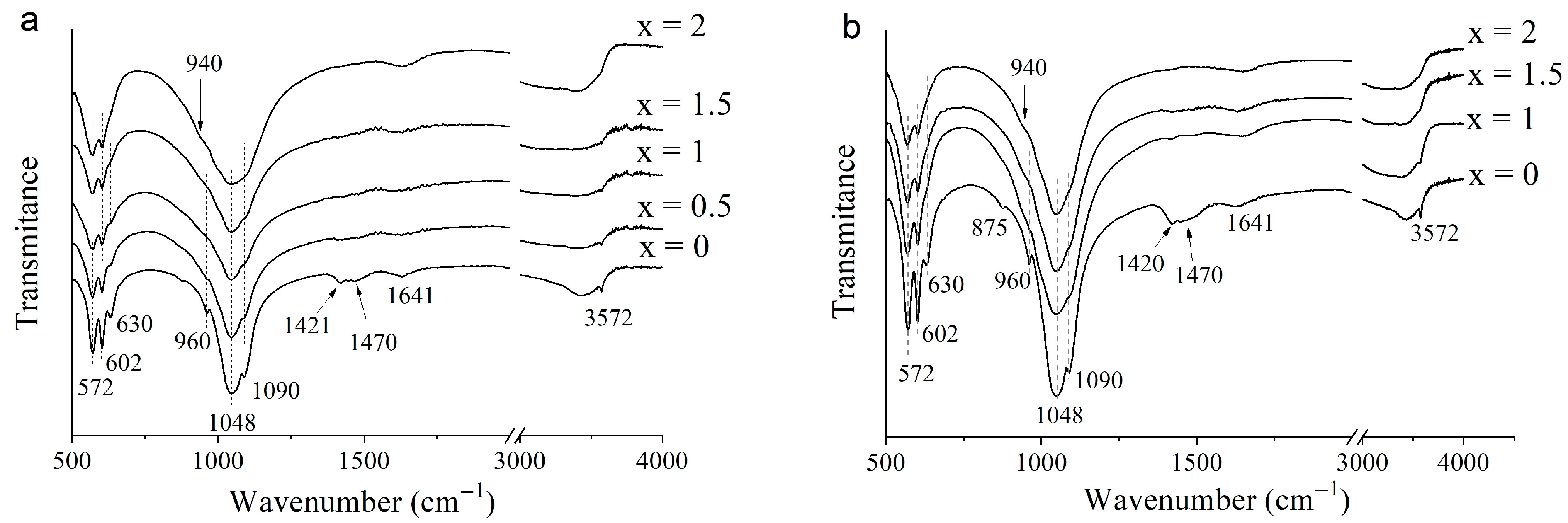
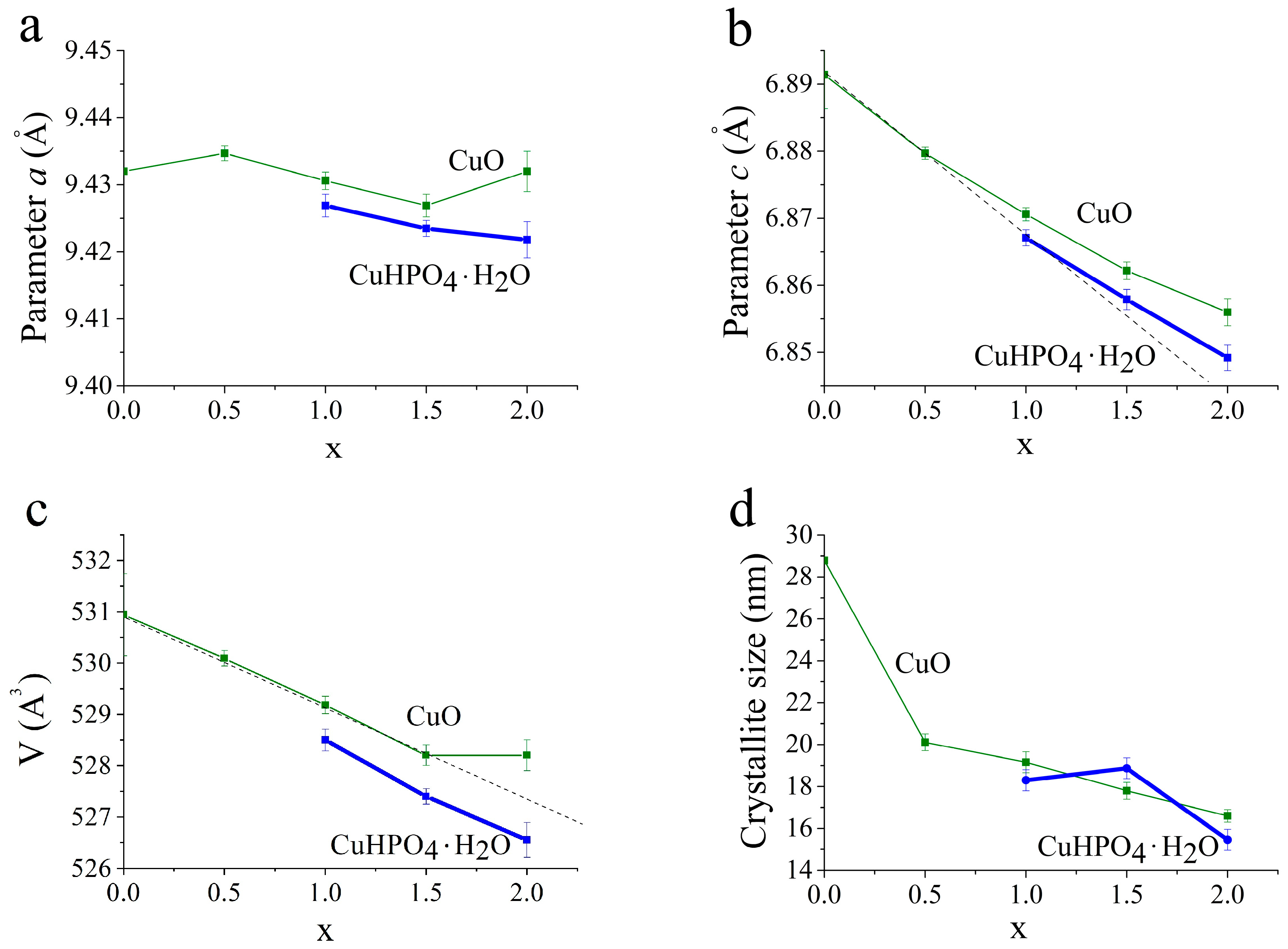
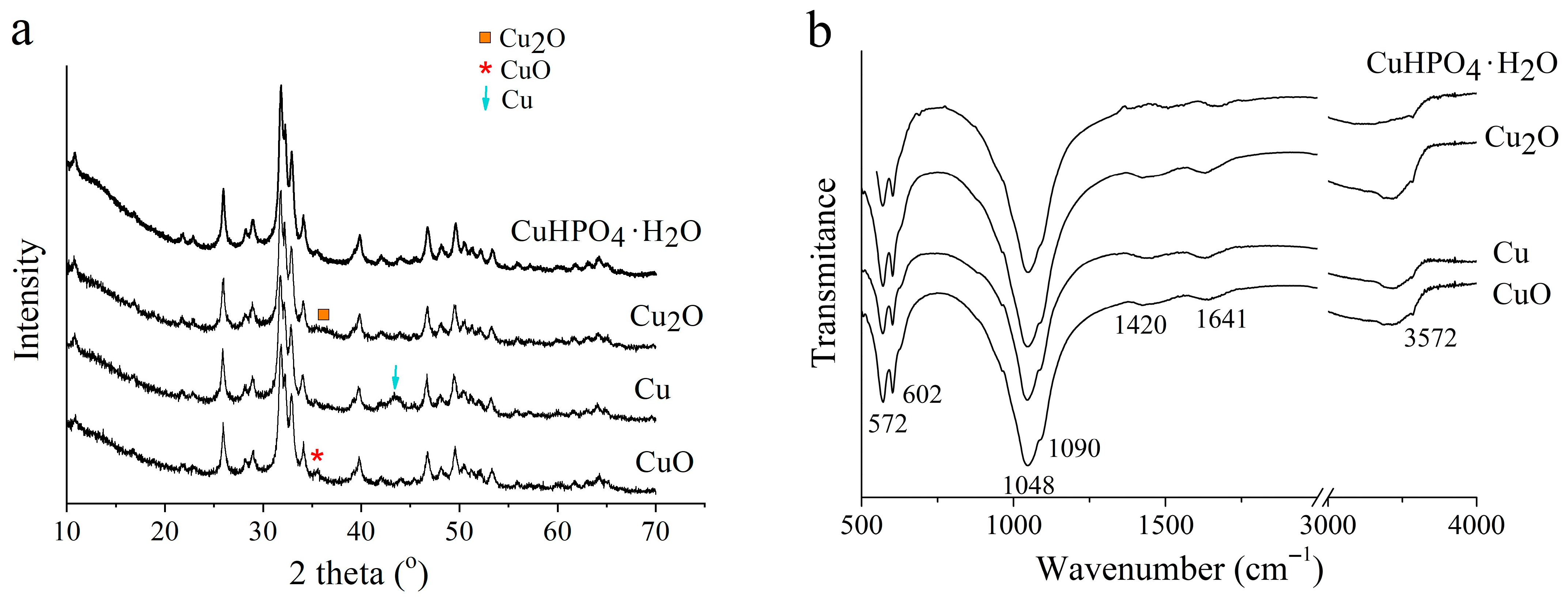
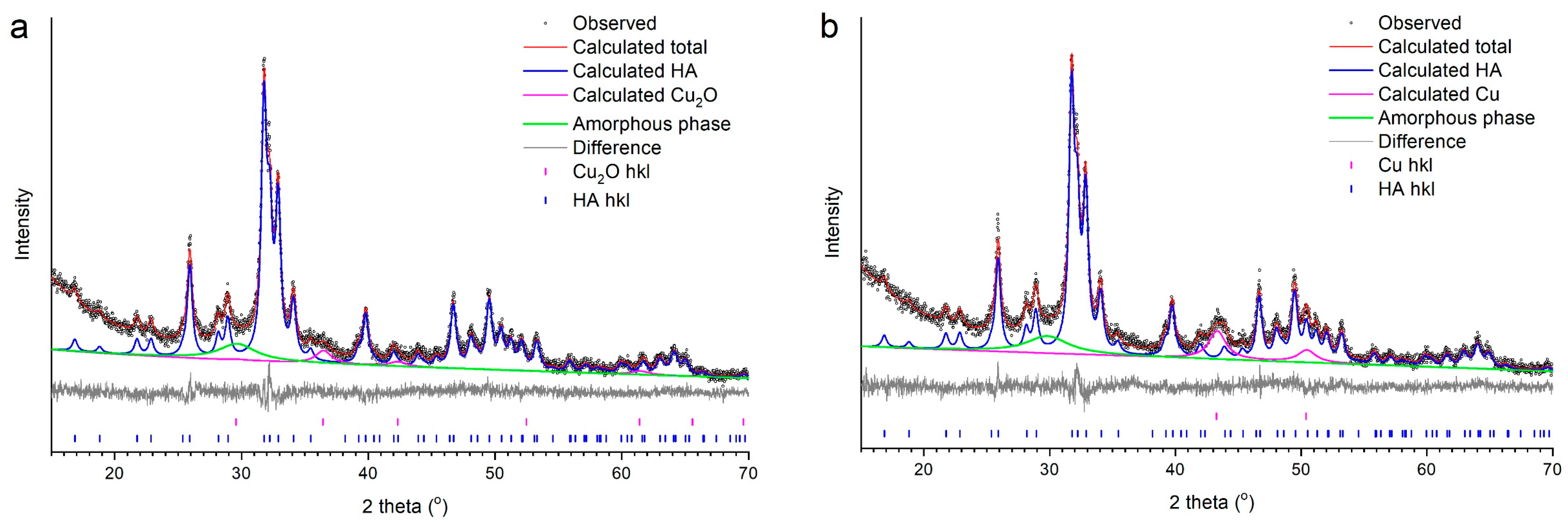
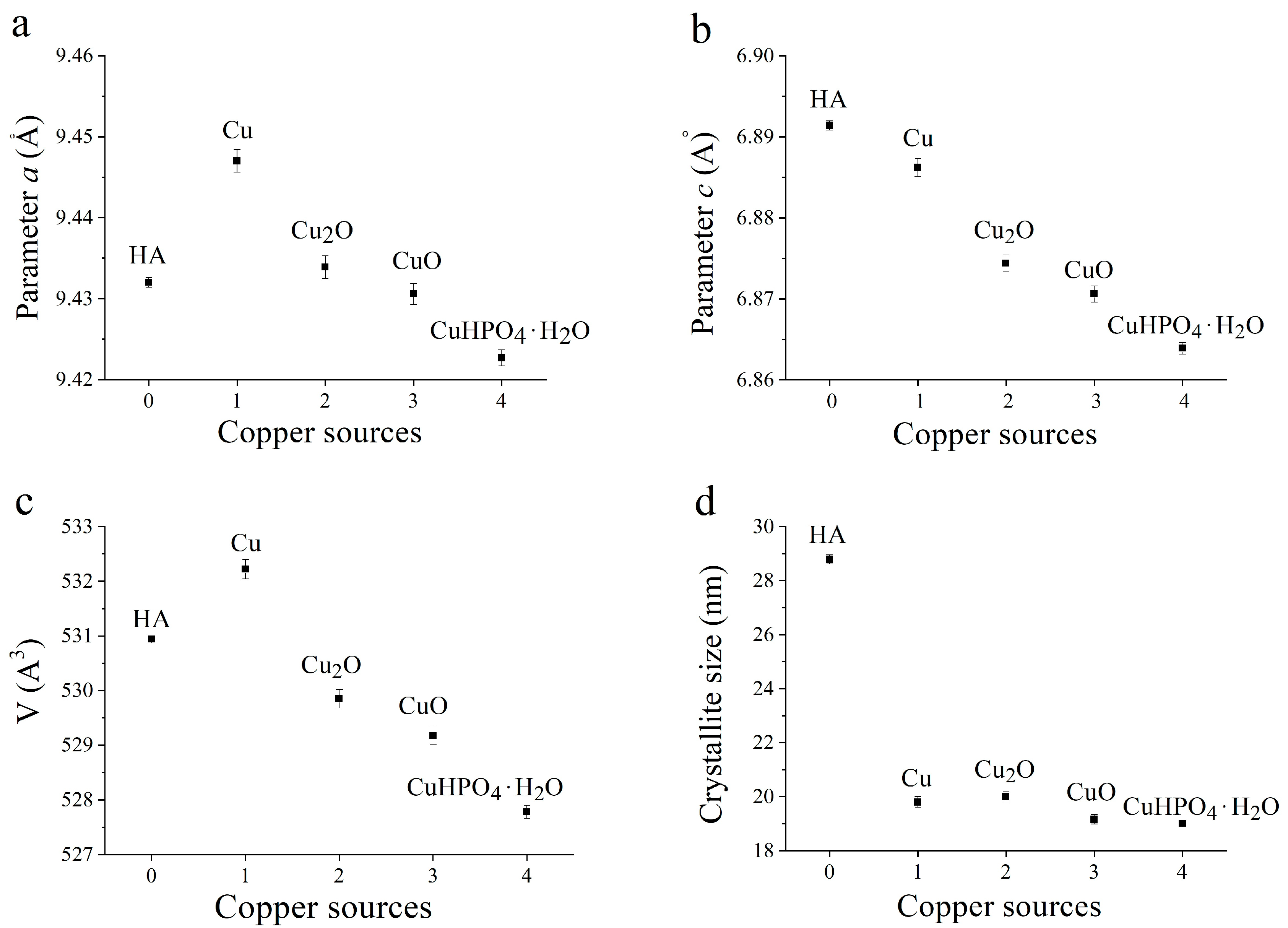
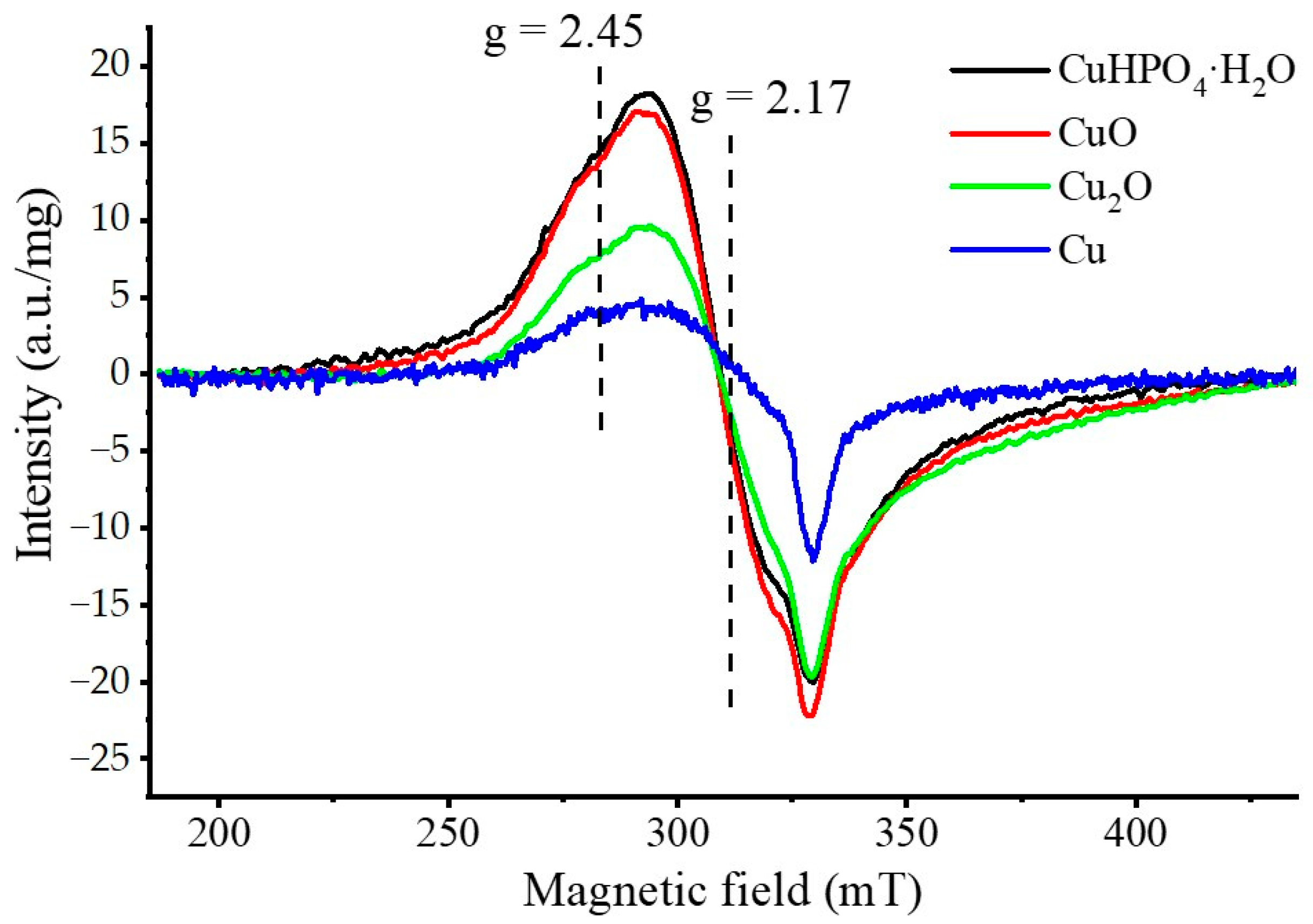
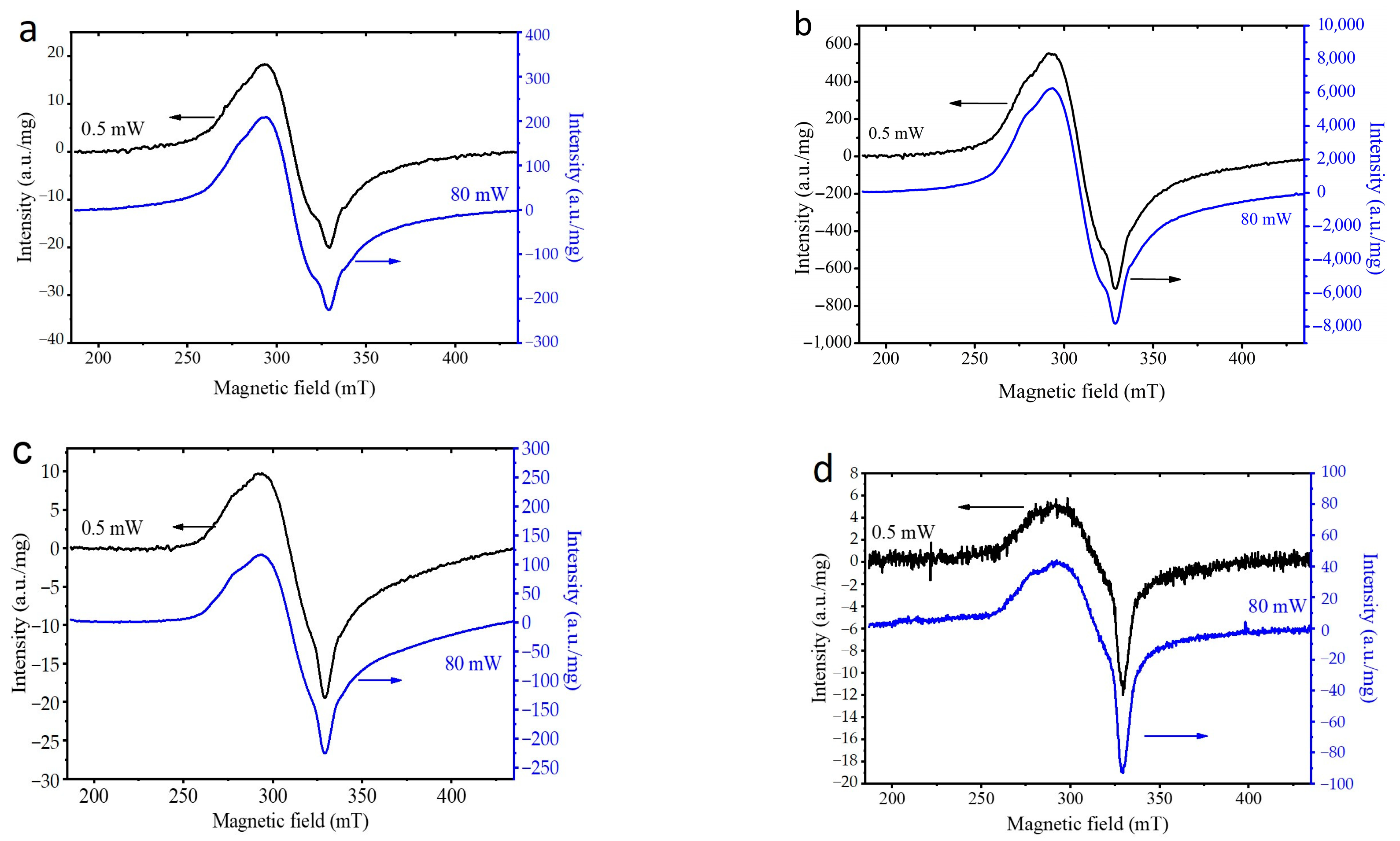
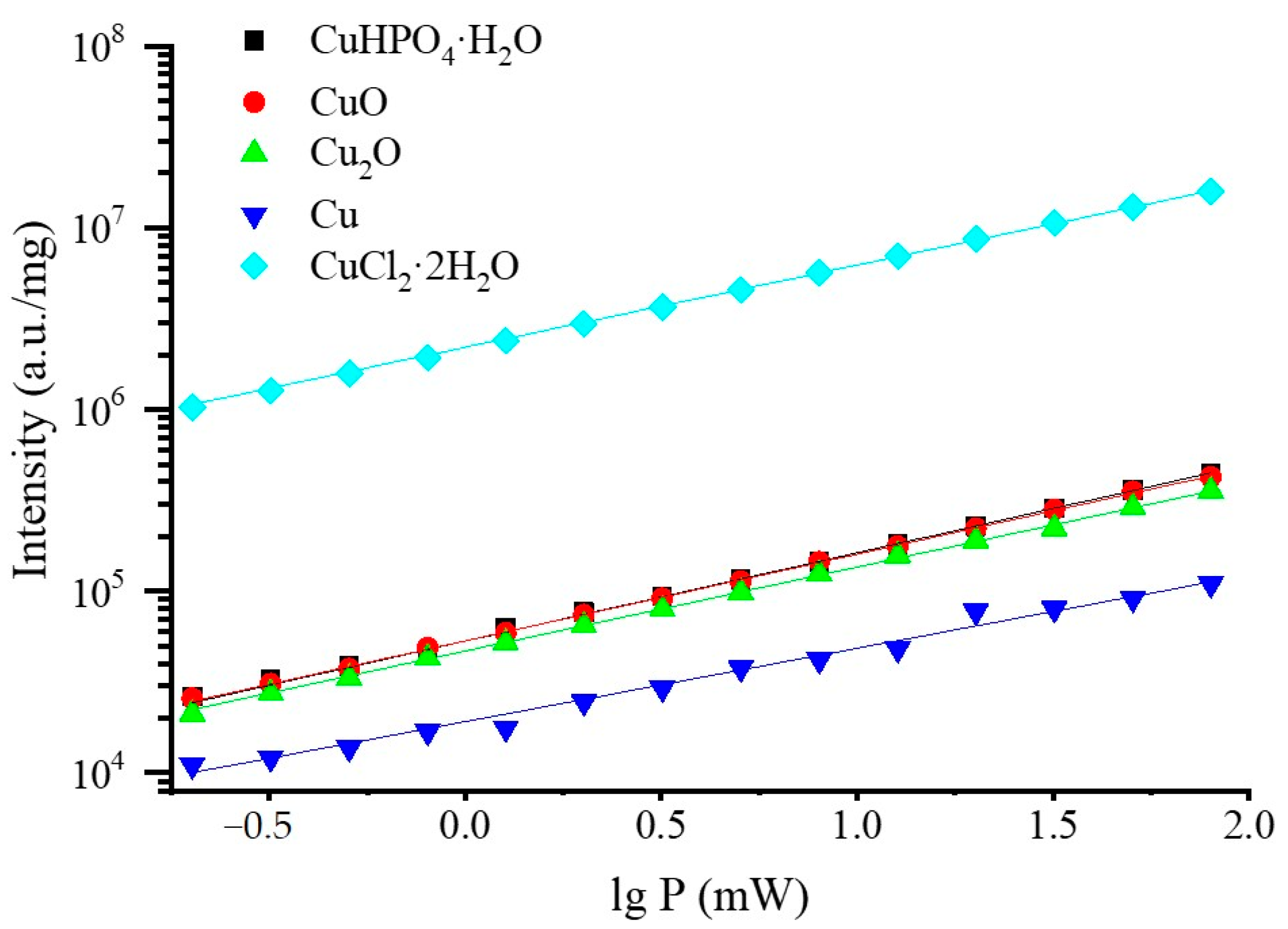
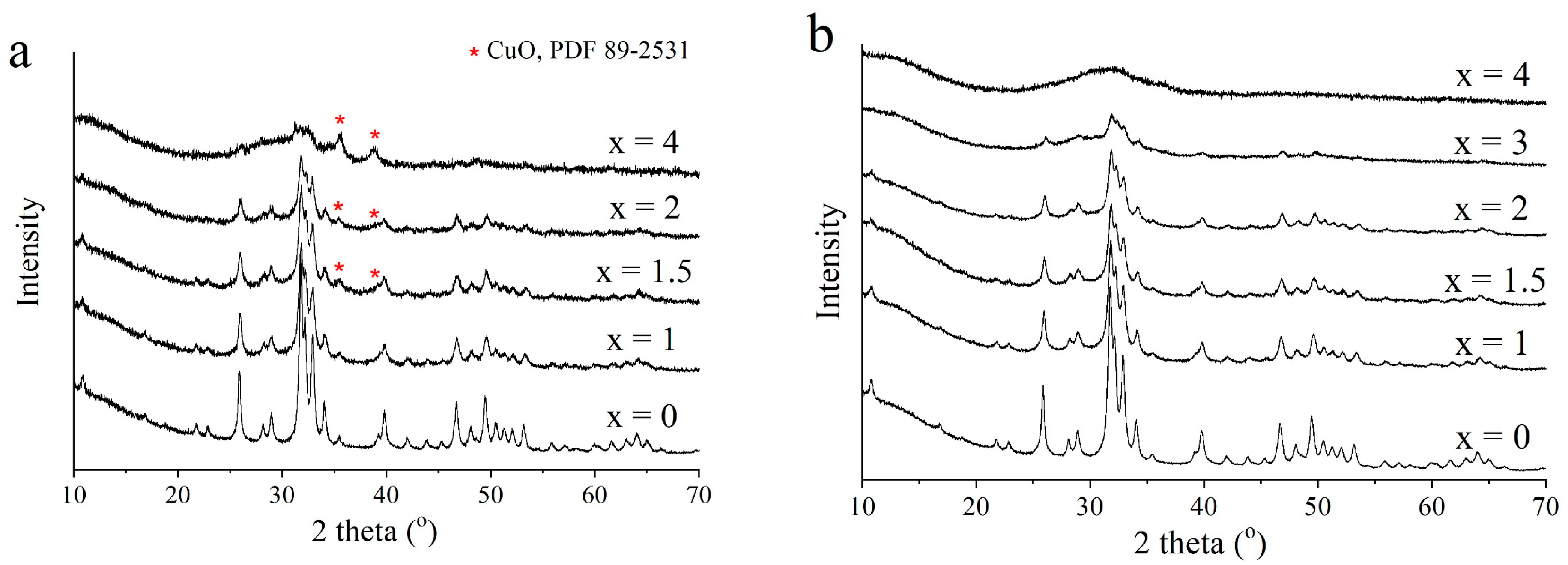
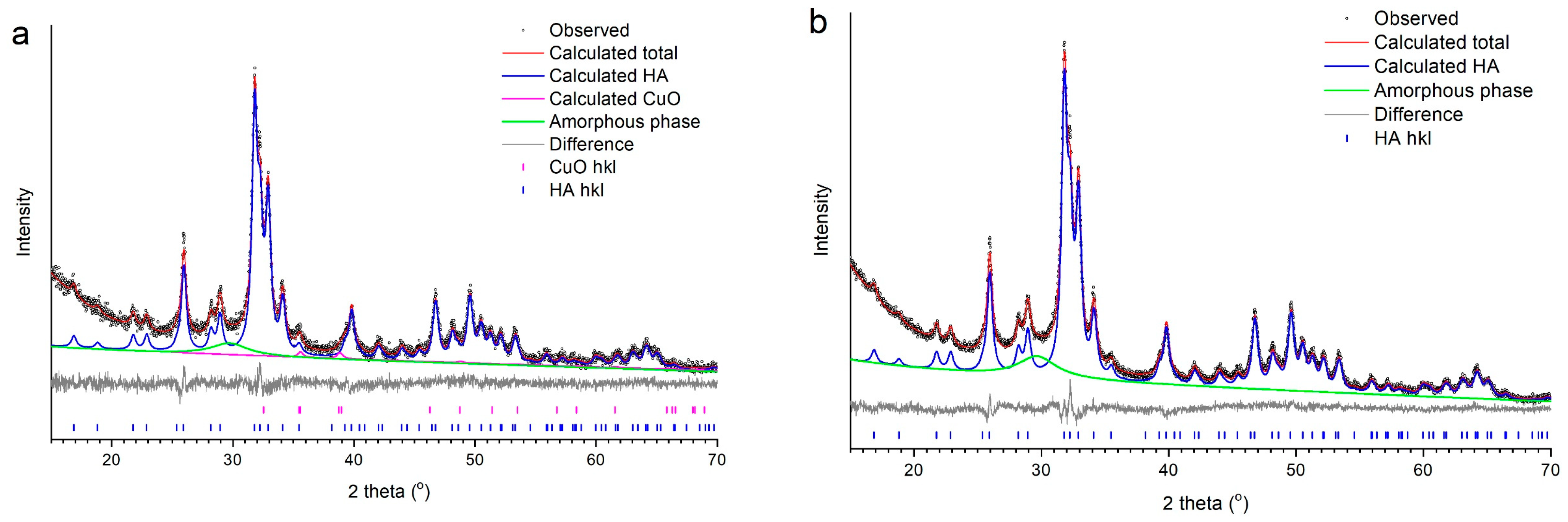
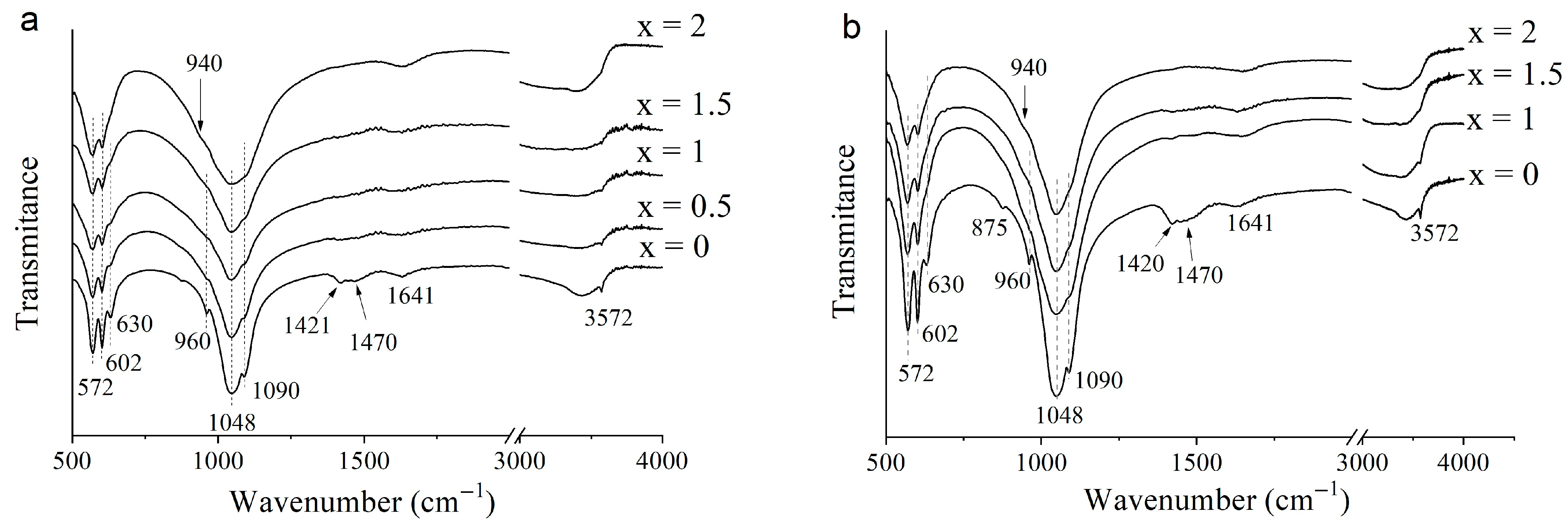
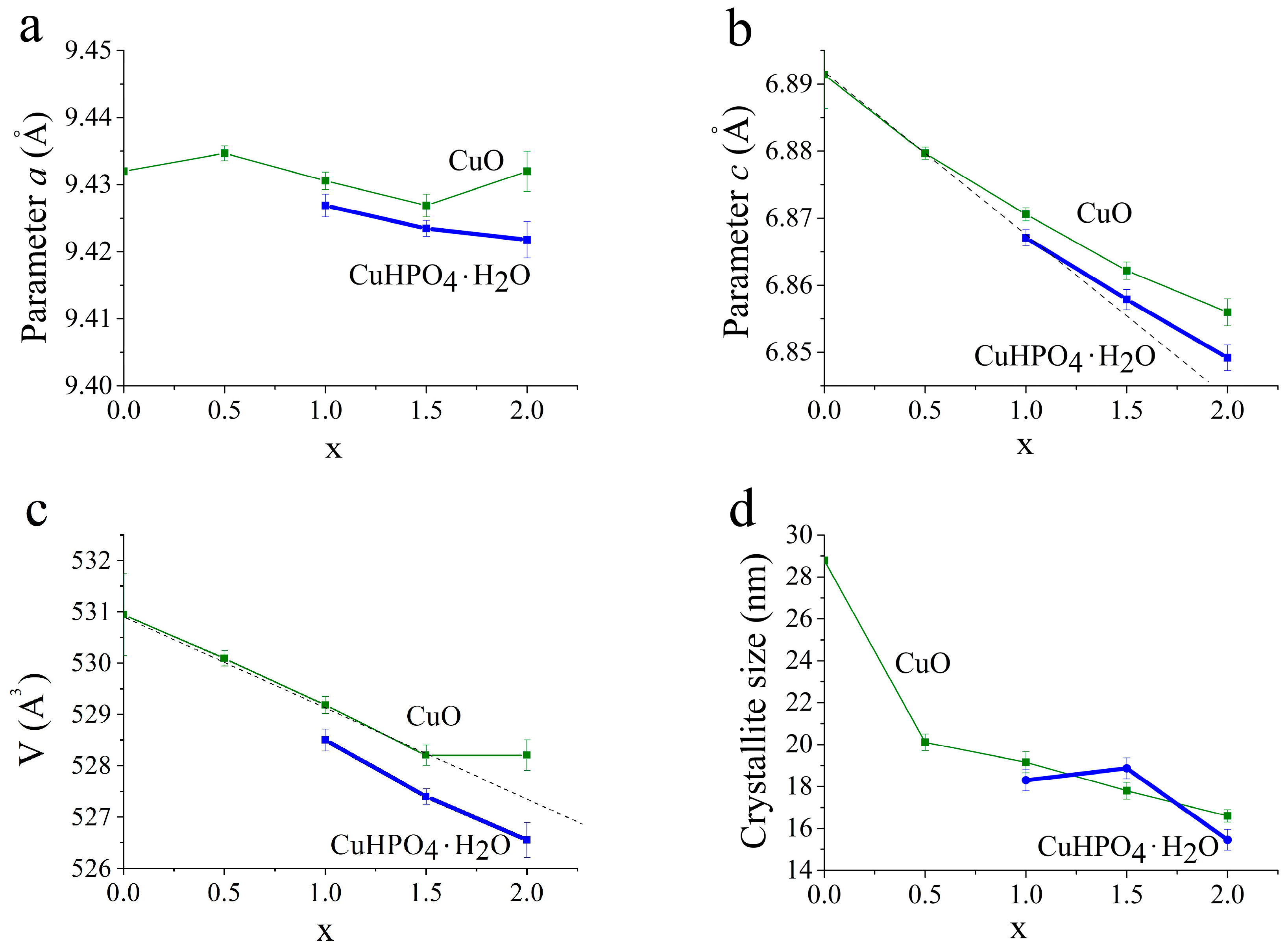
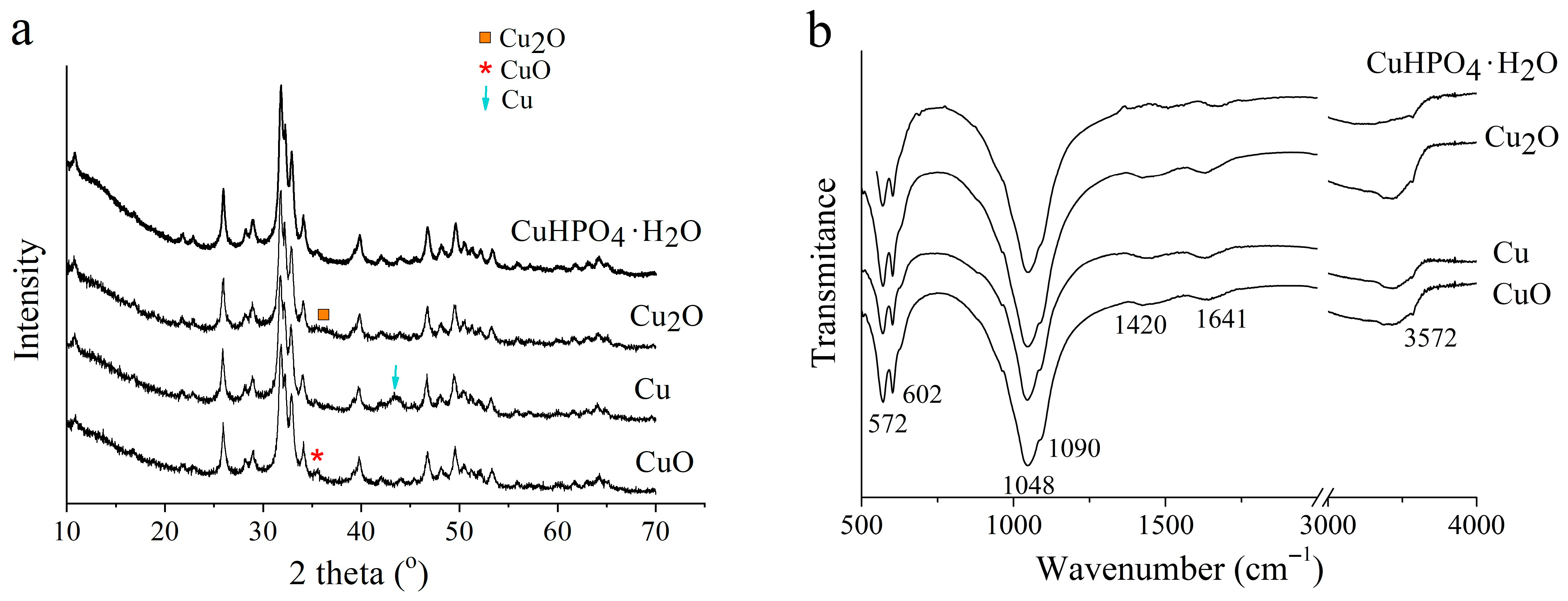
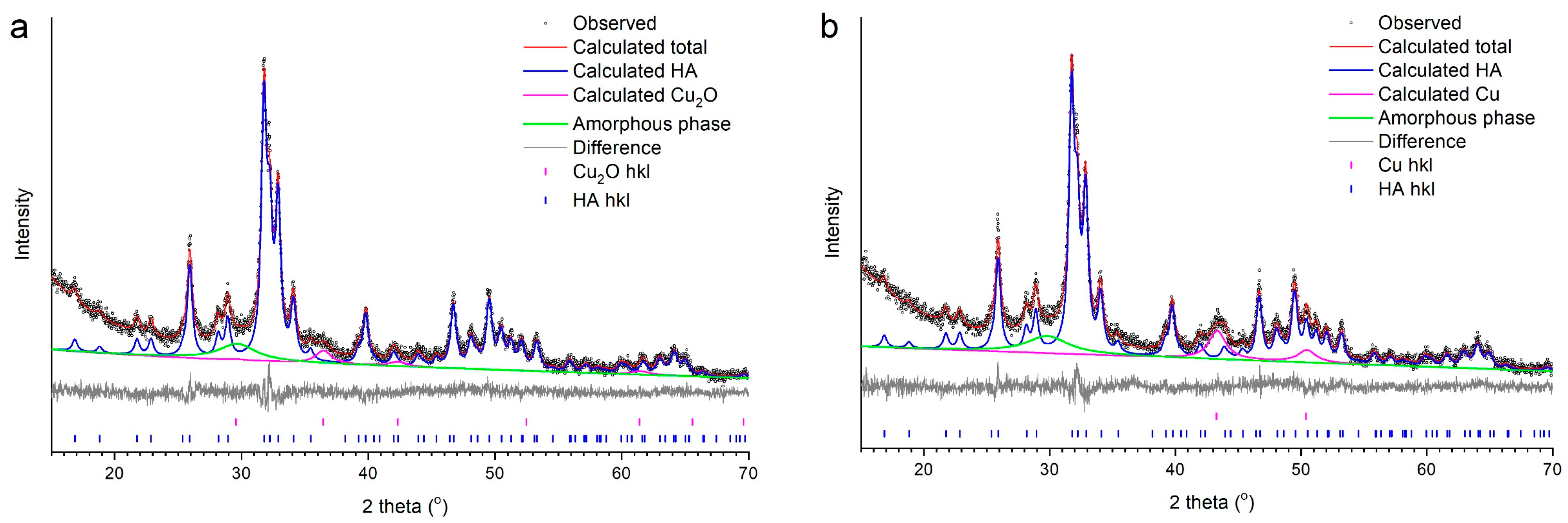
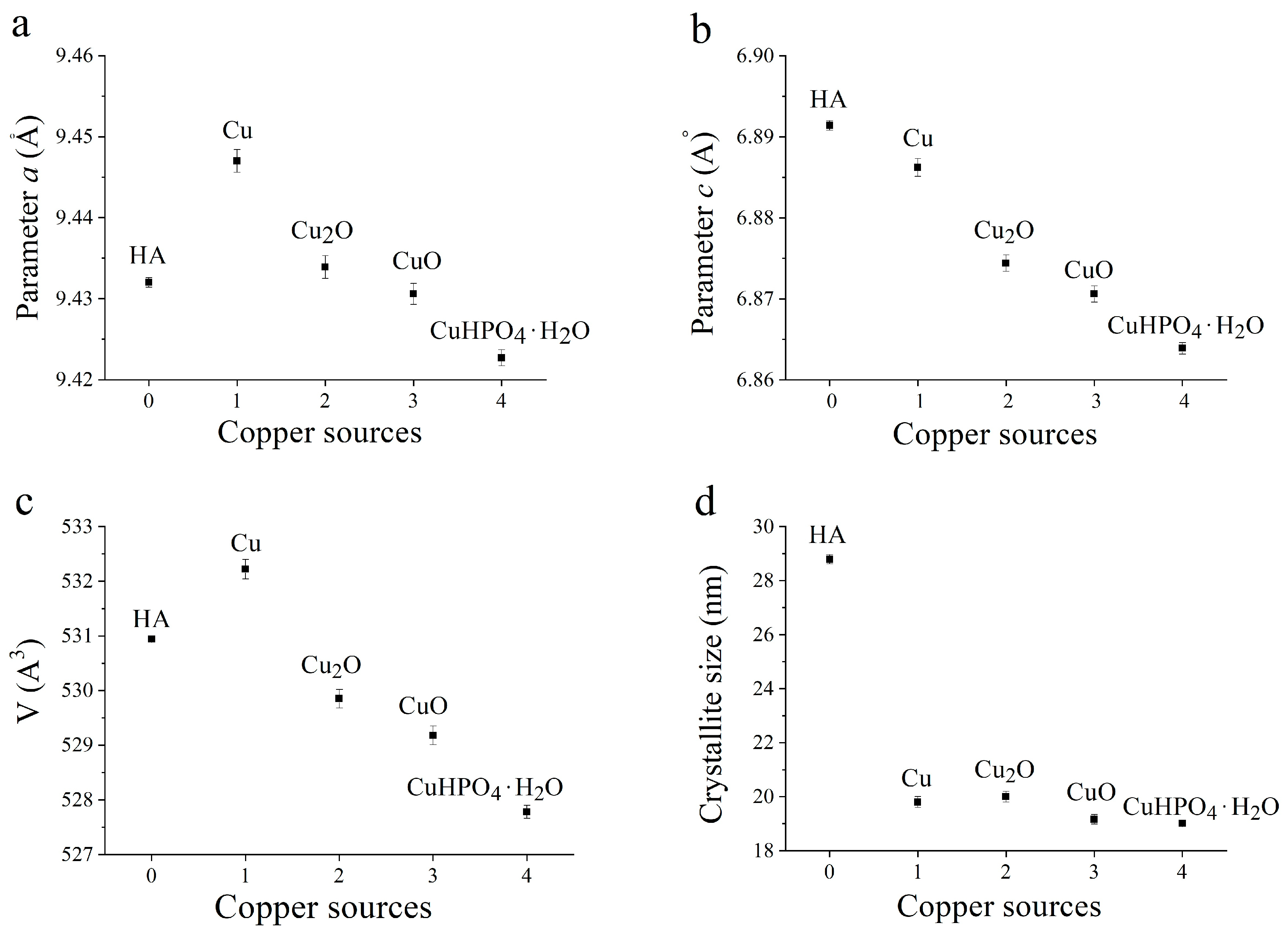
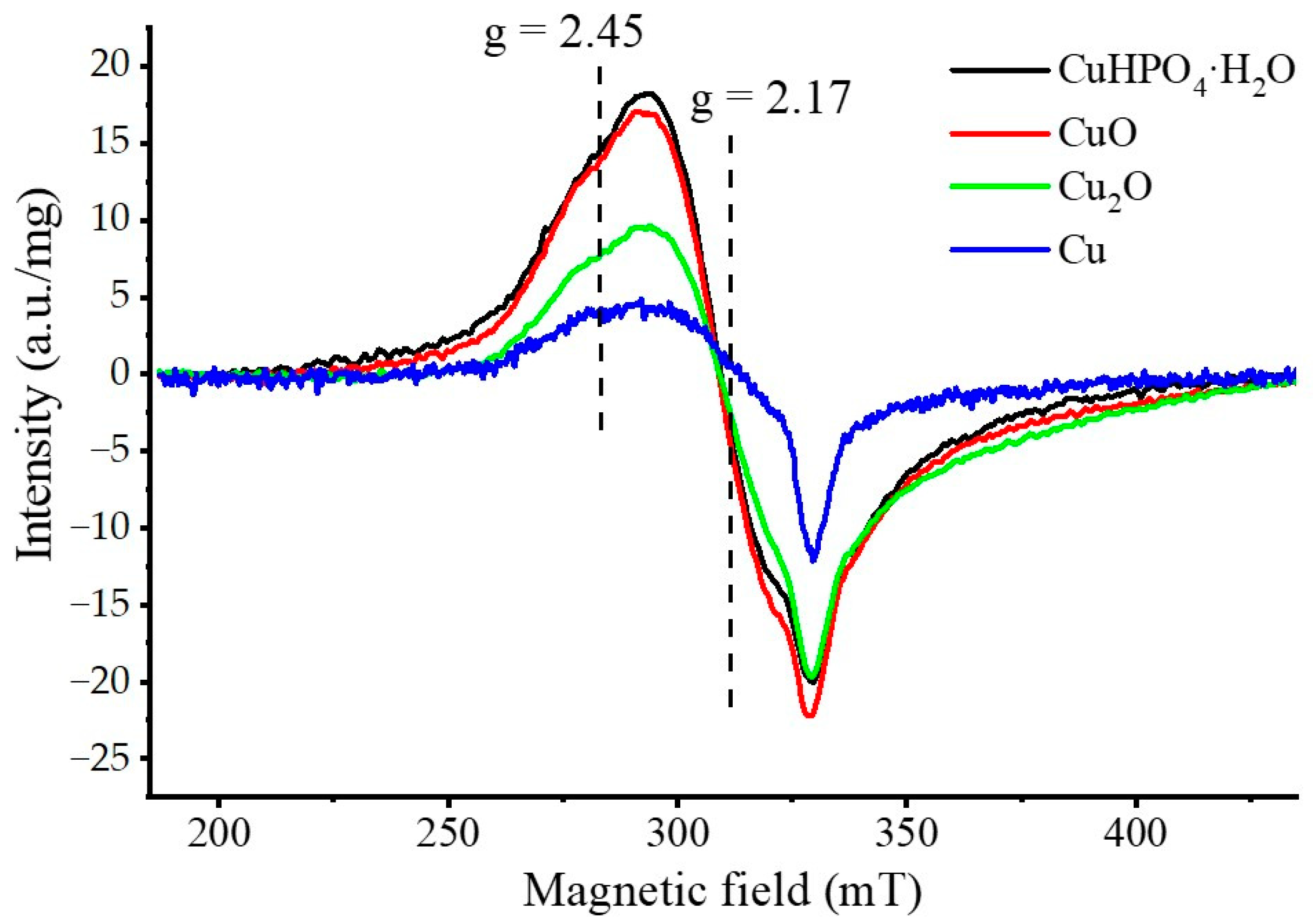
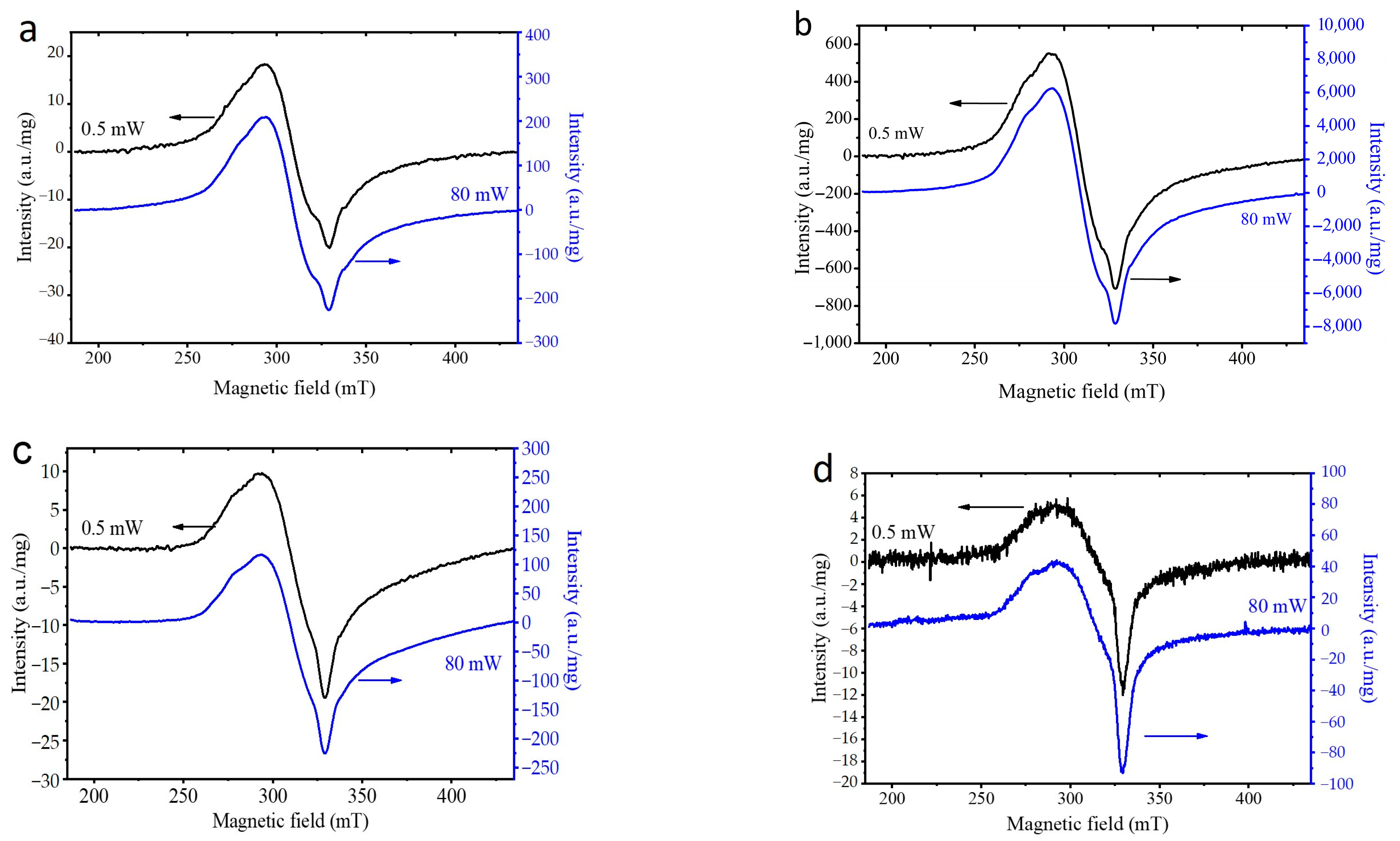
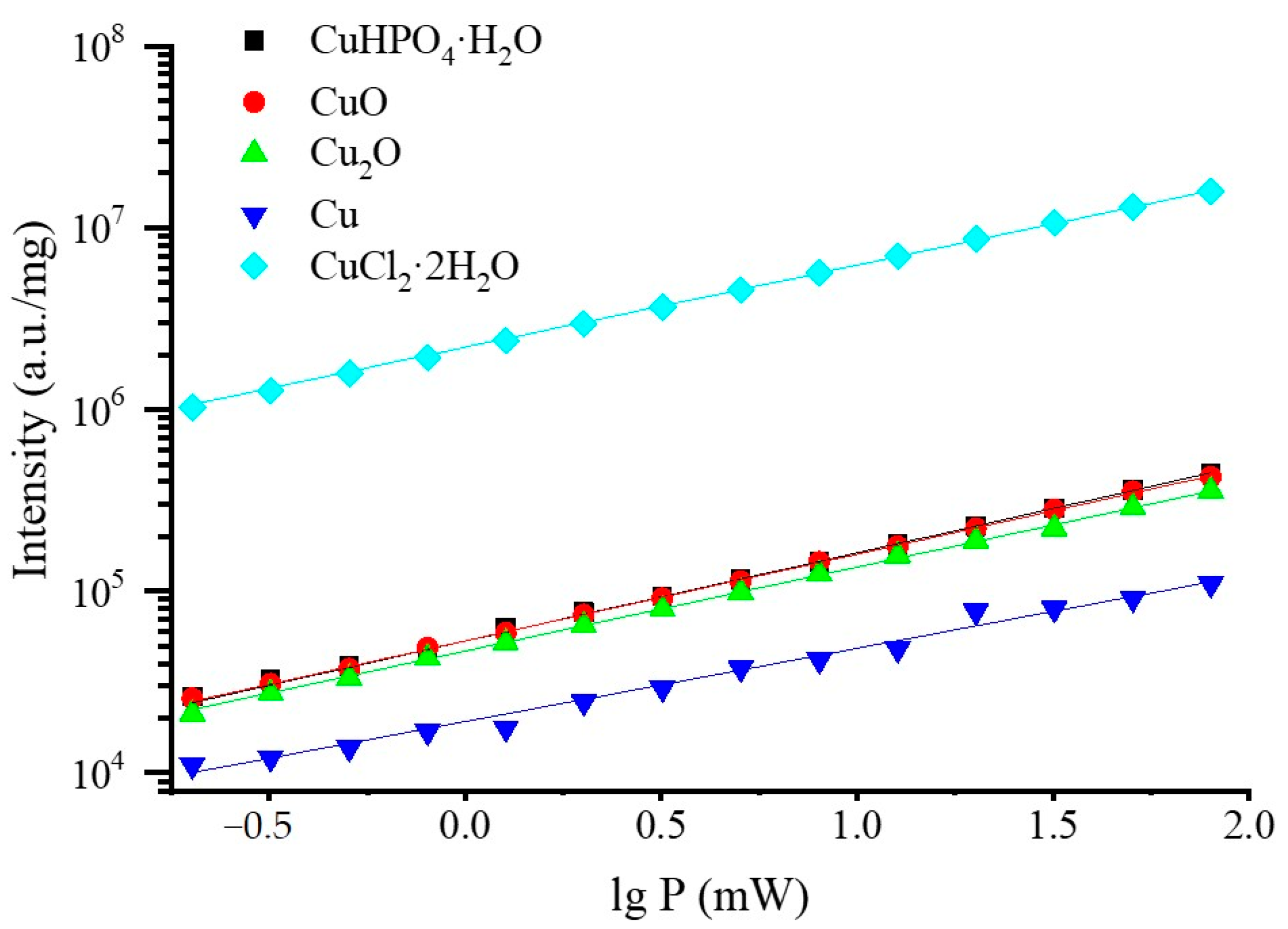
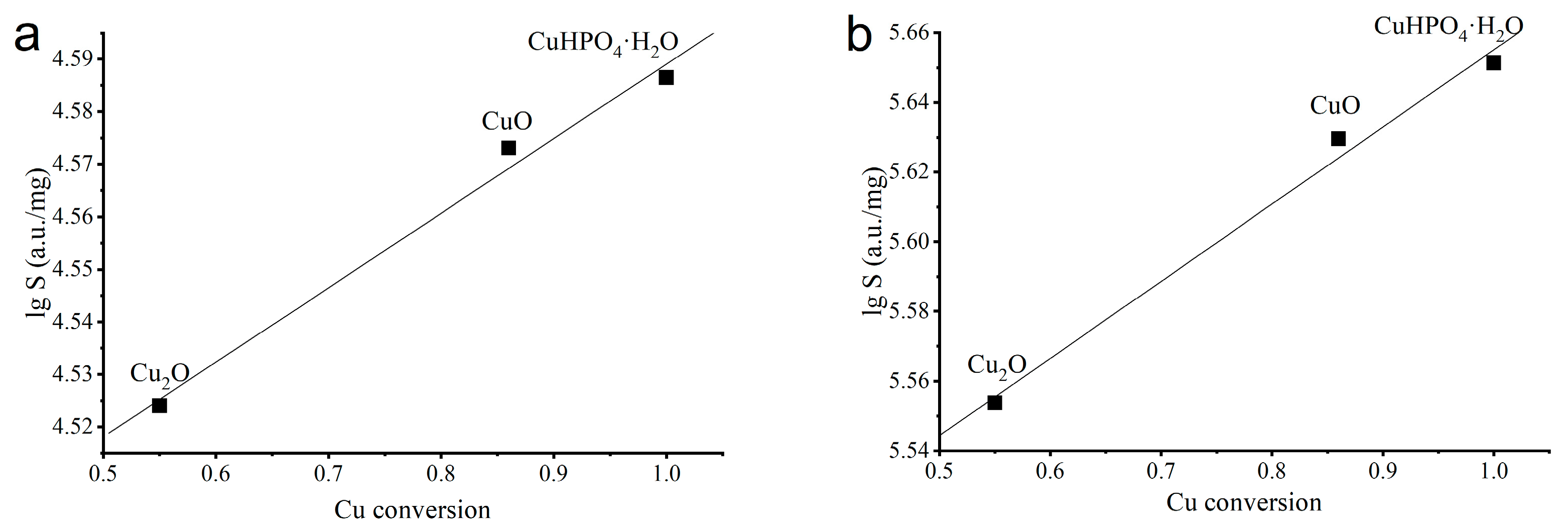
3.2. Selection of the Best Copper Source
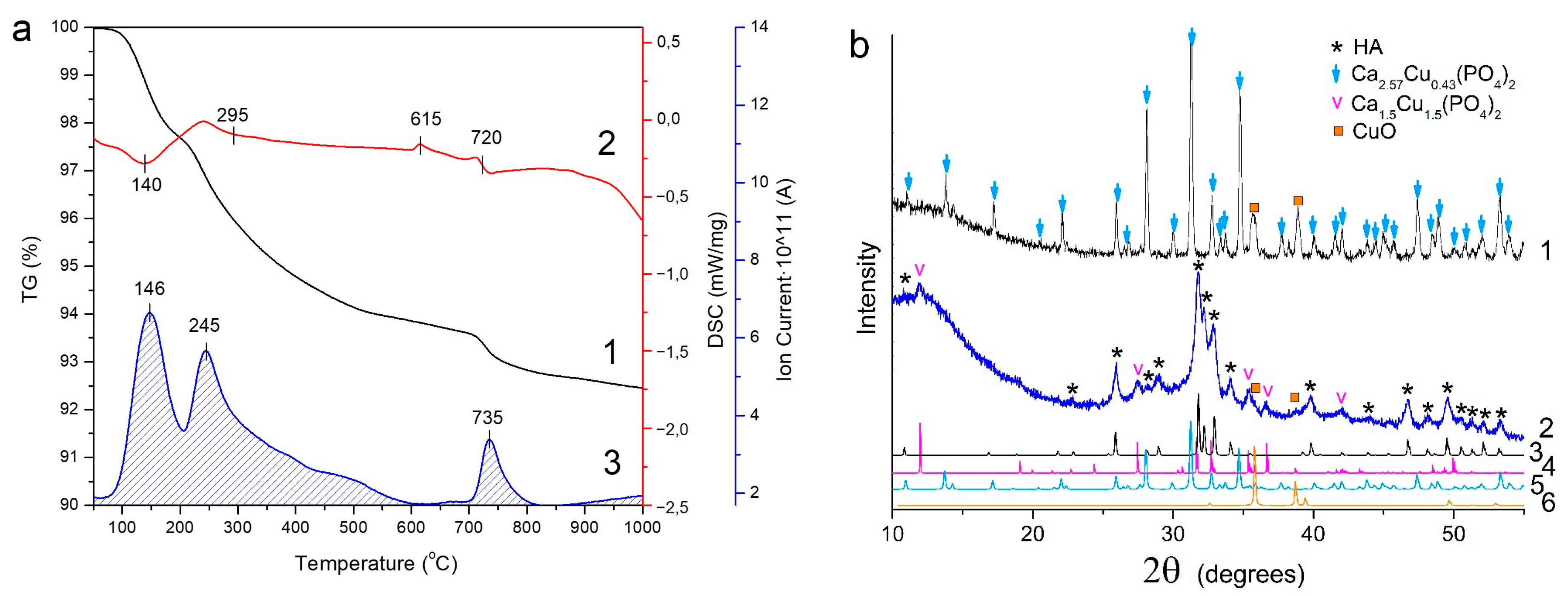
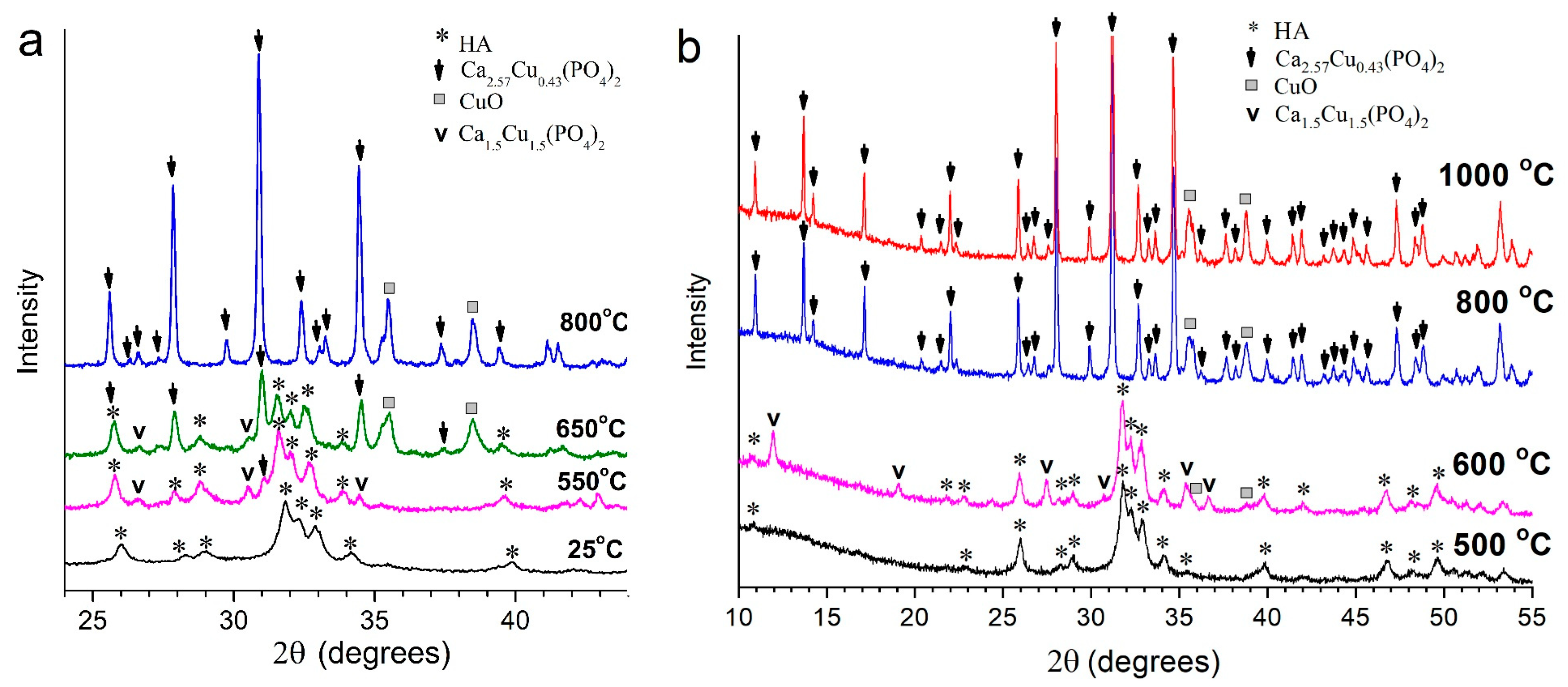
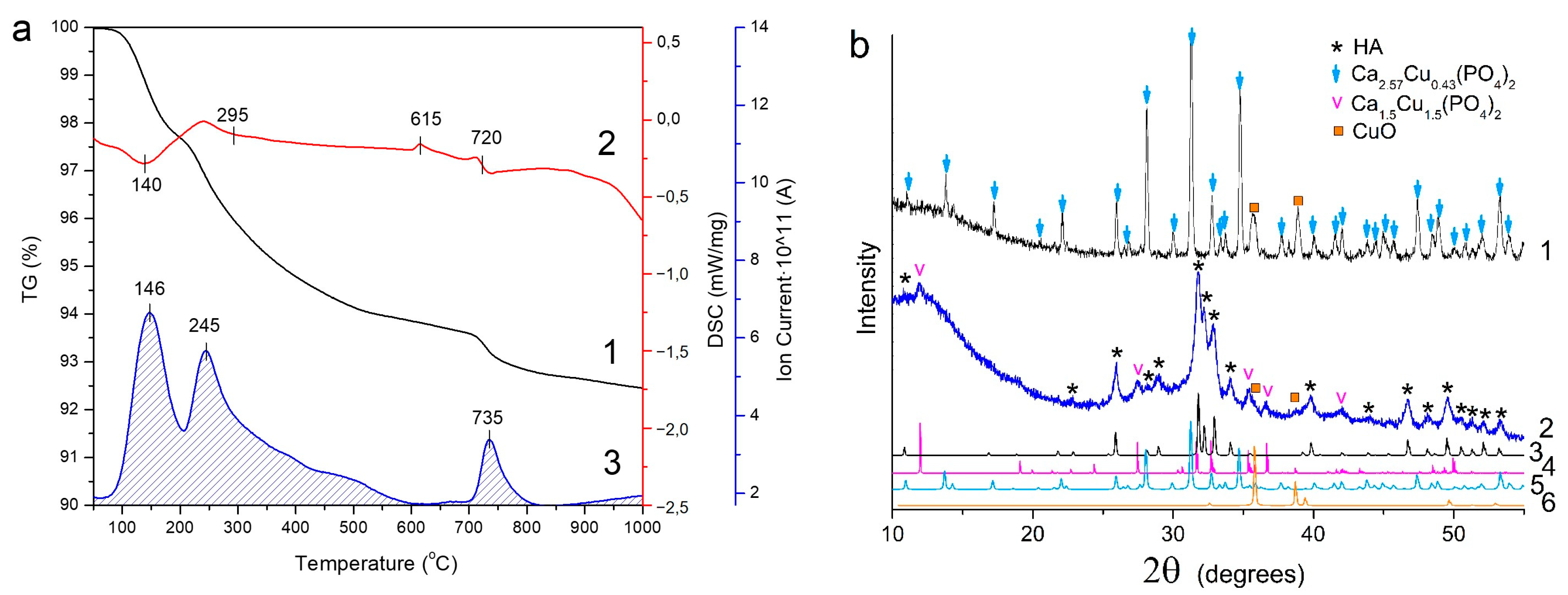
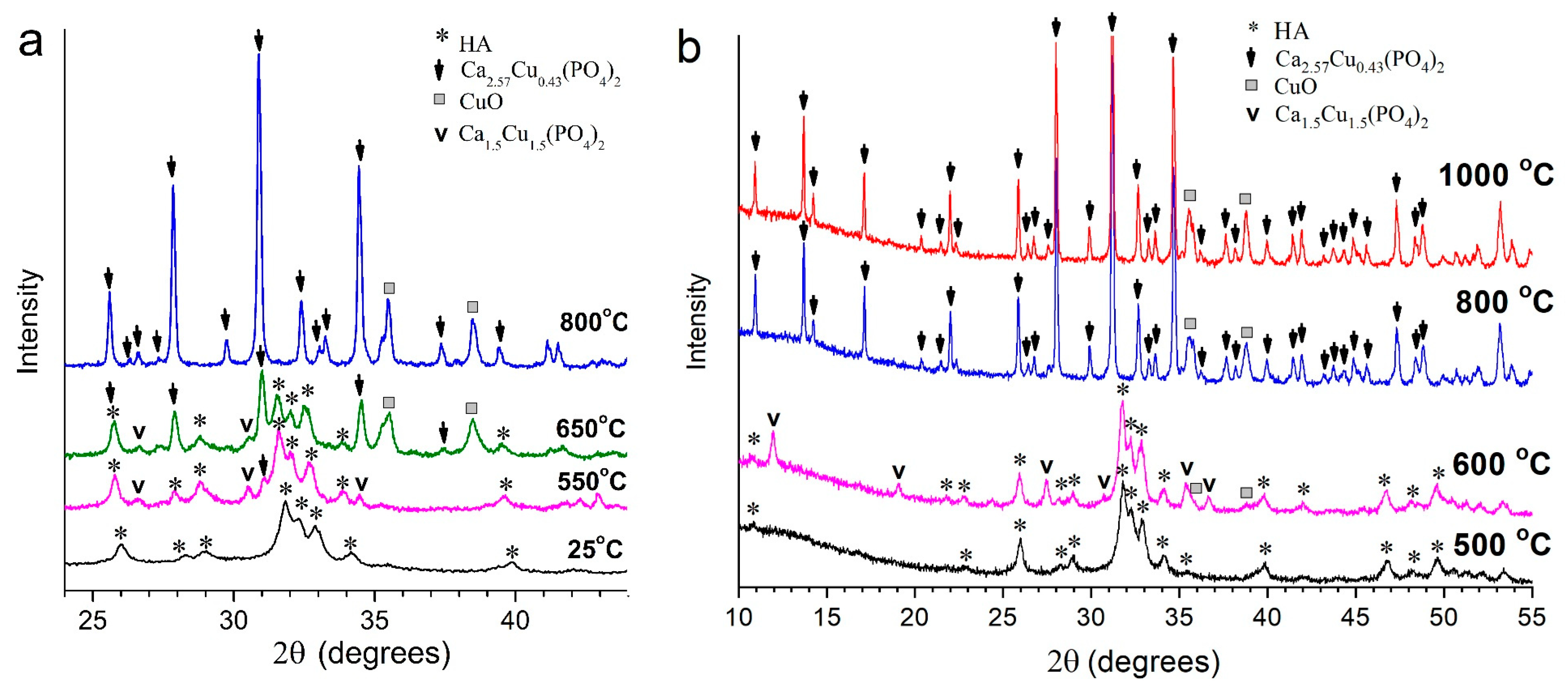
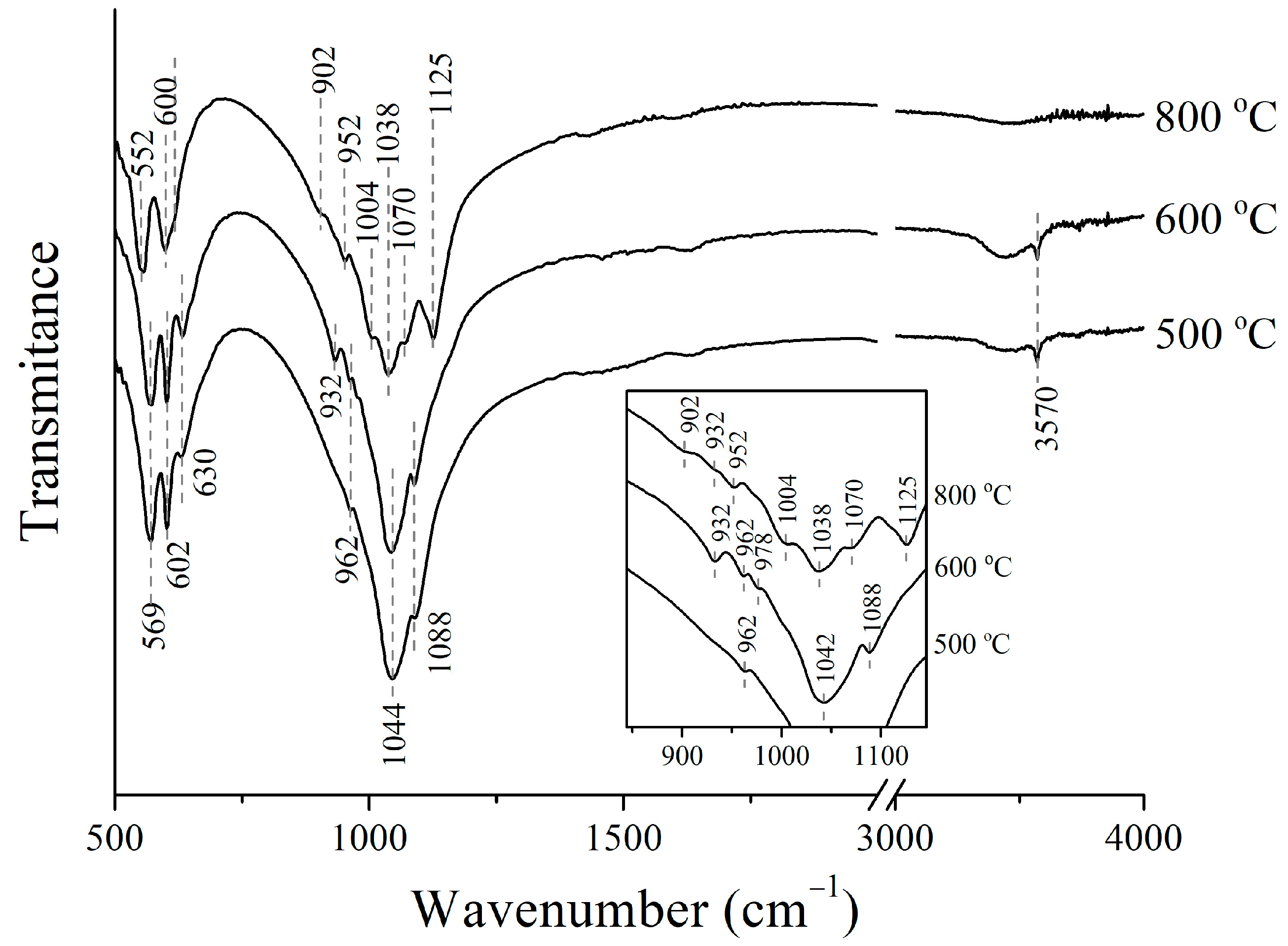
3.3. Thermal Stability of Cu-Substituted Hydroxyapatite
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mucalo, M. Hydroxyapatite (HAp) for Biomedical Applications; Woodhead Publishing Series in Biomaterials; Woodhead Publishing: Cambridge, UK, 2015. [Google Scholar]
- Thirumalai, J. Hydroxyapatite: Advances in Composite Nanomaterials, Biomedical Applications and Its Technological Facets; Intech Open: London, UK, 2018. [Google Scholar]
- Kumar, A.; Kargozar, S.; Baino, F.; Han, S. Additive Manufacturing Methods for Producing Hydroxyapatite and Hydroxyapatite-Based Composite Scaffolds: A review. Front. Mater. 2019, 6, 313. [Google Scholar] [CrossRef]
- Elliott, J.C. Structure and Chemistry of the Apatites and Other Calcium Orthophosphates; Elsevier Science: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Dorozhkin, S.V. Calcium orthophosphate (CaPO4)-based bioceramics: Preparation, properties, and applications. Coatings 2022, 12, 1380. [Google Scholar] [CrossRef]
- Okada, M.; Matsumoto, T. Synthesis and modification of apatite nanoparticles for use in dental and medical applications. Jpn. Dent. Sci. Rev. 2015, 51, 85–95. [Google Scholar] [CrossRef]
- Azari, A.; Nikzad, S.; Yazdani, A.; Atri, F.; Anvari-Yazdi, A.F. Deposition of crystalline hydroxyapatite nano-particle on zirconia ceramic: A potential solution for the poor bonding characteristic of zirconia ceramics to resin cement. J. Mater. Sci. Mater. Med. 2017, 28, 111. [Google Scholar] [CrossRef]
- Yang, X.; Tian, Z.; Guo, K.; Lu, T.; Ji, J. Preparation and mechanism of hydroxyapatite hollow microspheres with different surface charge by biomimetic method. J. Mater. Sci. Mater. Med. 2020, 31, 47. [Google Scholar] [CrossRef]
- Ressler, A.; Žužić, A.; Ivanišević, I.; Kamboj, N.; Ivanković, H. Ionic substituted hydroxyapatite for bone regeneration applications: A review. Open Ceram. 2021, 6, 100122. [Google Scholar] [CrossRef]
- Lu, J.; Yu, H.; Chen, C. Biological properties of calcium phosphate biomaterials for bone repair: A review. RSC Adv. 2018, 8, 2015–2033. [Google Scholar] [CrossRef]
- Supova, M. Substituted hydroxyapatites for biomedical applications: A review. Ceram. Int. 2015, 41, 9203–9231. [Google Scholar] [CrossRef]
- Tite, T.; Popa, A.-C.; Balescu, L.M.; Bogdan, I.M.; Pasuk, I.; Ferreira, J.M.F.; Stan, G.E. Cationic substitutions in hydroxyapatite: Current status of the derived biofunctional effects and their in vitro interrogation methods. Materials 2018, 11, 2081. [Google Scholar] [CrossRef]
- Jiang, Y.; Yuan, Z.; Huang, J. Substituted hydroxyapatite: A recent development. Mater. Technol. 2019, 35, 785–796. [Google Scholar] [CrossRef]
- White, T.J.; Dong, Z.L. Structural derivation and crystal chemistry of apatites. Acta Cryst. 2003, 59, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, X.; Zhao, R.; Mao, H.; Yan, Y.; Pang, X. Antibacterial efficacy, corrosion resistance, and cytotoxicity studies of copper-substituted carbonated hydroxyapatite coating on titanium substrate. J. Mater. Sci. 2015, 50, 1688–1700. [Google Scholar] [CrossRef]
- Jacobs, A.; Renaudin, G.; Forestier, C.; Nedelec, J.-M.; Descamps, S. Biological properties of copper-doped biomaterials for orthopedic applications: A review of antibacterial, angiogenic and osteogenic aspects. Acta Biomater. 2020, 117, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Kolmas, J.; Groszyk, E.; Kwiatkowska-Różycka, D. Substituted hydroxyapatites with antibacterial properties. BioMed Res. Int. 2014, 2014, 178123. [Google Scholar] [CrossRef]
- Barralet, J.; Gbureck, U.; Habibovic, P.; Vorndran, E.; Gerard, C.; Doillon, C.J. Angiogenesis in calcium phosphate scaffolds by inorganic copper ion release. Tissue Eng. 2009, 15, 1601–1609. [Google Scholar] [CrossRef]
- Shi, F.; Liu, Y.; Zhi, W.; Xiao, D.; Li, H.; Duan, K.; Qu, S.; Weng, J. The synergistic effect of micro/nano-structured and Cu2+-doped hydroxyapatite particles to promote osteoblast viability and antibacterial activity. Biomed. Mater. 2017, 12, 035006. [Google Scholar] [CrossRef]
- Renaudin, G.; Gomes, S.; Nedelec, J.M. First-row transition metal doping in calcium phosphate. Bioceramics: A detailed crystallographic study. Materials 2017, 10, 92. [Google Scholar] [CrossRef]
- Gomes, S.; Vichery, C.; Descamps, S.; Martinez, H.; Kaur, A.; Jacobs, A.; Nedelec, J.M.; Renaudin, G. Cu-doping of calcium phosphate bioceramics: From mechanism to the control of cytotoxicity. Acta Biomater. 2018, 65, 462–474. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Fang, Y.; Hooper, T.J.N.; Kelly, N.L.; Gupta, D.; Balani, K.; Manna, I.; Baikie, T.; Bishop, P.T.; White, T.J.; et al. Crystal chemistry and antibacterial properties of cupriferous hydroxyapatite. Materials 2019, 12, 1814. [Google Scholar] [CrossRef]
- Karpov, A.S.; Nuss, J.; Jansen, M.; Kazin, P.E.; Tretyakov, Y.D. Synthesis, crystal structure and properties of calcium and barium hydroxyapatites containing copper ions in hexagonal channels. Solid State Sci. 2003, 5, 1277–1283. [Google Scholar] [CrossRef]
- Imrie, F.E.; Skakle, J.M.S.; Gibson, I.R. Preparation of copper-doped hydroxyapatite with varying x in the composition Ca10(PO4)6CuxOyHz. Bioceram. Dev. Appl. S 2013, 1, 2013. Available online: https://www.semanticscholar.org/paper/Preparation-of-Copper-Doped-Hydroxyapatite-with-x-Imrie-Skakle/81a51347233550839197327f45f889e414b91754?p2df (accessed on 20 April 2023). [CrossRef]
- Pogosova, M.A.; Provotorov, D.I.; Eliseev, A.A.; Kazin, P.E.; Jansen, M. Synthesis and characterization of the Bi-for-Ca substituted copper-based apatite pigments. Dye. Pigment. 2015, 113, 96–101. [Google Scholar] [CrossRef]
- Othmani, M.; Bachoua, H.; Ghandour, Y.; Aissa, A.; Debbabi, M. Synthesis, characterization and catalytic properties of copper-substituted hydroxyapatite nanocrystals. Mater. Res. Bull. 2018, 97, 560–566. [Google Scholar] [CrossRef]
- Bulina, N.; Vinokurova, O.; Eremina, N.; Prosanov, I.; Khusnutdinov, V.; Chaikina, M. Features of solid-phase mechanochemical synthesis of hydroxyapatite doped by copper and zinc ions. J. Solid State Chem. 2021, 296. [Google Scholar] [CrossRef]
- Chaikina, M.V.; Bulina, N.V.; Prosanov, I.Y.; Vinokurova, O.B.; Ischenko, A.V. Structure formation of zinc-substituted hydroxyapatite during mechanochemical synthesis. Inorg. Mater. 2020, 56, 402–408. [Google Scholar] [CrossRef]
- Avvakumov, E.G.; Potkin, A.R.; Samarin, O.I. Planetary Mill. USSR Patent 975068, 26 June 1981. [Google Scholar]
- Rietveld, H.M. A Profile Refinement Method for Nuclear and Magnetic Structures. J. Appl. Cryst. 1969, 2, 65–70. [Google Scholar] [CrossRef]
- Chaikina, M.V.; Bulina, N.V.; Vinokurova, O.B.; Gerasimov, K.B.; Prosanov, I.Y.; Kompankov, N.B.; Lapina, O.B.; Papulovskiy, E.S.; Ischenko, A.V.; Makarova, S.V. Possibilities of mechanochemical synthesis of apatites with different Ca/P ratios. Ceramics 2022, 5, 404–422. [Google Scholar] [CrossRef]
- Chaikina, M.V.; Bulina, N.V.; Vinokurova, O.B.; Prosanov, I.Y.; Dudina, D.V. Interaction of calcium phosphates with calcium oxide or calcium hydroxide during the “soft” mechanochemical synthesis of hydroxyapatite. Ceram. Int. 2019, 45, 16927–16933. [Google Scholar] [CrossRef]
- Sutter, B.; Wasowicz, T.; Howard, T.; Hossner, L.R.; Ming, D.W. Characterization of iron, manganese, and copper synthetic hydroxyapatites by electron paramagnetic resonance spectroscopy. Soil Sci. Soc. Am. J. 2002, 66, 1359–1366. [Google Scholar] [CrossRef]
- Bulina, N.V.; Eremina, N.V.; Vinokurova, O.B.; Ishchenko, A.V.; Chaikina, M.V. Diffusion of copper ions in the lattice of substituted hydroxyapatite during heat treatment. Materials 2022, 15, 5759. [Google Scholar] [CrossRef]
- Ruszala, F.A.; Kostiner, E. The hydrothermal synthesis and crystal growth of various whitlockites and a manganese containing graftonite. J. Cryst. Growth 1980, 48, 473–474. [Google Scholar] [CrossRef]
- Bulina, N.V.; Chaikina, M.V.; Prosanov, I.Y. Mechanochemical synthesis of Sr-substituted hydroxyapatite. Inorg. Mater. 2018, 54, 820–825. [Google Scholar] [CrossRef]
- Chaair, H.; Labjar, H.; Britel, O. Synthesis of β-tricalcium phosphate. Morphologie 2017, 101, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Berzina-Cimdina, L.; Borodajenko, N. Research of calcium phosphates using fourier transform infrared spectroscopy. Infrared Spectrosc. 2012. [Google Scholar] [CrossRef]
- Tõnsuaadu, K.; Gross, K.A.; Plūduma, L.; Veiderma, M. A review on the thermal stability of calcium apatites. J. Therm. Anal. Calorim. 2012, 110, 647–659. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source of Copper | x | Concentration (wt%) | Crystallinity (wt%) | Rwp | χ2 | |||
|---|---|---|---|---|---|---|---|---|
| HA | CuO | Cu2O | Cu | |||||
| CuO | 0.5 | 100 | – | – | – | 98 | 4.3 | 1.2 |
| 0.75 | 99.2 | 0.8 | – | – | 97 | 4.2 | 1.3 | |
| 1.0 | 99 | 1 | – | – | 95 | 4.7 | 1.1 | |
| 1.5 2.0 | 97 95 | 3 5 | – – | – – | 91 83 | 4.3 4.2 | 1.1 1.1 | |
| CuHPO4·H2O | 1.0 | 100 | – | – | – | 92 | 2.8 | 1.4 |
| 1.5 2.0 | 100 100 | – – | – – | – – | 86 54 | 2.2 2.0 | 1.1 1.2 | |
| 3.0 | 100 | – | – | – | 29 | 1.8 | 1.1 | |
| Cu2O | 1.0 | 97 | – | 3 | – | 94 | 4.8 | 1.1 |
| Cu | 1.0 | 94 | – | – | 6 | 93 | 4.8 | 1.1 |
| Sample | t |
|---|---|
| Cu-HA from CuHPO4·H2O | 0.8929 ± 0.005 |
| Cu-HA from CuO | 0.91044 ± 0.008 |
| Cu-HA from Cu2O | 0.93951 ± 0.012 |
| Cu-HA from Cu | 1.07505 ± 0.052 |
| CuCl2·2H2O | 0.95962 ± 0.006 |
| Type of Experiment | Temperature (°C) | Concentration (wt%) | |||
|---|---|---|---|---|---|
| Cu–HA | Ca1.5Cu1.5(PO4)2 (Monoclinic) | Ca2.57Cu0.43(PO4)2 (Rhombohedral) | CuO | ||
| STA measurement | 640 | 86 | 13 | – | >1 |
| 1000 | – | – | 91 | 9 | |
| In situ diffraction | 550 | 86 | 7 | 7 | – |
| 650 | 47 | 5 | 42 | 12 | |
| 800 | – | – | 93 | 7 | |
| Heating in the furnace | 500 | 100 | – | – | – |
| 600 | 76 | 22 | – | 3 | |
| 800 | – | – | 92 | 8 | |
| 1000 | – | – | 92 | 8 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eremina, N.V.; Bulina, N.V.; Mikhailenko, M.A.; Vinokurova, O.B.; Prosanov, I.Y.; Chaikina, M.V. Cu-Substituted Hydroxyapatite Powder: Mechanochemical Synthesis Using Different Copper Sources and Thermal Stability. Powders 2023, 2, 678-696. https://doi.org/10.3390/powders2040042
Eremina NV, Bulina NV, Mikhailenko MA, Vinokurova OB, Prosanov IY, Chaikina MV. Cu-Substituted Hydroxyapatite Powder: Mechanochemical Synthesis Using Different Copper Sources and Thermal Stability. Powders. 2023; 2(4):678-696. https://doi.org/10.3390/powders2040042
Chicago/Turabian StyleEremina, Natalya V., Natalia V. Bulina, Mikhail A. Mikhailenko, Olga B. Vinokurova, Igor Y. Prosanov, and Marina V. Chaikina. 2023. "Cu-Substituted Hydroxyapatite Powder: Mechanochemical Synthesis Using Different Copper Sources and Thermal Stability" Powders 2, no. 4: 678-696. https://doi.org/10.3390/powders2040042
APA StyleEremina, N. V., Bulina, N. V., Mikhailenko, M. A., Vinokurova, O. B., Prosanov, I. Y., & Chaikina, M. V. (2023). Cu-Substituted Hydroxyapatite Powder: Mechanochemical Synthesis Using Different Copper Sources and Thermal Stability. Powders, 2(4), 678-696. https://doi.org/10.3390/powders2040042