Abstract
Antimicrobial resistance (AMR) is a silent pandemic that presents an urgent threat to human health. Recently, polymyxins have been revived as a last-line therapeutic option, despite their toxicity. As such, there is a need for fast and reliable approaches to devise novel polymyxin analogues. In this work, machine learning was employed to devise a semi-quantitative model of the activity of polymyxin-like molecules. Four learning algorithms and ten families of molecular descriptors were explored. Top performance was observed for an AdaBoost model using the Kier and Hall topological indexes, allowing for the exploration of the systematic changes in the structure of polymyxin B.
1. Introduction
Antimicrobial resistance represents one of the largest current health threats worldwide, whose impact has been heightened by the escalation of multidrug-resistant (MDR) pathogens, with the Gram-negative bacteria Pseudomonas aeruginosa, Acinetobacter baumannii, and Klebsiella pneumoniae heading the World Health Organization’s list of priority pathogens not responding to front-line antibiotics. Polymyxins (PMs) B and E are the two most-studied and utilized variants of the antimicrobial peptide PM group and are used in last-resort treatments for Gram-negative bacterial infections [1] due to their nephrotoxic and neurotoxic side effects. Nevertheless, improved dosing regimens and the rise of Gram-negative MDR strains has led to a renaissance of their clinical use [2]. Sadly, PM resistance has also emerged [3]. This, along with PM’s poor bioavailability, toxicity, and narrow-spectrum activity, compromises what is already the last available treatment option.
In this work, we endeavor to explore several approaches to model the antimicrobial activity of PM-like molecules towards an assortment of microbial species using different Machine Learning (ML) strategies, and the best-performing model is further explored in terms of its response to systematic mutations of the PM-B structure to gain new insights into the most preponderant features of highly active PM derivatives.
2. Methodology
A dataset containing the Minimum Inhibitory Concentration (MIC) for 408 molecule/microorganism pairs was collected from PubChem [4]. The information regarding the targeted microorganism was condensed into two variables, namely its taxonomic genus (TxG) and a broader classification of the type of microorganism (MTyp): Gram-negative bacteria, Gram-positive bacteria, or fungi. Several families of molecular descriptors were calculated using the RDKit software package: H-bond donor and acceptor counts (Hb), Kier and Hall topological indexes (CPK), functional group counts (FG), two-dimensional autocorrelation functins (AC2D), eigenvalues of the adjacency matrix weighted by the van der Waals volume (BCUT2D), as well as van der Waals Surface Area (VSA) contributions to charge distribution (PEOE_VSA), refractive index (SMR_VSA), Log(P) (SlogP_VSA), and molecular electrostatic potential (EState_VSA). Four learning algorithms were used: logistic regression, decision tree, random forest, and AdaBoost, as implemented in the Scikit-learn package, version 1.0.2. The variables TxG and MTyp were added to each set of molecular descriptors to form the feature set used by each model. Each algorithm/descriptor set pair was trained, targeting a multi-class prediction of the MIC quartile (among the full data) using a 65:35 split between the training and the testing data. All numerical fields were first scaled to zero mean and unit standard deviation and TxG and MTyp were codified using one-hot encoding. A 5-fold cross validation scheme was used to optimize some hyper-parameters of the random forest models (number of estimators, nest, fraction of samples, ns, and features, nf, considered by each tree) and of the AdaBoost models (nest, as well as the depth of each tree, dest, and the learning rate, rL).
3. Results and Discussion
Of the 399 data points that were collected, 287 were related to Gram-negative bacteria, 79 to Gram-positive bacteria, and 33 entries were related to antifungal activity. Among the bacteria, the most represented genera were Escherichia, Pseudomonas, Salmonella, and Staphylococcus, making up about 82.5% of the bacterial data. The collected MIC values were quite asymmetrical, with the boundary of the first quartile (Q1) located at 1.25 µM and the upper limit of the third quartile (Q3) at 32.0 µM, with a median of 4.0 µM.
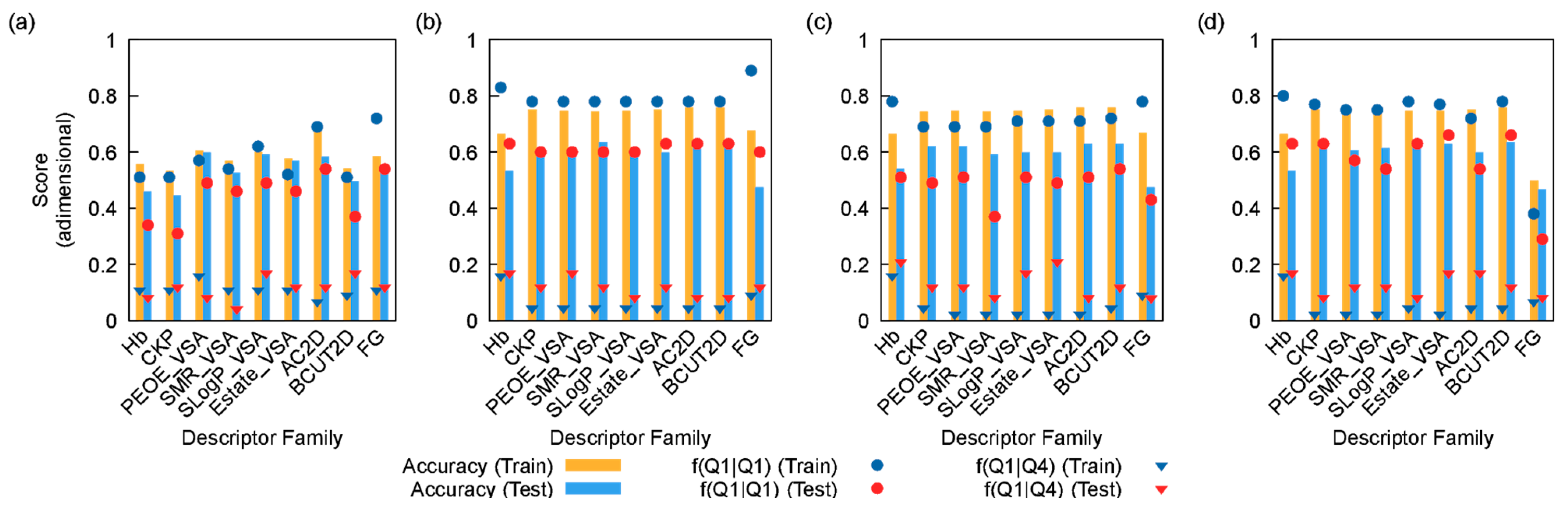
Figure 1 depicts the behavior of the 40 models considered in this work. Overall, the logistic regression models performed the worst (Figure 1a). Likewise, the decision tree models showed considerable over-fitting behavior, as well as low f(Q1|Q1) scores (i.e., the correct prediction of a molecule/target combination belonging to Q1, Figure 1b). On the other hand, both the random forest and the AdaBoost models yielded better scores (Figure 1c and 1d, respectively). In particular, the AdaBoost model trained using the CKP set showed a good overall accuracy, high f(Q1|Q1) and very low f(Q1|Q4) scores. This prompted the AdaBoost model devised using the CKP set of descriptors to be selected for further studying. Indeed, the Kier and Hall descriptors forming the CPK set are widely used for the analysis of the biological activity of compounds, mainly due to their lipophilic and hydrophilic affinity [5,6]. The good performance of this set of descriptors in this particular application is consistent with these compounds’ established mode of action [2].
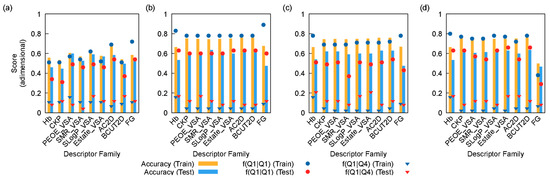
Figure 1.
Values of different scores (overall accuracy, f(Q1|Q1) and f(Q1|Q4) scores) for each family of descriptors and algorithms: (a) logistic regression; (b) decision tree; (c) random forest, and (d) AdaBoost. The f(Q1|Q1) score refers to the fraction of data points belonging to Q1 rightfully classified as Q1, whereas f(Q1|Q4) refers to the fraction of data points in the Q4 range wrongfully classified as Q1.
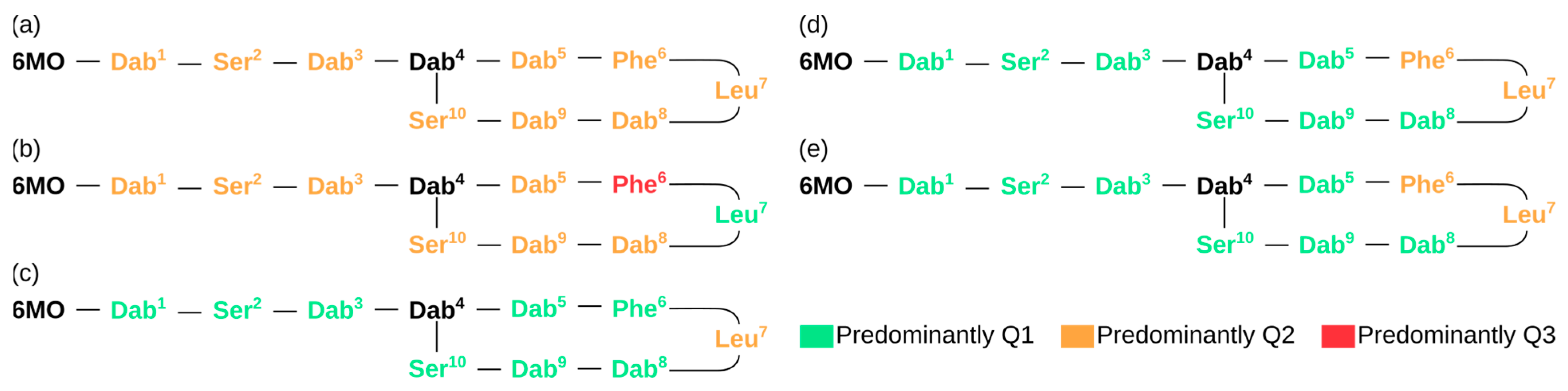
To obtain a more immediate sense of the model’s response, the structure of PM-B was systematically mutated in positions 1 to 3 and 5 to 10 using glycine (Gly, Figure 2b), leucine (Leu, Figure 2c), lysine (Lys, Figure 2d), and glutamic acid (Glu, Figure 2e). The predicted MIC of these “mutations” against P. aeruginosa are shown in Figure 2. Considering that the combination PM/P. aeruginosa is ranked Q2 (Figure 2a), the substitution of Leu7 by Gly appears to improve the antimicrobial activity, whereas the contrary is observed when replacing Phe6 with Gly. Replacing each amino acid residue with Leu, Lys, or Glu usually resulted in a more optimistic prediction of antimicrobial activity. The major exception to this trend was Leu7, for which the model predicted no substantial improvement over the original PM-B structure. Overall, the model’s predictions may be linked to an increase in lipophilicity, perceived by the model via the increase in the amino acid side chains.

Figure 2.
Most probable classification regarding the antimicrobial activity towards P. aeruginosa of PM-B (a) and the mutated variants of PM-B upon systematically changing each amino acid residue for: Gly (b), Leu (c), Lys (d), and Glu (e).
4. Conclusions
In this work, we applied the AdaBoost algorithm to generate a semi-quantitative model of the antimicrobial activity of PM-B analogs using well-established and readily available molecular descriptors, which can be used to rapidly determine whether a proposed structure can be considered a viable candidate for novel PM-derived antibiotics. Moreover, exploration of the model’s predictions using systematic mutations of the PM-B framework proved valuable for discerning which would be the most favorable modification to this molecular scaffold. Future work will be carried out to apply novel game strategies to suggest optimal PM-B derivatives effective against a determined bacterial genus.
Author Contributions
Conceptualization, F.T.; methodology, F.T. and P.J.; data curation, J.I. and F.T.; writing—original draft preparation, I.M.; writing—review and editing, F.T. and P.J.; visualization, J.I. and F.T.; supervision, F.T. and P.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by FCT—Fundação para a Ciência e Tecnologia—in the framework of the project POLYmix-POLYmic (2022.06595.PTDC), of the programmatic funding to CQUM (UID/QUI/00686/2020), of the strategic funding to CEB (UIDB/04469/2020), and of the contracts 2020.00194.CEECIND and CEECINST/00156/2018/CP1642/CT0011.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The following supporting information can be downloaded at https://doi.org/10.5281/zenodo.8346365 (accessed on 20 September 2023): labeling convention used for the sets of molecular descriptors used in this work; optimized hyper-parameters of the random forest and AdaBoost models; and curated data used in this work.
Conflicts of Interest
The authors declare no conflict of interest and that the funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Garg, S.K.; Singh, O.; Juneja, D.; Tyagi, N.; Khurana, A.S.; Qamra, A.; Motlekar, S.; Barkate, H. Resurgence of Polymyxin B for MDR/XDR Gram-Negative Infections: An Overview of Current Evidence. Crit. Care Res. Pract. 2017, 2017, 3635609. [Google Scholar] [CrossRef] [PubMed]
- Manioglu, S.; Modaresi, S.M.; Ritzmann, N.; Thoma, J.; Overall, S.A.; Harms, A.; Upert, G.; Luther, A.; Barnes, A.B.; Obrecht, D.; et al. Antibiotic polymyxin arranges lipopolysaccharide into crystalline structures to solidify the bacterial membrane. Nat. Commun. 2022, 13, 6195. [Google Scholar] [CrossRef] [PubMed]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure-Activity Relationships of Polymyxin Antibiotics. J. Med. Chem. 2010, 53, 1898–1916. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2022, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.H.; Kier, L.B. The Molecular Connectivity Chi Indexes and Kappa Shape Indexes in Structure-Property Modeling. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; John Wiley & Sons, Inc.: Weinheim, Germany, 1991; Volume 2, pp. 367–422. [Google Scholar]
- Kier, L.B. An Index of Molecular Flexibility from Kappa Shape Attributes. Quant. Struct.-Act. Relat. 1989, 8, 221–224. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).