
Harnessing Nitrous Oxide for Sustainable Methane Activation: A Computational Exploration of CNC-Ligated Iron Catalysts

Abstract
1. Introduction
- Electronic Flexibility: The CNC framework offers strong σ-donation and π-acceptance, stabilizing high-valent iron–oxo intermediates crucial for OAT and methane C–H activation. The absence of BR2 groups eliminates potential electronic interferences, allowing the study to focus on primary metal–ligand interactions [52].
- Steric Profile: Excluding bulky BR2 substituents ensures a compact coordination environment around the iron center. This steric simplicity reduces hindrance at the active site, facilitating efficient catalytic turnover [44].
- Catalyst Modularity: The simplified ligand design enables direct comparisons with previously studied systems that include BR2 groups, isolating the effects of the primary coordination sphere. This approach provides clearer mechanistic insights into the role of the Fe center [53].
- Suitability for High-Valent Oxo Complexes: The CNC ligand’s electronic properties support the stabilization of FeIV=O intermediates, which are critical for methane C–H activation. Excluding BR2 substituents avoids secondary interactions that might introduce competing pathways, simplifying mechanistic investigations [31,54,55,56,57,58].
Through Computational Modeling, This Research Aims to Achieve the Following
- Examine the impact of spin states on methane activation pathways.
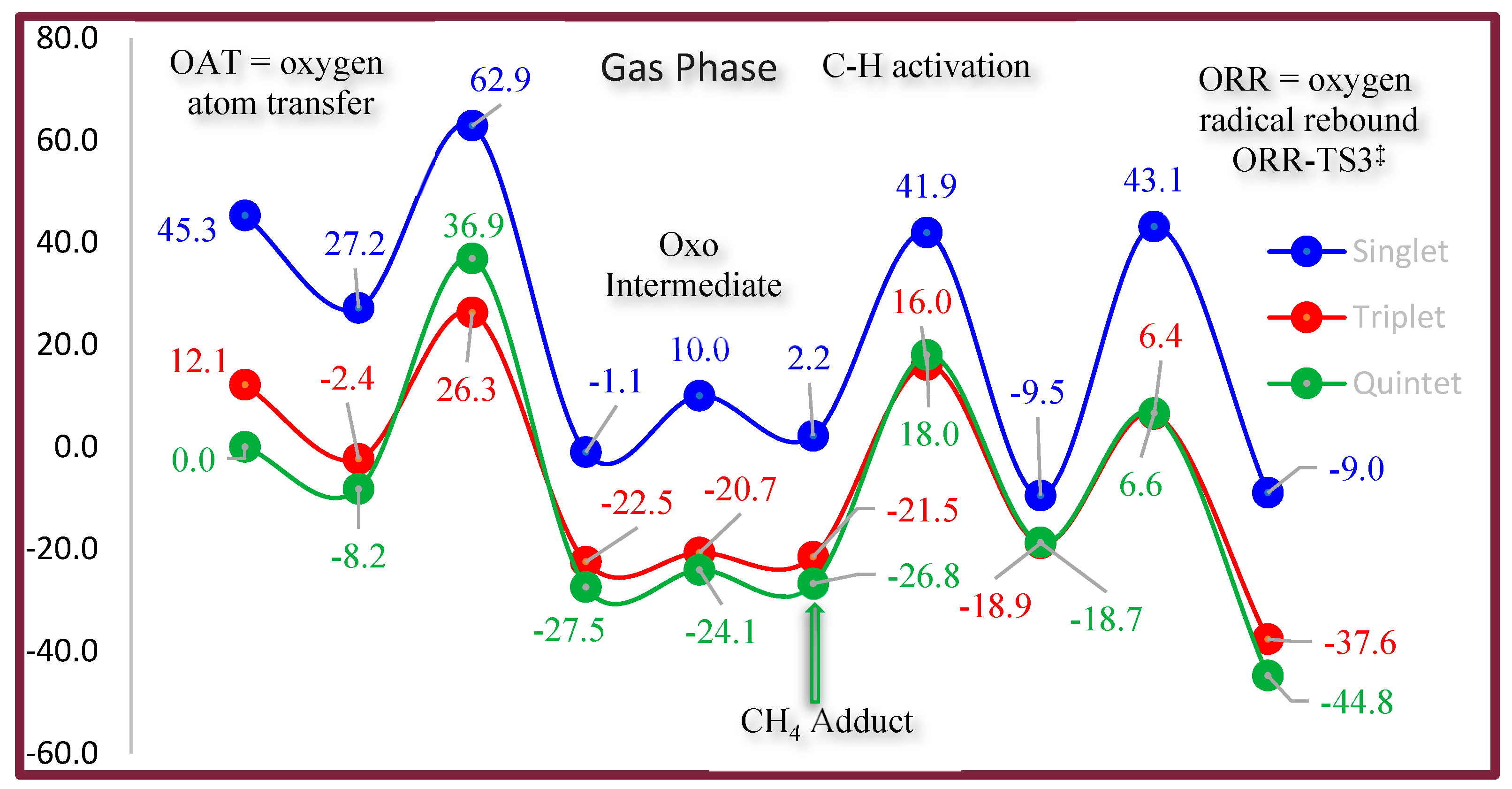
- Assess the activation barriers for the OAT, HAA, and ORR steps.
- Explore the role of CNC ligands in stabilizing reactive intermediates and facilitating efficient catalysis.
2. Computational Methods
2.1. Level of Theory
2.2. Geometric Optimization and Frequency Analysis
2.3. Spin-State Considerations
2.4. Energetic Calculations
2.5. Computational Model
2.6. Reaction Pathways Investigated
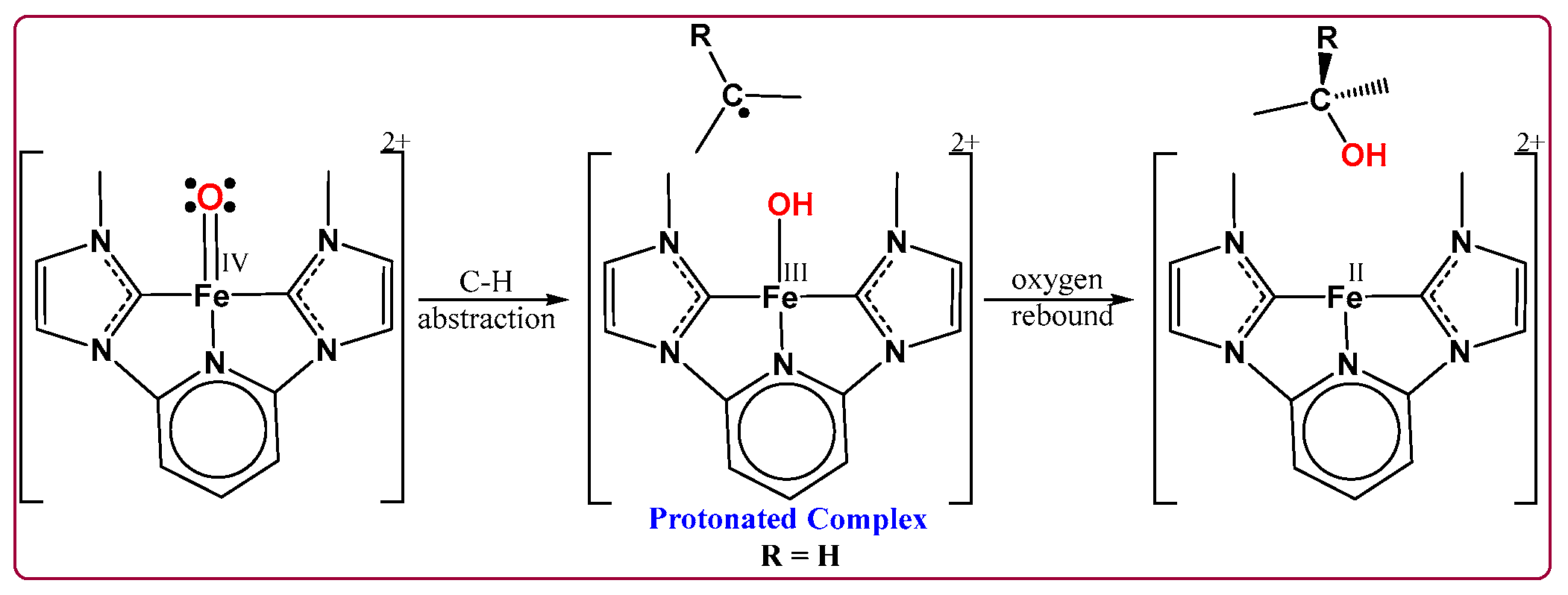
- Oxygen Atom Transfer (OAT): Coordination of N2O to the Fe center and oxygen atom transfer to form the high-valent FeIV=O intermediate.
- Hydrogen Atom Abstraction (HAA): Interaction of the FeIV=O species with methane to cleave the C–H bond.
- Oxygen Radical Rebound (ORR): Rebound of the hydroxyl group with the methyl radical to yield methanol.
2.7. Software and Computational Resources
3. Results and Discussion
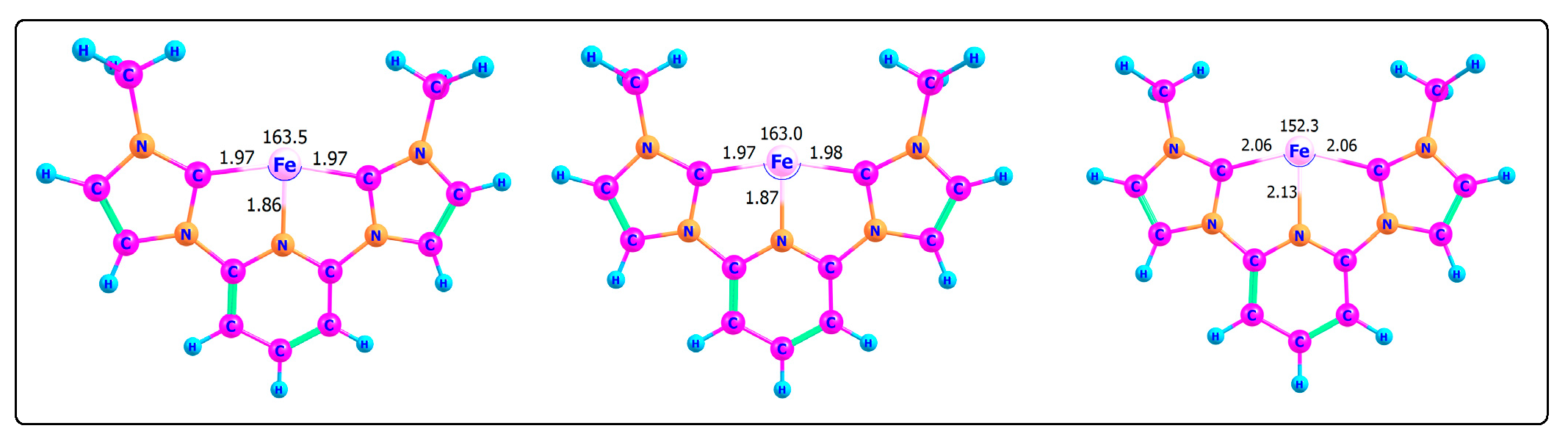
3.1. [(CNC)FeII]2+ Cation Complexes
3.2. Geometric and Spin-State Analysis
3.3. Electronic Structure and Reactivity Implications
3.4. Conclusion for [(CNC)FeII]2+ Cation Complexes
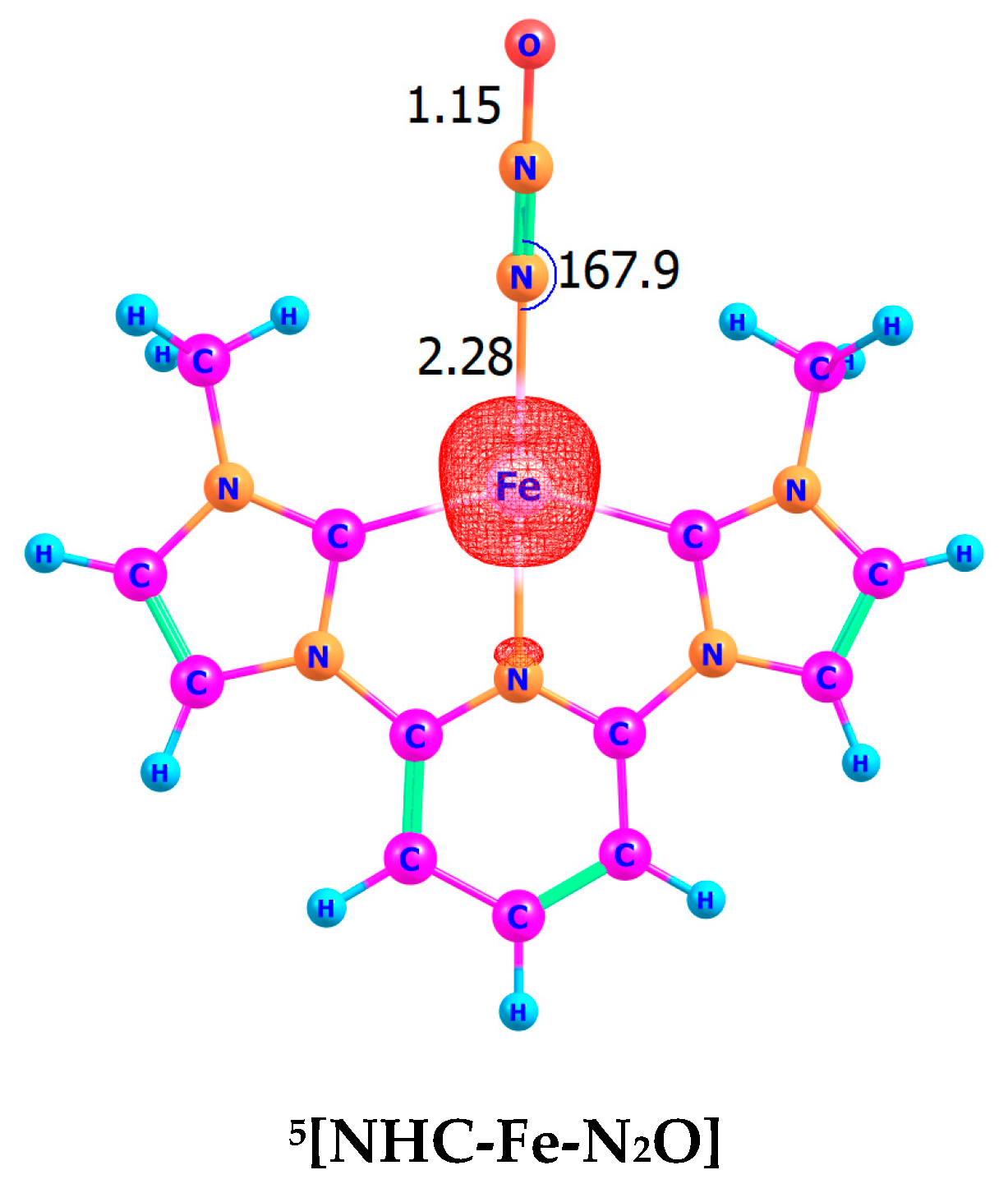
4. Coordination and Energetics of N2O to [(κ3-CNC)FeII]2+ Cation Complex
4.1. Geometric Features of the Quintet State
4.2. Conclusion for N2O Coordination
5. Oxygen Atom Transfer of N2O to Form Oxo FeIV Cation
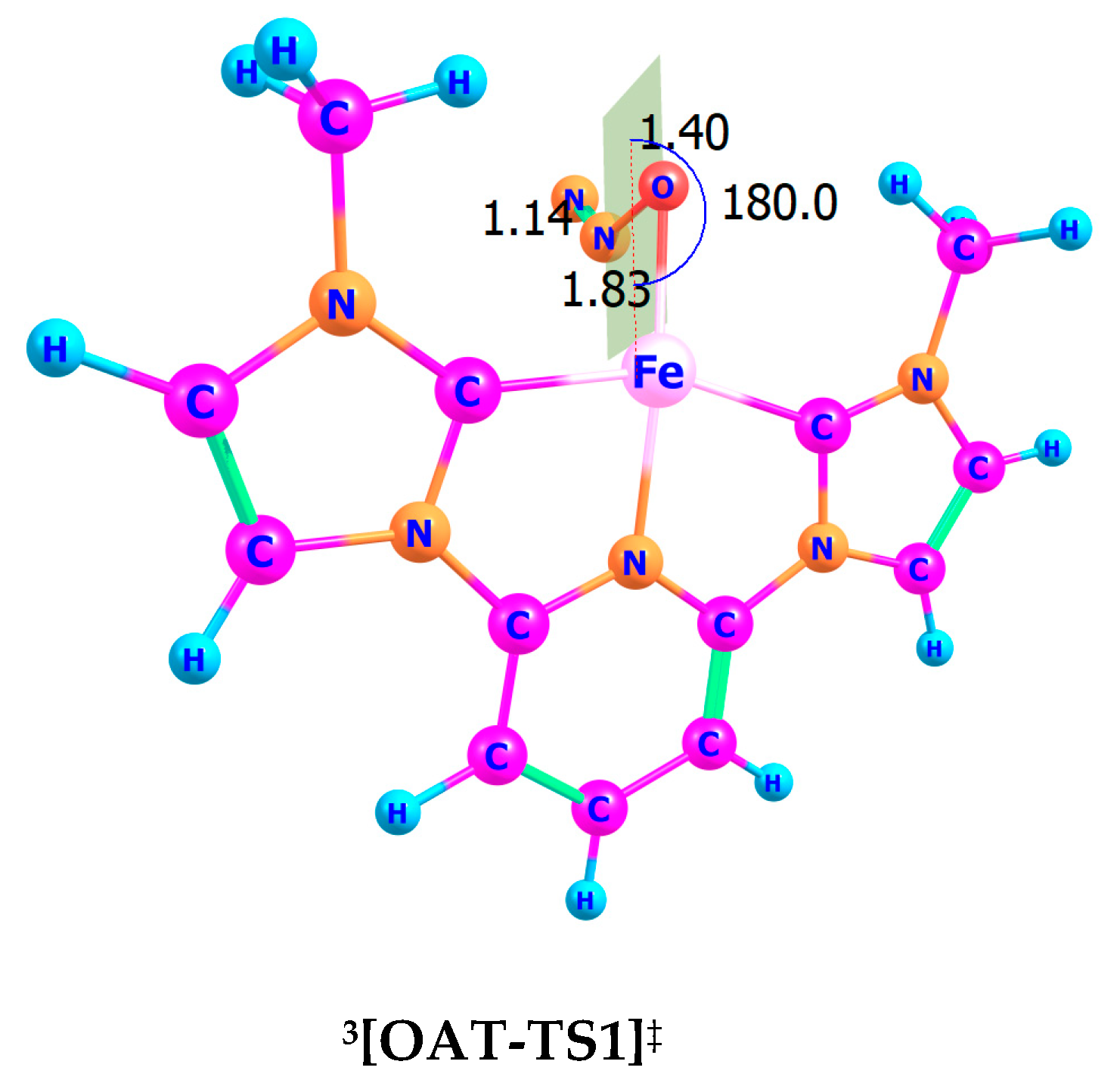
5.1. Geometry and Transition State of OAT
- Fe–O Bond Length: 1.83 Å, reflecting the partial formation of the Fe–O bond.
- O–N Bond Length: 1.40 Å, showing significant elongation as the N–O bond undergoes cleavage.
- N–N Bond Length: 1.14 Å, consistent with the release of N2.
- Fe–O–N–N Bond Angle: 180.0°, indicative of a linear arrangement facilitating efficient oxygen transfer and nitrogen release.
5.2. Spin-State Dependence of OAT
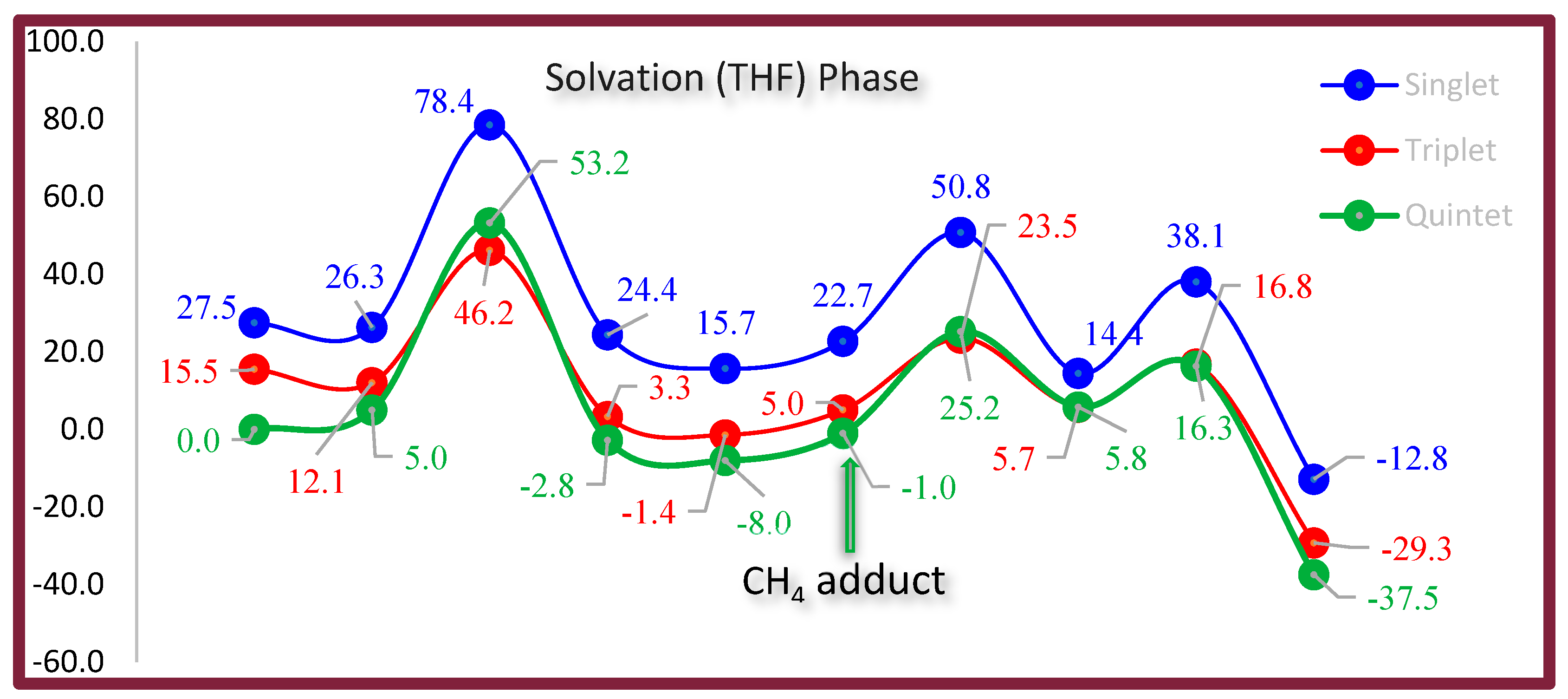
5.3. Solvent Effects and Alternative Solvent Recommendations
5.4. Alternative Solvents
- 1.
- Polar Aprotic Solvents:
- Acetonitrile (CH3CN): A highly polar, non-coordinating solvent that minimizes interactions with the Fe center while providing the strong stabilization of polar intermediates and transition states. Previous studies have shown that acetonitrile improves reaction kinetics by facilitating charge separation and stabilizing high-valent metal–oxo species [87].
- Dimethylformamide (DMF): A high-polarity solvent that enhances the stability of charged species but may introduce undesired ligand competition with the Fe center. Studies indicate that DMF can reduce transition state energy barriers for oxidative transformations in Fe-based catalysis.
- 2.
- Dimethyl Sulfoxide (DMSO):
- DMSO effectively stabilizes charged intermediates due to its high dielectric constant. However, its coordinating nature may lead to direct interactions with Fe, potentially altering the catalytic pathways [87]. Some Fe-based systems show enhanced selectivity in DMSO, while others experience competitive ligand effects [56].
- 3.
- Moderate Polarity Solvents:
- Dichloromethane (DCM) and ethyl acetate (EtOAc): These solvents provide a balance between polarity and inertness, stabilizing key intermediates without significant coordination with the Fe center. Their ability to influence reaction rates and selectivity has been demonstrated in transition-metal-catalyzed oxidation reactions [58].
- 4.
- Nonpolar Solvents:
- Toluene and other nonpolar solvents may reduce solvation effects and lower transition state energies, particularly in systems where polar solvents over-stabilize reactants. Previous research suggests that nonpolar solvents improve Fe-catalyzed hydrocarbon oxidations by favoring reactive high-spin states [67].
- Co-solvent Strategies: Combining nonpolar solvents with small amounts of polar co-solvents could fine-tune the solvation effects, optimizing catalytic performance [69].
5.5. Conclusion of OAT Mechanism
6. Structure and Properties of the FeIV=O Complex
Geometric Characterization of Oxo/Oxyl Complex
- Fe=O Bond Length: 1.62 Å, indicative of a strong double bond with significant oxo character.
- CNC Ligand Coordination: The CNC ligand maintains a distorted square planar geometry around the Fe center, with the oxo ligand occupying an axial position.
- Spin-State Configurations: The geometric variations between the triplet (S = 1) and quintet (S = 2) states highlight the influence of the spin state on bond lengths and the coordination environment.
7. Spin-State Energetics
- Quintet (S = 2) State: The quintet spin state is the ground-state configuration for the FeIV=O species. The high spin density (3.16 e− on Fe and 0.87 e− on O) reflects the distribution of unpaired electrons, which enhances the oxyl radical character of the oxygen atom. This configuration is stabilized by the strong σ-donation from the oxo group and π-backbonding interactions with the CNC ligand.
- Triplet (S = 1) State: While the triplet state plays a critical role during the OAT step, it lies higher in terms of energy (by ~3–7 kcal/mol depending on the environment phase) compared to the quintet state for the FeIV=O species. This energy gap facilitates a spin flip to the quintet state after OAT, enabling the complex to adopt its thermodynamically preferred configuration.
7.1. Electronic Structure
- Iron Center (Fe): Spin density of 3.17 e− in the quintet state, consistent with its d4 configuration.
- Oxo Ligand (O): Partial spin density of 0.87 e−, indicative of an oxyl radical. This feature is critical for activating methane via HAA.
7.2. Reactivity Implications
- Oxo Radical Rebound: The oxyl radical character facilitates rapid rebound with the methyl radical during the oxygen radical rebound (ORR) step, ensuring the efficient formation of methanol.
- Hydrogen Atom Abstraction (HAA): The FeIV=O species initiates HAA by abstracting a hydrogen atom from methane, driven by its strong oxidizing potential and spin-state reactivity.
7.3. Conclusion Oxo/Oxyl Characterization
8. Methane C–H Activation by Oxo/Oxyl Cation Complex
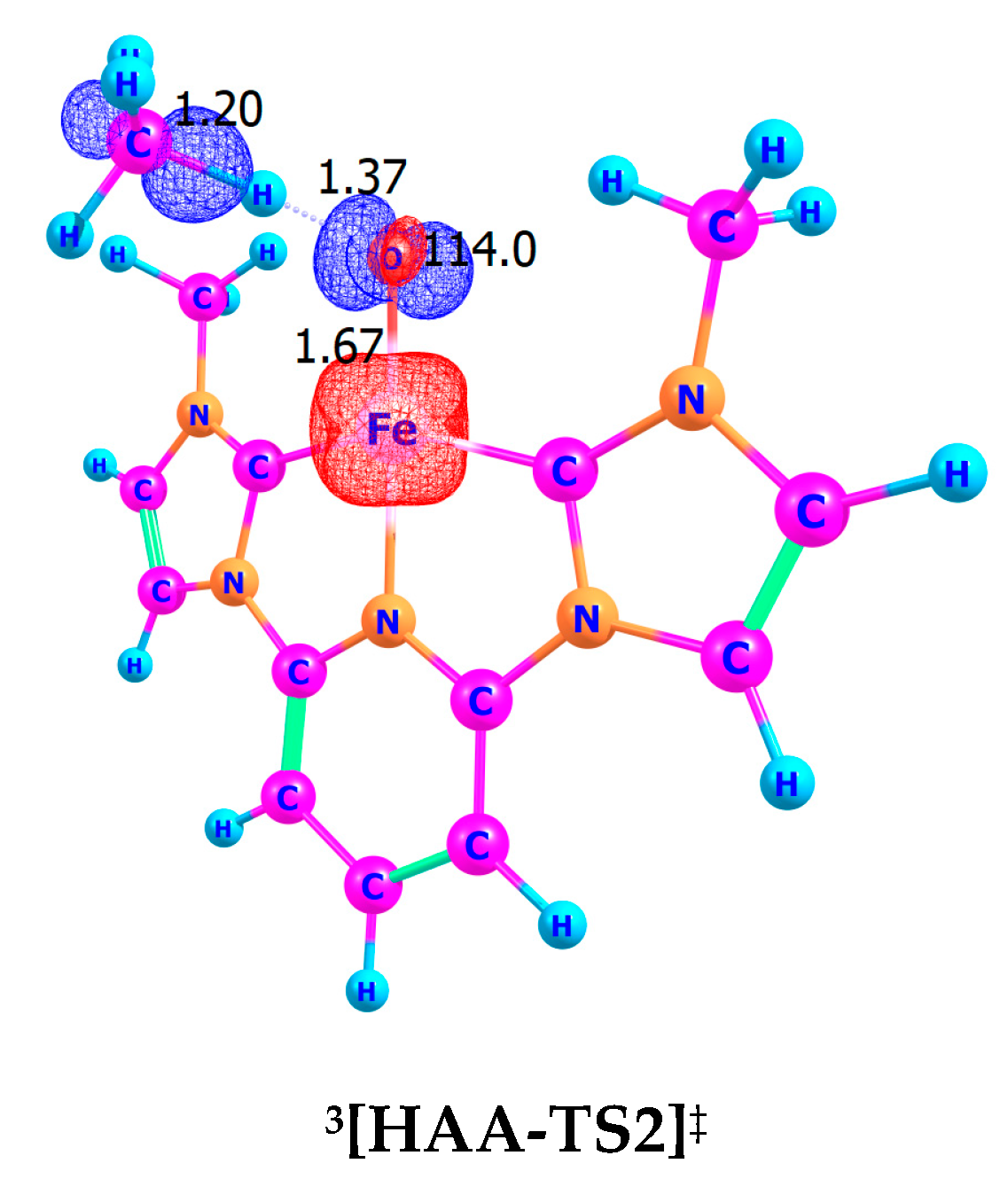
8.1. Hydrogen Atom Abstraction (HAA)
8.2. Geometric Descriptions of Figure 5 and Figure 6
8.3. The Quintet and Triplet State Spin Densities
8.4. Energetic Barrier of Figure 5
9. Oxygen Radical Rebound (ORR)
9.1. Geometric Features of ORR TS Structure
9.2. Spin Densities of the ORR TS Structure
9.3. Energetic Barrier of Figure 6
9.4. Conclusions
10. Summary and Conclusions
- The CNC-ligated iron complexes effectively stabilize high-valent Fe intermediates, facilitating methane C–H activation under mild conditions.
- The triplet (S = 1) and quintet (S = 2) spin states play distinct but complementary roles in the catalytic cycle, with intersystem crossing (ISC) enhancing reactivity.
- The OAT step is the rate-determining step, with the energy barriers influenced by solvation effects and the electronic structure.
- The FeIV=O intermediate exhibits significant oxyl radical character, enabling efficient methane C–H activation via HAA.
- The ORR step proceeds with moderate energy barriers, ensuring methanol formation is feasible.
11. Prospectus
- Experimental Validation: The computationally derived mechanisms and spin-state energetics provide a foundation for experimental efforts to synthesize and characterize CNC-ligated iron complexes under conditions analogous to those modeled computationally.
- Ligand Optimization: Future work could explore alternative CNC ligand scaffolds or substitutions to enhance stability, selectivity, and catalytic turnover, especially for systems involving polar or nonpolar solvent environments.
- Solvent Engineering: The demonstrated impact of solvation on OAT and methane activation highlights the need for further exploration of mixed solvent systems or highly nonpolar environments to optimize solvation-induced energy barriers.
- Broader Substrate Scope: Beyond methane, this catalytic approach could be extended to other hydrocarbons or small molecules, broadening its applicability in chemical synthesis.
- Industrial Scale-Up: Developing scalable processes for methanol production using N2O as an oxidant could reduce the reliance on conventional, energy-intensive methods like steam methane reforming, aligning with global sustainability goals.
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, J.; Xu, Y.; Zhou, X.; Gong, J.; Yin, Y.; Zheng, H.; Guo, H. Direct Oxidation of Methane to Hydrogen Peroxide and Organic Oxygenates in a Double Dielectric Plasma Reactor. ChemSusChem 2011, 4, 1095. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.; Soorholtz, M.; Bilke, M.; Schüth, F. Selective methane oxidation catalyzed by platinum salts in oleum at turnover frequencies of large-scale industrial processes. J. Am. Chem. Soc. 2016, 138, 12395. [Google Scholar] [CrossRef]
- Zerella, M.; Mukhopadhyay, S.; Bell, A.T. Direct oxidation of methane to acetic acid catalyzed by Pd2+ and Cu2+ in the presence of molecular oxygen. Chem. Commun. 2004, 1948–1949. [Google Scholar] [CrossRef]
- Zerella, M.; Kahros, A.; Bell, A.T. Methane oxidation to acetic acid catalyzed by Pd2+ cations in the presence of oxygen. J. Catal. 2006, 237, 111. [Google Scholar] [CrossRef]
- Yang, H.; Hu, C.; Qin, S. Theoretical study on the reaction mechanism of CH4 with CaO. Chem. Phys. 2006, 330, 343–348. [Google Scholar] [CrossRef]
- Yamanaka, I.; Soma, M.; Oisuka, K. Oxidation of methane to methanol with oxygen catalysed by europium trichloride at room temperature. J. Chem. Soc. Chem. Commun. 1995, 2235–2236. [Google Scholar] [CrossRef]
- Yuan, J.; Wang, L.; Wang, Y. Direct Oxidation of Methane to a Methanol Derivative Using Molecular Oxygen. Ind. Eng. Chem. Res. 2011, 50, 6513. [Google Scholar] [CrossRef]
- Prince, B.M. A computational DFT study of methane CH and ammine NH activations by group 9 N-pyrrolyl complexes. Comput. Theor. Chem. 2019, 1162, 112503. [Google Scholar] [CrossRef]
- Prince, B.M.; Gunnoe, T.B.; Cundari, T.R. Oxy-functionalization of Group 9 and 10 transition metal methyl ligands: Use of pyridine-based hemi-labile ligands. Dalton Trans. 2014, 43, 7608–7614. [Google Scholar] [CrossRef]
- Prince, B.M.; Cundari, T.R.; Tymczak, C.J. DFT Study of the Reaction of a Two-Coordinate Iron(II) Dialkyl Complex with Molecular Oxygen. J. Phys. Chem. A 2014, 118, 11056–11061. [Google Scholar] [CrossRef]
- Prince, B.M.; Cundari, T.R. C–H Bond Activation of Methane by PtII–N-Heterocyclic Carbene Complexes. The Importance of Having the Ligands in the Right Place at the Right Time. Organometallics 2012, 31, 1042–1048. [Google Scholar] [CrossRef]
- Prince, B.M.; Cundari, T.R. Methane C-H Bond Activation by “Naked” Alkali Metal Imidyl and Alkaline Earth Metal Imide Complexes. The Role of Ligand Spin and Nucleophilicity. J. Phys. Chem. A 2013, 117, 9245–9251. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, S.; Biswas, B.; Sugimoto, M. Oxidative addition of a C–H σ bond to M(PH3)2 (M=Pd or Pt). An ab initio molecular orbital study on the chelate phosphine effect. J. Chem. Soc. Dalton Trans. 1997, 803–810. [Google Scholar] [CrossRef]
- Jursic, B.S.; Timberlake, J.W.; Engel, P.S. Computation of bond dissociation energies of substituted methanes with density functional theory. Tetrahedron Lett. 1996, 37, 6473–6474. [Google Scholar] [CrossRef]
- Blanksby, S.J.; Ellison, G.B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36, 255–263. [Google Scholar] [CrossRef]
- Gunsalus, N.J.; Koppaka, A.; Park, S.H.; Bischof, S.M.; Hashiguchi, B.G.; Periana, R.A. Homogeneous Functionalization of Methane. Chem. Rev. 2017, 117, 8521–8573. [Google Scholar] [CrossRef] [PubMed]
- Sobalík, Z.; Tabor, E.; Nováková, J.; Sathu, N.K.; Závěta, K. Role of active oxygen and NOx species in N2O decomposition over Fe-ferrierite. J. Catal. 2012, 289, 164–170. [Google Scholar] [CrossRef]
- Meng, X.; Cui, X.; Rajan, N.P.; Yu, L.; Deng, D.; Bao, X. Direct Methane Conversion under Mild Condition by Thermo-, Electro-, or Photocatalysis. Chem 2019, 5, 2296–2325. [Google Scholar] [CrossRef]
- Paul, A.; Musgrave, C.B. A Detailed Theoretical Study of the Mechanism and Energetics of Methane to Methanol Conversion by Cisplatin and Catalytica. Organometallics 2007, 26, 793–809. [Google Scholar] [CrossRef]
- Fifth Assessment Report—IPCC. Available online: https://www.ipcc.ch/assessment-report/ar5/ (accessed on 31 July 2024).
- Special Report on the Ocean and Cryosphere in a Changing Climate. Available online: https://www.ipcc.ch/srocc/ (accessed on 31 July 2024).
- Sixth Assessment Report—IPCC. Available online: https://www.ipcc.ch/assessment-report/ar6/ (accessed on 31 July 2024).
- Bahrami, M.; Pourfayaz, F.; Kasaeian, A. Low global warming potential (GWP) working fluids (WFs) for Organic Rankine Cycle (ORC) applications. Energy Rep. 2022, 8, 2976–2988. [Google Scholar] [CrossRef]
- Danny Harvey, L.D. A guide to global warming potentials (GWPs). Energy Policy 1993, 21, 24–34. [Google Scholar] [CrossRef]
- Sanz-Cobena, A.; Sánchez-Martín, L.; García-Torres, L.; Vallejo, A. Gaseous emissions of N2O and NO and NO3− leaching from urea applied with urease and nitrification inhibitors to a maize (Zea mays) crop. Agric. Ecosyst. Environ. 2012, 149, 64–73. [Google Scholar] [CrossRef]
- Wang, J.; Xiong, Z.; Yan, X. Fertilizer-induced emission factors and background emissions of N2O from vegetable fields in China. Atmos. Environ. 2011, 45, 6923–6929. [Google Scholar] [CrossRef]
- Tierling, J. The Effects of Different Mineral Nitrogen Fertilizer Forms on N2O Emissions from Arable Soils Under Aerobic Conditions; Cuvillier Verlag: Göttingen, Germany, 2017. [Google Scholar]
- Zhang, J.-S.; Zhang, F.-P.; Yang, J.-H.; Wang, J.-P.; Cai, M.-L.; Li, C.-F.; Cao, C.-G. Emissions of N2O and NH3, and nitrogen leaching from direct seeded rice under different tillage practices in central China. Agric. Ecosyst. Environ. 2011, 140, 164–173. [Google Scholar]
- Jurado, A.; Borges, A.; Brouyère, S. Dynamics and emissions of N2O in groundwater: A review. Sci. Total Environ. 2017, 584–585, 207–218. [Google Scholar] [CrossRef]
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. Beyond Oil and Gas the Methanol Economy; Wiley-VCH: Weinheim, Germany, 2018. [Google Scholar]
- Nam, W. High-Valent Iron(IV)–Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions. Acc. Chem. Res. 2007, 40, 522–531. [Google Scholar] [CrossRef]
- Matsunaga, P.T.; Mavropoulos, J.C.; Hillhouse, G.L. Oxygen-atom transfer from nitrous oxide (N=N=O) to nickel alkyls. Syntheses and reactions of nickel(II) alkoxides. Polyhedron 1995, 14, 175–185. [Google Scholar] [CrossRef]
- Mei, J.; Carsch, K.M.; Freitag, C.R.; Gunnoe, T.B.; Cundari, T.R. Variable Pathways for Oxygen Atom Insertion into Metal-Carbon Bonds: The Case of Cp*W(O)2(CH2SiMe3). J. Am. Chem. Soc. 2013, 135, 424–435. [Google Scholar] [CrossRef]
- Matsunaga, P.T.; Hillhouse, G.L.; Rheingold, A.L. Oxygen-atom transfer from nitrous oxide to a nickel metallacycle. Synthesis, structure, and reactions of [cyclic] (2,2′-bipyridine)Ni(OCH2CH2CH2CH2). J. Am. Chem. Soc. 1993, 115, 2075–2077. [Google Scholar] [CrossRef]
- Burch, R.; Daniells, S.T.; Breen, J.P.; Hu, P. A combined transient and computational study of the dissociation of N2O on platinum catalysts. J. Catal. 2004, 224, 252–260. [Google Scholar] [CrossRef]
- Martínez, A.; Goursot, A.; Coq, B.; Delahay, G. Theoretical Study of the Dissociation of N2O in a Transition Metal Ion-Catalyzed Reaction. J. Phys. Chem. B 2004, 108, 8823–8829. [Google Scholar] [CrossRef]
- Dibeler, V.H. N2O Bond Dissociation Energy by Photon Impact. J. Chem. Phys. 1967, 47, 2191–2192. [Google Scholar] [CrossRef]
- Danopoulos, A.; Pugh, D.; Smith, H.; Saßmannshausen, J. Structural and Reactivity Studies of “Pincer” Pyridine Dicarbene Complexes of Fe0: Experimental and Computational Comparison of the Phosphine and NHC Donors. Chem. A Eur. J. 2009, 15, 5491–5502. [Google Scholar] [CrossRef]
- Darmon, J.M.; Yu, R.P.; Semproni, S.P.; Turner, Z.R.; Stieber, S.C.; DeBeer, S.; Chirik, P.J. Electronic Structure Determination of Pyridine N-Heterocyclic Carbene Iron Dinitrogen Complexes and Neutral Ligand Derivatives. Organometallics 2014, 33, 5423–5433. [Google Scholar] [CrossRef]
- Scott, N.M.; Stevens, E.D.; Dorta, R.; Costabile, C.; Cavallo, L.; Hoff, C.D.; Nolan, S.P. Steric and Electronic Properties of N-Heterocyclic Carbenes (NHC): A Detailed Study on Their Interaction with Ni(CO)4. J. Am. Chem. Soc. 2005, 127, 2485–2495. [Google Scholar]
- Rosen, E.L.; Varnado, C.D.; Tennyson, A.G.; Khramov, D.M.; Kamplain, J.W.; Sung, D.H.; Cresswell, P.T.; Lynch, V.M.; Bielawski, C.W. Redox-Active N-Heterocyclic Carbenes: Design, Synthesis, and Evaluation of Their Electronic Properties. Organometallics 2009, 28, 6695–6706. [Google Scholar] [CrossRef]
- Peìrez, P.J.; DiÌaz-Requejo, M.M.; Nolan, S.P. N-Heterocyclic Carbenes in Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Muehlhofer, M.; Strassner, T.; Herrmann, W.A. New Catalyst Systems for the Catalytic Conversion of Methane into Methanol. Angew. Chem. Int. Ed. 2002, 41, 1745–1747. [Google Scholar] [CrossRef]
- Kiernicki, J.J.; Zeller, M.; Szymczak, N.K. Requirements for Lewis Acid-Mediated Capture and N–N Bond Cleavage of Hydrazine at Iron. Inorg. Chem. 2019, 58, 1147–1154. [Google Scholar] [CrossRef]
- Zimmer, P.; Burkhardt, L.; Friedrich, A.; Steube, J.; Neuba, A.; Schepper, R.; Müller, P.; Flörke, U.; Huber, M.; Lochbrunner, S.; et al. The Connection between NHC Ligand Count and Photophysical Properties in Fe(II) Photosensitizers: An Experimental Study. Inorg. Chem. 2018, 57, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Danopoulos, A.A.; Wright, J.A.; Motherwell, W.B.; Ellwood, S. N-Heterocyclic “Pincer” Dicarbene Complexes of Cobalt(I), Cobalt(II), and Cobalt(III). Organometallics 2004, 23, 4807. [Google Scholar] [CrossRef]
- Arduengo, A.J.; Gamper, S.F.; Calabrese, J.C.; Davidson, F. Low-Coordinate Carbene Complexes of Nickel(0) and Platinum(0). J. Am. Chem. Soc. 1994, 116, 4391–4394. [Google Scholar] [CrossRef]
- Arduengo, A.J. Looking for Stable Carbenes: The Difficulty in Starting Anew. Acc. Chem. Res. 1999, 32, 913–921. [Google Scholar] [CrossRef]
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Zimmer, P.; Müller, P.; Burkhardt, L.; Schepper, R.; Neuba, A.; Steube, J.; Dietrich, F.; Flörke, U.; Mangold, S.; Gerhards, M. N-Heterocyclic Carbene Complexes of Iron as Photosensitizers for Light-Induced Water Reduction. Eur. J. Inorg. Chem. 2017, 2017, 1504–1509. [Google Scholar] [CrossRef]
- Danopoulos, A.A.; Simler, T.; Braunstein, P. N-Heterocyclic Carbene Complexes of Copper, Nickel, and Cobalt. Chem. Rev. 2019, 119, 3730–3961. [Google Scholar] [CrossRef] [PubMed]
- Dröge, T.; Glorius, F. The Measure of All Rings—N-Heterocyclic Carbenes. Angew. Chem. Int. Ed. 2010, 49, 6940–6952. [Google Scholar] [CrossRef] [PubMed]
- Dalton, T.; Faber, T.; Glorius, F. C–H Activation: Toward Sustainability and Applications. ACS Cent. Sci. 2021, 7, 245–261. [Google Scholar] [CrossRef]
- Groves, J.T. High-valent iron in chemical and biological oxidations. J. Inorg. Biochem. 2006, 100, 434–447. [Google Scholar] [CrossRef]
- Moody, P.C.E.; Raven, E.L. The Nature and Reactivity of Ferryl Heme in Compounds I and II. Acc. Chem. Res. 2018, 51, 427–435. [Google Scholar] [CrossRef]
- Que, L.; Tolman, W.B. Biologically inspired oxidation catalysis. Nature 2008, 455, 333–340. [Google Scholar] [CrossRef]
- McDonald, A.R.; Que, L. High-valent nonheme iron-oxo complexes: Synthesis, structure, and spectroscopy. Coord. Chem. Rev. 2013, 257, 414–428. [Google Scholar] [CrossRef]
- Krebs, C.; Galonić Fujimori, D.; Walsh, C.T.; Bollinger, J.M. Non-Heme Fe(IV)–Oxo Intermediates. Acc. Chem. Res. 2007, 40, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Olankitwanit, A.; Rajca, S.; Rajca, A. Intramolecular Hydrogen Atom Transfer in Aminyl Radical at Room Temperature with Large Kinetic Isotope Effect. J. Am. Chem. Soc. 2017, 139, 7144–7147. [Google Scholar] [CrossRef]
- Valgimigli, L.; Banks, J.T.; Ingold, K.U.; Lusztyk, J. Kinetic Solvent Effects on Hydroxylic Hydrogen Atom Abstractions Are Independent of the Nature of the Abstracting Radical. Two Extreme Tests Using Vitamin E and Phenol. J. Am. Chem. Soc. 1995, 117, 9966–9971. [Google Scholar] [CrossRef]
- Seal, P.; Oyedepo, G.; Truhlar, D.G. Kinetics of the Hydrogen Atom Abstraction Reactions from 1-Butanol by Hydroxyl Radical: Theory Matches Experiment and More. J. Phys. Chem. A 2013, 117, 275. [Google Scholar] [CrossRef] [PubMed]
- Renaud, P.; Beaufils, F.; Feray, L.; Schenk, K. Diastereoselective Radical-Mediated Hydrogen-Atom Abstraction. Angew. Chem. Int. Ed. 2003, 42, 4230–4233. [Google Scholar] [CrossRef] [PubMed]
- Neidig, M.L.; Decker, A.; Choroba, O.W.; Huang, F.; Kavana, M.; Moran, G.R.; Spencer, J.B.; Solomon, E.I. Spectroscopic and electronic structure studies of aromatic electrophilic attack and hydrogen-atom abstraction by non-heme iron enzymes. Proc. Natl. Acad. Sci. USA 2006, 103, 12966. [Google Scholar] [CrossRef]
- Mayer, J.M. Hydrogen Atom Abstraction by Metal−Oxo Complexes: Understanding the Analogy with Organic Radical Reactions. Acc. Chem. Res. 1998, 31, 441–450. [Google Scholar] [CrossRef]
- Mai, B.K.; Kim, Y. Is It Fe(III)-Oxyl Radical That Abstracts Hydrogen in the C–H Activation of TauD? A Theoretical Study Based on the DFT Potential Energy Surfaces. Inorg. Chem. 2016, 55, 3844–3852. [Google Scholar] [CrossRef]
- Shubin, A.A.; Ruzankin, S.P.; Zilberberg, I.L.; Parmon, V.N. Distinct activity of the oxyl FeIII–O group in the methane dissociation by activated iron hydroxide: DFT predictions. Chem. Phys. Lett. 2015, 640, 94–100. [Google Scholar] [CrossRef]
- Dietl, N.; Schlangen, M.; Schwarz, H. Thermal Hydrogen-Atom Transfer from Methane: The Role of Radicals and Spin States in Oxo-Cluster Chemistry. Angew. Chem. Int. Ed. 2012, 51, 5544–5555. [Google Scholar] [CrossRef]
- Pan, J.; Wenger, E.S.; Matthews, M.L.; Pollock, C.J.; Bhardwaj, M.; Kim, A.J.; Allen, B.D.; Grossman, R.B.; Krebs, C.; Bollinger, J.M. Evidence for Modulation of Oxygen Rebound Rate in Control of Outcome by Iron(II)- and 2-Oxoglutarate-Dependent Oxygenases. J. Am. Chem. Soc. 2019, 141, 15153–15165. [Google Scholar] [CrossRef]
- Simons, M.C.; Prinslow, S.D.; Babucci, M.; Hoffman, A.S.; Hong, J.; Vitillo, J.G.; Bare, S.R.; Gates, B.C.; Lu, C.C.; Gagliardi, L.; et al. Beyond Radical Rebound: Methane Oxidation to Methanol Catalyzed by Iron Species in Metal–Organic Framework Nodes. J. Am. Chem. Soc. 2021, 143, 12165–12174. [Google Scholar] [CrossRef]
- Bach, R.D. The DMDO Hydroxylation of Hydrocarbons via the Oxygen Rebound Mechanism. J. Phys. Chem. A 2016, 120, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, K.I.; Goldman, A.S. (Eds.) Activation and Functionalization of C–H Bonds; American Chemical Society: Washington, DC, USA, 2004. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Austin, A.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssell, K. A Density Functional with Spherical Atom Dispersion Terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef]
- Stevens, W.J.; Krauss, M.; Basch, H.; Jasien, P.G. Relativistic compact effective potentials and efficient, shared-exponent basis sets for the third-, fourth-, and fifth-row atoms. Can. J. Chem. 1992, 70, 612–630. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215. [Google Scholar]
- Bosch, E.; Moreno, M.; Lluch, J.M.; Bertrán, J. Intrinsic reaction coordinate calculations for reaction paths possessing branching points. Chem. Phys. Lett. 1989, 160, 543–548. [Google Scholar] [CrossRef]
- Coskun, D.; Jerome, S.V.; Friesner, R.A. Evaluation of the Performance of the B3LYP, PBE0, and M06 DFT Functionals, and DBLOC-Corrected Versions, in the Calculation of Redox Potentials and Spin Splittings for Transition Metal Containing Systems. J. Chem. Theory Comput. 2016, 12, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; Gordon, M.S.; Dupuis, M. The intrinsic reaction coordinate and the rotational barrier in silaethylene. J. Am. Chem. Soc. 1985, 107, 2585–2589. [Google Scholar] [CrossRef]
- Santini, C.; Marinelli, M.; Pellei, M. Boron-Centered Scorpionate-Type NHC-Based Ligands and Their Metal Complexes. Eur. J. Inorg. Chem. 2016, 2016, 2312–2331. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Databas, ConQuest Version 2.0.1; CCDC: Cambridge, UK, 2021.
- Wurzenberger, X.; Piotrowski, H.; Klüfers, P. A Stable Molecular Entity Derived from Rare Iron(II) Minerals: The Square-Planar High-Spin-d6 FeIIO4 Chromophore. Angew. Chem. Int. Ed. 2011, 50, 4974–4978. [Google Scholar] [CrossRef]
- Cao, A.; Nørskov, J.K. Spin Effects in Chemisorption and Catalysis. ACS Catal. 2023, 13, 3456–3462. [Google Scholar] [CrossRef]
- Understanding the Effect of Catalyst Spin State on Reaction. Available online: https://phys.org/news/2024-01-effect-catalyst-state-reaction.html (accessed on 16 December 2024).
- Rajca, A.; Olankitwanit, A.; Wang, Y.; Boratyński, P.J.; Pink, M.; Rajca, S. High-Spin S = 2 Ground State Aminyl Tetraradicals. J. Am. Chem. Soc. 2013, 135, 18205–18215. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spin State | Fe–C (Å) | Fe–N (Å) | C–Fe–C (°) | Spin Density (Fe, e−) |
|---|---|---|---|---|
| Singlet (S = 0) | 1.97 | 1.86 | 163.5 | 0.00 |
| Triplet (S = 1) | 1.97 | 1.87 | 163.0 | 2.17 |
| Quintet (S = 2) | 2.06 | 2.13 | 152.3 | 4.00 |
| Parameter | Gas Phase (Triplet) | Gas Phase (Quintet) | THF Solvent (Triplet) | THF Solvent (Quintet) |
|---|---|---|---|---|
| ΔG‡ HAA (kcal/mol) | 16.0 | 18.0 | 23.5 | 25.2 |
| ΔG‡ ORR (kcal/mol) | 6.4 | 6.6 | 16.8 | 16.3 |
| Fe–O (HAA, Å) | 1.67 | 1.72 | 1.67 | 1.72 |
| O–H (HAA, Å) | 1.37 | 1.22 | 1.37 | 1.22 |
| C–H (HAA, Å) | 1.20 | 1.33 | 1.20 | 1.33 |
| Fe–O–H (HAA, °) | 114.0 | 94.1 | 114.0 | 94.1 |
| Fe–O (ORR, Å) | 1.82 | 1.82 | 1.80 | 1.82 |
| O–C (ORR, Å) | 2.08 | 2.17 | 2.08 | 2.17 |
| Fe–O–C (ORR, °) | 74.2 | 71.4 | 74.2 | 71.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prince, B.M. Harnessing Nitrous Oxide for Sustainable Methane Activation: A Computational Exploration of CNC-Ligated Iron Catalysts. Methane 2025, 4, 6. https://doi.org/10.3390/methane4010006
Prince BM. Harnessing Nitrous Oxide for Sustainable Methane Activation: A Computational Exploration of CNC-Ligated Iron Catalysts. Methane. 2025; 4(1):6. https://doi.org/10.3390/methane4010006
Chicago/Turabian StylePrince, Bruce M. 2025. "Harnessing Nitrous Oxide for Sustainable Methane Activation: A Computational Exploration of CNC-Ligated Iron Catalysts" Methane 4, no. 1: 6. https://doi.org/10.3390/methane4010006
APA StylePrince, B. M. (2025). Harnessing Nitrous Oxide for Sustainable Methane Activation: A Computational Exploration of CNC-Ligated Iron Catalysts. Methane, 4(1), 6. https://doi.org/10.3390/methane4010006