Abstract
Childhood (pediatric) obesity is becoming increasingly common at an alarming rate. Obese children are more likely to develop insulin resistance, relative insulin insufficiency, and type 2 diabetes. Recent research suggests that mitochondrial dysfunction is related to, and may be predictive of, insulin resistance among adult relatives of type 2 diabetes patients. Mitochondria produce ATP, which is used to create energy, especially in muscle tissue, and they play a role in glucose and fat metabolism. Mitochondrial dysfunction plays a role in the development of metabolic diseases. Affected tissues include adipose, liver, and skeletal muscle, which all contribute to food metabolism. Since cells require a balance between mitochondrial ATP generation through oxidative phosphorylation (OXPHOS) and proton gradient dissipation to avoid damage caused by reactive oxygen species (ROS), abnormal mitochondrial function leads to fat buildup and insulin resistance. Obesity, insulin resistance, and type 2 diabetes (T2D) are all caused by growth and transcription factors that influence mitochondrial gene expression. On the other hand, obesity and hypertension both impair heart mitochondrial biogenesis and function. By promoting the expression of chaperones, SIRT1, and antioxidants, moderate weight reduction reduces systemic inflammation and improves mitochondrial dysfunction. In this review, the variables that relate mitochondrial dysfunction to pediatric obesity are discussed.
1. Introduction
Obesity is a multifactorial disease characterized by a low-grade chronic inflammatory state caused by an excess of malfunctioning adipose tissue [1]. This low-grade chronic inflammatory state is strongly and persistently associated with excess body fat mass and is characterized by pro-inflammatory cytokine and chemokine release as a result of pro-inflammatory macrophage and other immune cell infiltration and activation [2]. A recent study from the USA found that by 2030, at least 55–60% of today’s children will be obese [3]. A nutrient imbalance between energy input and output causes secondary mitochondrial dysfunction in patients affected by this pathological condition [4], which may contribute to an increased risk of developing obesity-related diseases [5].
Mitochondria are key organelles for several aspects of cellular homeostasis, including cellular energy production via oxidative phosphorylation, apoptosis, and calcium regulation. Due to these various activities within cells, mitochondrial function is critical to cellular and metabolic health. Changes in mitochondrial function can cause a variety of disorders, including neurological, cardiovascular, muscular, hepatic, endocrine, and reproductive disorders [6].
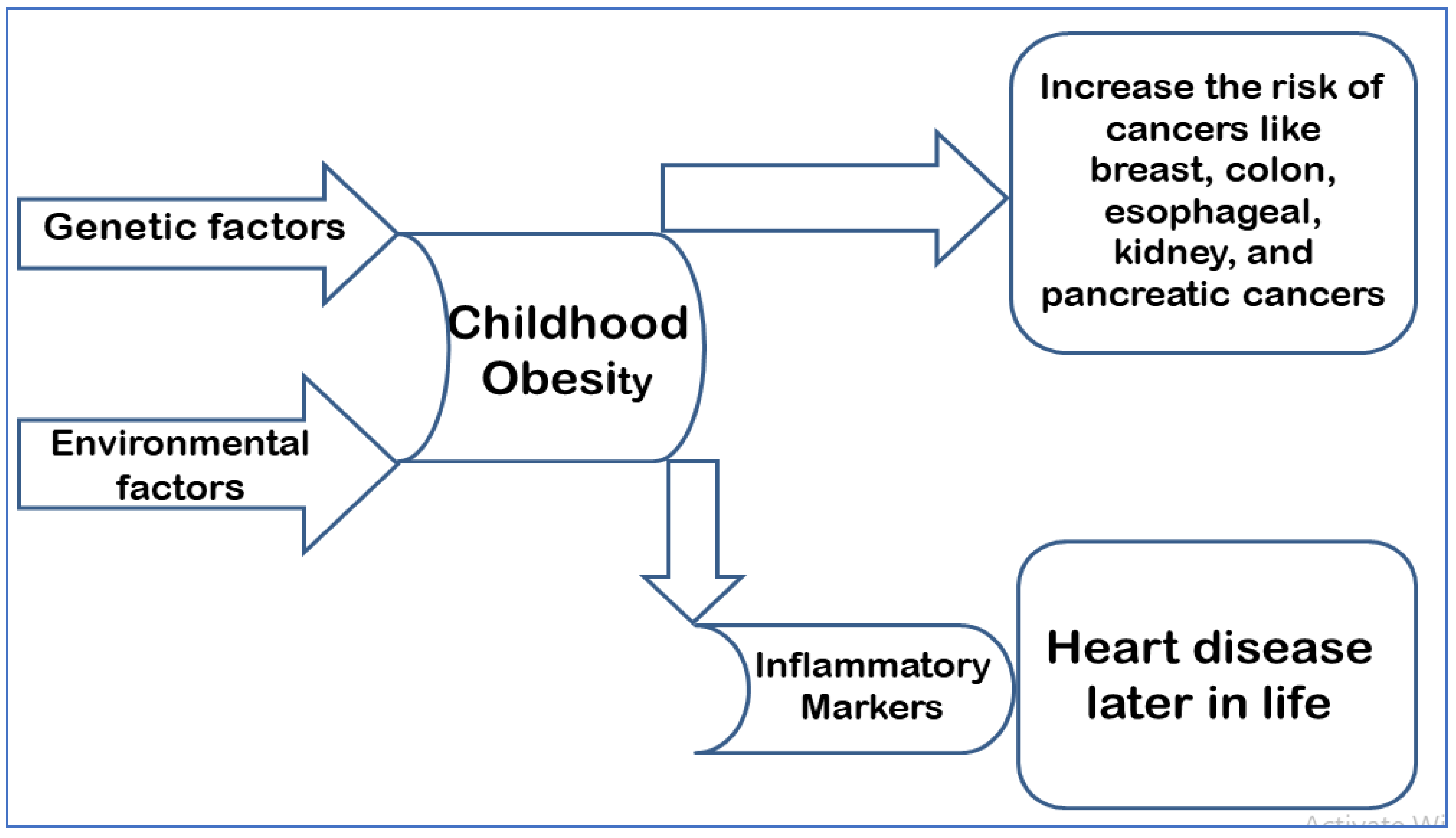
Childhood obesity, also known as pediatric obesity, is caused by genetic predisposition and some environmental factors such as the consumption of fatty foods and a high-sugar diet, cigarette smoking, and a lack of exercise. Obesity affects 34% of children in the United States, and it is a major public health concern due to the high associated rates of illness and mortality. Furthermore, obesity-related medical costs have risen, accounting for 40% of the healthcare budget in 2006 [7]. Children with obesity who were followed-up to adulthood were much more likely to suffer from cardiovascular and digestive diseases. In addition, the increase in body fat also predisposes such children to an increased risk of developing numerous forms of cancer, such as breast, colon, esophageal, kidney, and pancreatic cancer [7]. The incidence of pediatric obesity has more than doubled among children and quadrupled among adolescents globally in the last 30 years [8]. Children with obesity have elevated levels of inflammatory markers as early as three years of age [9]. This has been linked to heart disease later in life [10]. The long-term effects of such findings can result in cumulative vascular damage, which correlates with increased weight status [11] (Figure 1).
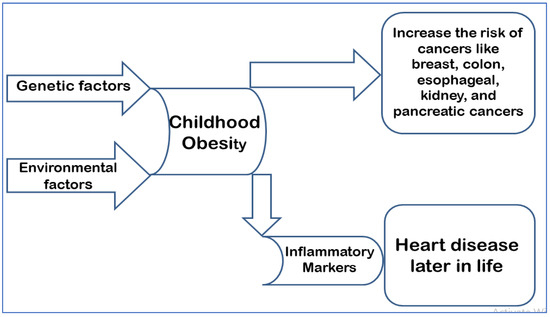
Figure 1.
Depicts the pathophysiology of childhood obesity and its consequences in adulthood.
Mitochondrial dysfunction plays a role in the pathophysiology of metabolic diseases. Affected tissues include those involved in metabolism, such as adipose, liver, and skeletal muscle. As previously mentioned, the cells require a balance between mitochondrial ATP generation via oxidative phosphorylation (OXPHOS) and proton gradient dissipation to avoid damage caused by reactive oxygen species (ROS). Abnormal mitochondrial function results in fat buildup and insulin resistance. The pathophysiology of obesity, insulin resistance, and type 2 diabetes is influenced by growth and transcription factors that regulate mitochondrial gene expression [4].
The main purpose of this overview is to examine the link between mitochondrial dysfunction and childhood obesity.
2. Mitochondria (Anatomy and Physiology)
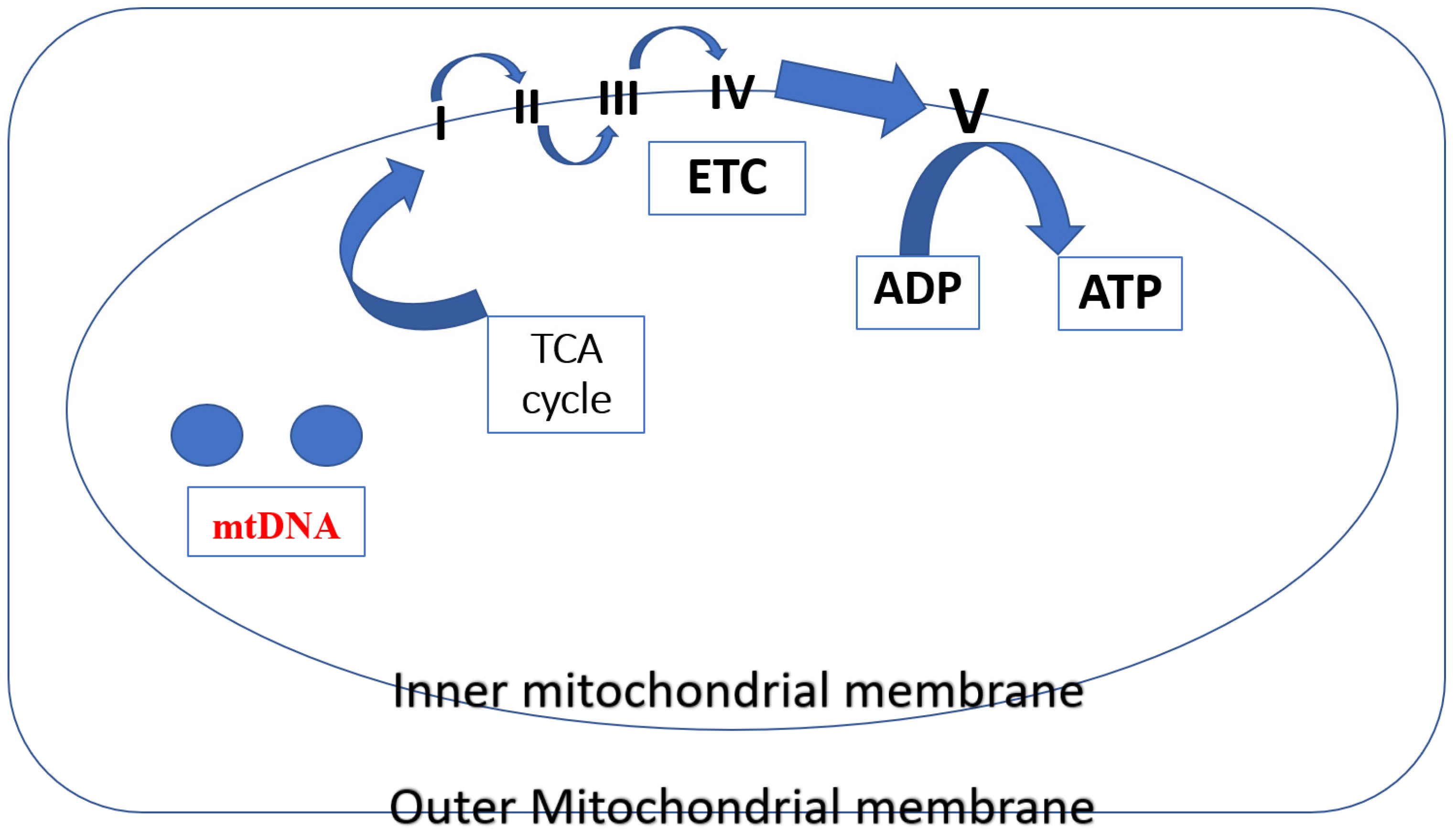
The TCA cycle and the electron transport chain enable mitochondria to produce most of the body’s cellular energy (>90%) in the form of ATP. Five multi-subunit enzyme complexes make up mitochondrial ETC, and they are situated in the inner mitochondrial membrane (I, II, III, IV, and V). The ETC receives electron donations from NADH and FADH2 produced through the TCA cycle at Complex I (NADH:ubiquinone oxidoreductase) and Complex II (succinate dehydrogenase), respectively. After passing through Complex III (coenzyme Q: cytochrome c reductase), electrons are subsequently transferred to cytochrome c. Molecular oxygen binds and is reduced to water in Complex IV (cytochrome c oxidase) via the mobile electron carrier cytochrome c after it has undergone reduction. The mitochondrial membrane potential () is an electrochemical proton gradient that is produced when ten protons (H+)—two from Complex III and four each from Complex I and Complex IV—are pushed from the matrix into the intermembrane space in response to electron transport. Since it connects electron transport (complexes I–IV) (and oxygen consumption) to the activity of Complex V (ATP synthase), where protons re-enter the matrix to dissipate the proton gradient, the protonmotive force (p) is a crucial part of the process of energy storage during OXPHOS [12] (Figure 2).
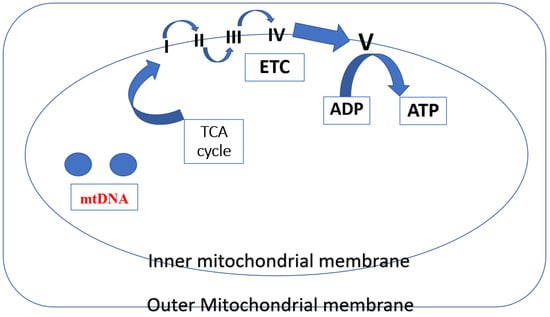
Figure 2.
In mitochondria, several different protein complexes are involved in mitochondrial respiration, i.e., the inner mitochondrial membrane contains I–IV complexes. These complexes produce a proton gradient (H+) by successively moving electrons (e). This gradient is subsequently utilized by Complex V (ATP synthase) to phosphorylate ADP to ATP.
ROS are continuously produced by mitochondria because of oxygen metabolism and excessive free radical generation. The antioxidant enzymes superoxide dismutase, catalase, glutathione reductase, and glutathione peroxidase are all part of the enzymatic defense system and act as a protective mechanism against this oxidative stress. Antioxidant compounds (e.g., glutathione (GSH), vitamins E and C, and many carotenoids and flavonoids) shield cells from oxidative damage. However, when the antioxidant defenses are overpowered, there is an excess of ROS, which results in oxidative damage to the mitochondria’s proteins, DNA, and lipids [13]. Excessive ROS may damage DNA, RNA, lipids, and proteins under pathological circumstances, which causes their interactions to result in functional harm or irreversible alterations to the targets. ROS are currently thought to be the main agents of cell destruction. Additionally, oxidative stress can harm cells and possibly support the development of cancer [14].
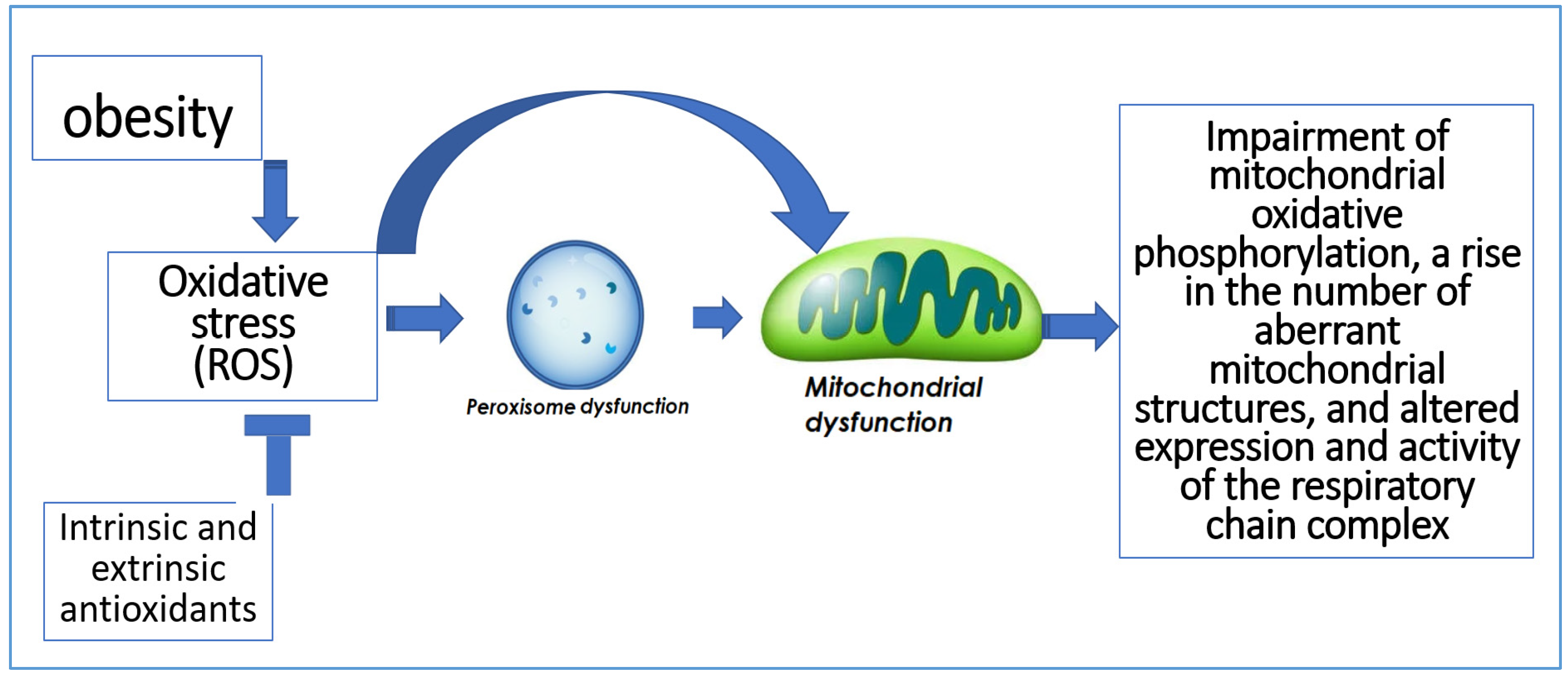
In addition to being the primary producers of ROS, mitochondria are also the primary targets of oxidative damage, which can decrease mitochondrial effectiveness and increase ROS production [15]. Additionally, peroxisomes play a role in the generation and removal of ROS. Numerous oxygen-consuming metabolic processes take place in peroxisomes. Hydrogen peroxide, which can oxidize several compounds, is produced through oxygen consumption in peroxisomes [16]. Impairment of mitochondrial oxidative phosphorylation, a rise in the number of aberrant mitochondrial structures, and altered expression and activity of the respiratory chain complex are all consequences of peroxisome dysfunction [17] (Figure 3).
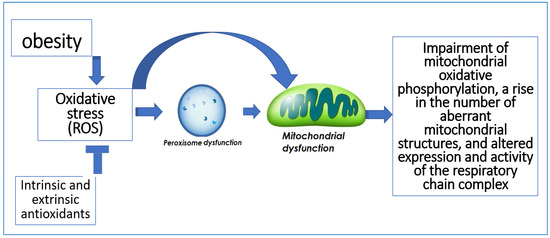
Figure 3.
The relation between oxidative stress and peroxisomal and mitochondrial dysfunction.
3. Pathophysiology of Childhood Obesity
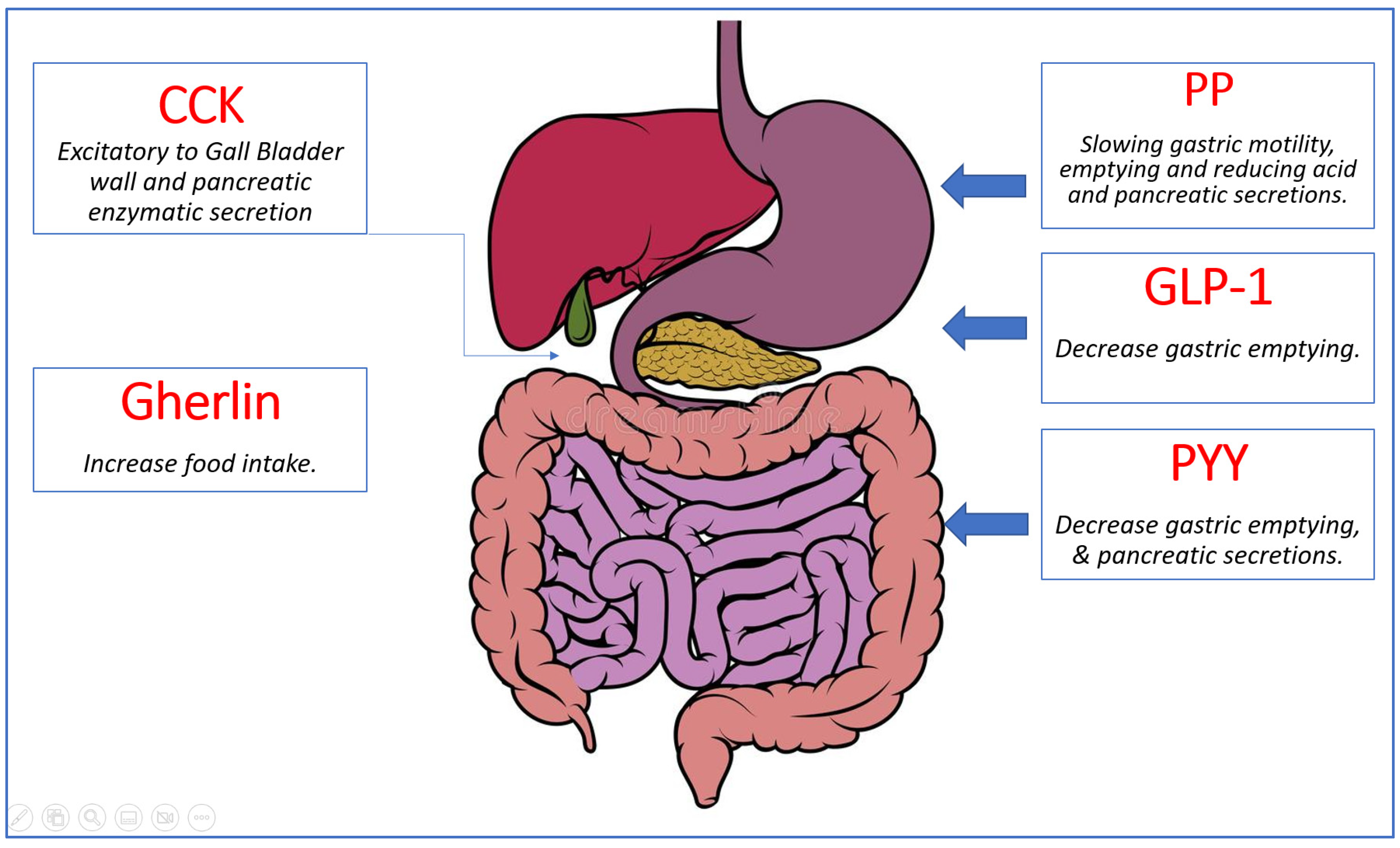
A great variety of hormones are released by the gastrointestinal system and involved in appetite control and energy balance [18]. Ghrelin is the sole known appetite-stimulating (orexigenic) gut hormone. It is produced by the stomach’s oxyntic glands. Shortly before a meal, ghrelin levels rise. Other gut hormones that have been discovered so far are anorexigenic (i.e., they decrease appetite and food intake). Peptide tyrosine tyrosine (PYY), pancreatic polypeptide, oxyntomodulin, amylin, glucagon, glucagon-like peptide-1 (GLP-1), and GLP-2 are but a few of them. PYY, for example, is a satiety signal and rises within 15 min after eating, resulting in a reduction in food consumption. The gastrointestinal tract is the body’s largest endocrine organ, releasing hormones that have critical sensing and signaling functions in energy balance management [19] (Figure 4).
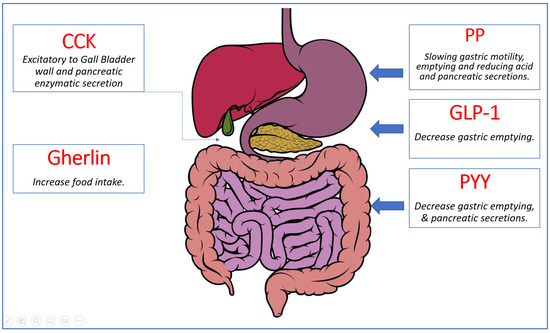
Figure 4.
Summary of gut hormones involved in appetite regulation. Ghrelin, the orexigenic hormone presented in green, is released from the stomach and stimulates food intake. The anorexigenic hormones shown in red (Cholecystokinin (CCK), Pancreatic Polypeptide (PP), Peptide YY (PYY), and Glucagon-Like Peptide 1 (GLP-1)) are released from various organs in the gastrointestinal system and work synergistically to decrease food intake by slowing gastric motility and emptying and reducing acid and pancreatic secretions [18].
Obesity in children is a multifaceted disorder caused by genetic and non-genetic factors as well as their complex interactions. Energy intake and expenditure are also influenced by genetics and societal variables (including socioeconomic position, race/ethnicity, media and marketing, and the physical environment). Excess body fat appears to be the outcome of a complicated interaction between the environment and the body’s genetic and epigenetic susceptibility to obesity. The development of childhood overweight or obesity is influenced by a number of variables or risk factors [20].
In populations at high risk of developing obesity and diabetes, gestational diabetes exposure is associated with an elevated risk of childhood and early adult obesity in children [21]. Some studies have found that severe variation in maternal adiposity (achieved, for example, via bariatric surgery) impacts childhood obesity. The frequency of overweight and obesity among children of women who lost a great deal of weight after surgery was comparable to that of the general population, with no rise in underweight [22]. Furthermore, higher birth weight is linked to both increased fat and increased lean mass in the progeny. Babies that are small for their gestational age and yet develop quickly may be at risk of childhood obesity [23] (Table 1).

Table 1.
Summary of the common factors contributing in the development of pediatric obesity.
4. Gestational Diabetes, Childhood Obesity, and Mitochondrial Dysfunction
Gestational diabetes mellitus (GDM) is a frequent condition in pregnancies in which hyperglycemia develops spontaneously during the pregnancy [24]. Maternal overweight or obesity, a westernized diet with nutritional deficiencies, maternal age, and a family history of insulin resistance and/or diabetes are all risk factors. While GDM normally subsides after delivery, it can have long-term health implications for the mother, such as an increased risk of type 2 diabetes (T2DM) and cardiovascular disease (CVD) as well as future obesity, CVD, T2DM, and/or GDM for the child [25].
GDM has both short and long-term consequences. The fetus’s endogenous synthesis of insulin and insulin-like growth factor 1 (IGF-1) is stimulated by the increase in the placental transport of glucose, amino acids, and fatty acids stated earlier. These factors can combine to induce fetal enlargement, which can lead to macrosomia at birth [26]. Babies born from GDM pregnancies have a higher risk of obesity, T2DM, CVD, and other metabolic illnesses later in life. Even after controlling for variables such as maternal BMI, children born to diabetic women have nearly double the chance of developing childhood obesity as children born to nondiabetic women [27]. As a result, females are more prone to have GDM during their own pregnancies, thus adding to a vicious intergenerational cycle of GDM affliction [28].
5. Dysregulation of and Biogenesis Modulators in Adipose Tissue of Obese Children
Energy storage is dependent on adipose tissue. It is also a metabolically active organ that plays a part in glucose homeostasis and endocrine activities [29]. Obesity, on the other hand, can alter adipose tissue functions and cause an increase in the release of fatty acids, hormones, and proinflammatory molecules, all of which can contribute to obesity-related complications [30]. Several studies have shown that obesity reduces adipose tissue mitochondrial respiration, suggesting mitochondrial dysfunction [31]. Obesity and other metabolic disorders such as type 2 diabetes and metabolic syndrome are linked with a mitochondrial overload of glucose and fatty acids, which results in incomplete substrate oxidation and increased production of intermediate products such as diacylglycerol, which stimulates the production of reactive oxygen species (ROS) [32]. Reduced fatty acid oxidation has been reported in animal models and in individuals with obesity as another manifestation of mitochondrial dysfunction [33].
It has been reported that changes in mitochondrial structure and function may have a role in the pathophysiology of obesity-related diseases [34]. Reduced mitochondrial mass and mitochondrial biogenesis (MB) genes, structural damage, and altered mitochondrial morphology were observed in cases of severe obesity along with mitochondrial protein hyperacetylation [35], increased inflammation, ROS generation, and oxidative stress [36].
The major transcription factor PPARƔ regulates MB in adipocytes [37]. The PGC-1 transcriptional factor family also has a role in the dynamic modulation of MB and respiratory performance [38]. Post-translational modifications provoked by intracellular energy-state sensors such as cAMP-activated protein kinase (AMPK) and NAD+-dependent deacetylase SIRT1 modulate the activity of PGC-1α [39]. SIRT1 stimulates the generation of adiponectin in adipose tissue, which improves metabolic efficiency [40]. SIRT1 expression is significantly decreased in adipose tissue from obese people, which has a negative influence on adipocytes’ energy status [38]. SIRT1 interacts closely with AMPK and deacetylates several lysine residues in PGC-1α under normal circumstances, resulting in the promotion of lipid catabolism while inhibiting the “inflammation” of white adipose tissue (WAT) [41]. The expression and function of MB regulators such as NRF1, NRF2, and TFAM, which are involved in the replication and transcription of mtDNA, are decreased as a result of lower SIRT1 expression and an unregulated downstream effector [42].
In mice with cardiomyopathy, obesity leads to metabolic abnormalities, such as insulin resistance and intracellular low-grade inflammation, and increased oxidative stress and mitochondrial dysfunction [43,44]. Only diabetic mice had low amounts of ATP synthase and complexes II and III in adipocytes when the expression of mitochondrial proteins in the liver, muscle, and adipocytes of normal, obese, and diabetic mice was compared. Furthermore, abnormal mitochondrial shape, reduced mtDNA content, and elevated rates of oxidation and respiration were observed in obese and diabetic mouse adipocytes, indicating a significant mitochondrial malfunction. Obese mice had skeletal muscle that displayed altered mitochondrial dynamic behavior, including decreased mitochondrial respiratory capacity, low ATP content, increased fission (increased Fis1 and Drp1 protein concentrations), and reduced fusion (increased Mfn1 and Mfn2 protein concentrations) [45] (Table 2).
Table 2.
Effects of obesity on mitochondrial structure and function in humans.
6. Bariatric Surgery for Pediatric Patients with Severe Obesity
Severe childhood and adolescent obesity is a growing public health concern in the United States. Unfortunately, there are few effective treatments for severe obesity. In the pediatric population, the use of metabolic and bariatric surgery constitutes an evidence-based, effective treatment of extreme obesity and concomitant comorbid disorders [46]. Bariatric surgery has emerged as one of the most successful and long-lasting treatment options for morbid obesity and associated comorbidities in terms of weight loss and glycemic control [47].
Surprisingly, weight loss induced by bariatric surgery has been shown to promote mitochondrial biogenesis, whereas weight loss mediated by diet has not been proven to improve mitochondrial dysfunction [31]. Furthermore, bariatric surgery may be linked to a reduction in renal tubular injury as measured by the level of kidney injury molecule-1 in obese patients [48]. Several studies have also found that bariatric surgery is superior to non-surgical therapies with respect to improving the glycemic and metabolic characteristics of obese people with T2DM [49]. In addition, in an obese rat model, bariatric surgery greatly reduced chromosomal damage [50].
Author Contributions
R.A.S. and H.M.Q. contributed to the conception of this manuscript; R.A.S. drafted the manuscript; H.M.Q. contributed to manuscript writing and revision for intellectual content; R.A.S. and H.M.Q. conducted the literature search critically; R.A.S. and H.M.Q. provided constructive discussions and contributed to paper review. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of Adipose Tissue Inflammation to the Development of Type 2 Diabetes Mellitus. Compr. Physiol. 2018, 9, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Ward, Z.J.; Long, M.W.; Resch, S.C.; Giles, C.M.; Cradock, A.L.; Gortmaker, S.L. Simulation of Growth Trajectories of Childhood Obesity into Adulthood. N. Engl. J. Med. 2017, 377, 2145–2153. [Google Scholar] [CrossRef] [PubMed]
- Lahera, V.; Heras, N.D.L.; Farre, A.L.; Manucha, W.; Ferder, L. Role of Mitochondrial Dysfunction in Hypertension and Obesity. Curr. Hypertens. Rep. 2017, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, P.S.S.; Willemen, H.L.D.M.; Eijkelkamp, N. Mitochondria and sensory processing in inflammatory and neuropathic pain. Front. Pain Res. 2022, 3, 1013577. [Google Scholar] [CrossRef]
- Nicolson, G.L. Mitochondrial Dysfunction and Chronic Disease: Treatment With Natural Supplements. Integr. Med. 2014, 13, 35–43. [Google Scholar]
- Rome, E.S. Obesity Prevention and Treatment. Pediatr. Rev. 2011, 32, 363–373. [Google Scholar] [CrossRef]
- Sanyaolu, A.; Okorie, C.; Qi, X.; Locke, J.; Rehman, S. Childhood and Adolescent Obesity in the United States: A Public Health Concern. Glob. Pediatr. Health 2019, 6, 2333794X19891305. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Cornejo, M.P.; Hentges, S.T.; Maliqueo, M.; Coirini, H.; Becu-Villalobos, D.; Elias, C.F. Neuroendocrine Regulation of Metabolism. J. Neuroendocr. 2016, 28, 12395. [Google Scholar] [CrossRef]
- Lakshman, R.; Elks, C.E.; Ong, K.K. Childhood Obesity. Circulation 2012, 126, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Jurcău, M.C.; Andronie-Cioara, F.L.; Jurcău, A.; Marcu, F.; Ţiț, D.M.; Pașcalău, N.; Nistor-Cseppentö, D.C. The Link between Oxidative Stress, Mitochondrial Dysfunction and Neuroinflammation in the Pathophysiology of Alzheimer’s Disease: Therapeutic Implications and Future Perspectives. Antioxidants 2022, 11, 2167. [Google Scholar] [CrossRef] [PubMed]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef] [PubMed]
- Albano, G.D.; Gagliardo, R.P.; Montalbano, A.M.; Profita, M. Overview of the Mechanisms of Oxidative Stress: Impact in Inflammation of the Airway Diseases. Antioxidants 2022, 11, 2237. [Google Scholar] [CrossRef]
- Kong, Y.; Trabucco, S.E.; Zhang, H. Oxidative stress, mitochondrial dysfunction and the mitochondria theory of aging. Interdiscip. Top. Gerontol. 2014, 39, 86–107. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Griffin, A.; Qiang, Z.; Ren, J. Organelle-targeted therapies: A comprehensive review on system design for enabling precision oncology. Signal Transduct. Target. Ther. 2022, 7, 379. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef]
- Alhabeeb, H.; AlFaiz, A.; Kutbi, E.; AlShahrani, D.; Alsuhail, A.; AlRajhi, S.; Alotaibi, N.; Alotaibi, K.; AlAmri, S.; Alghamdi, S.; et al. Gut Hormones in Health and Obesity: The Upcoming Role of Short Chain Fatty Acids. Nutrients 2021, 13, 481. [Google Scholar] [CrossRef]
- Borer, K.T.; Lin, P.-J.; Wuorinen, E. Timing of Meals and Exercise Affects Hormonal Control of Glucoregulation, Insulin Resistance, Substrate Metabolism, and Gastrointestinal Hormones, but Has Little Effect on Appetite in Postmenopausal Women. Nutrients 2021, 13, 4342. [Google Scholar] [CrossRef]
- Kansra, A.R.; Lakkunarajah, S.; Jay, M.S. Childhood and Adolescent Obesity: A Review. Front. Pediatr. 2021, 8, 581461. [Google Scholar] [CrossRef]
- Voerman, E.; Santos, S.; Inskip, H.; Amiano, P.; Barros, H.; Charles, M.A.; Chatzi, L.; Chrousos, G.P.; Corpeleijn, E.; Crozier, S.; et al. Association of Gestational Weight Gain With Adverse Maternal and Infant Outcomes. JAMA 2019, 321, 1702–1715. [Google Scholar] [CrossRef] [PubMed]
- Kral, J.G.; Biron, S.; Simard, S.; Hould, F.-S.; Lebel, S.; Marceau, S.; Marceau, P. Large Maternal Weight Loss From Obesity Surgery Prevents Transmission of Obesity to Children Who Were Followed for 2 to 18 Years. Pediatrics 2006, 118, e1644–e1649. [Google Scholar] [CrossRef]
- Ong, K.K.; Loos, R. Rapid infancy weight gain and subsequent obesity: Systematic reviews and hopeful suggestions. Acta Paediatr. 2006, 95, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Alejandro, E.U.; Mamerto, T.P.; Chung, G.; Villavieja, A.; Gaus, N.L.; Morgan, E.; Pineda-Cortel, M.R.B. Gestational Diabetes Mellitus: A Harbinger of the Vicious Cycle of Diabetes. Int. J. Mol. Sci. 2020, 21, 5003. [Google Scholar] [CrossRef] [PubMed]
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.; Gruppuso, P.A.; Petzold, K.; Brambilla, D.; Hiilesmaa, V.; Teramo, K.A. Hyperinsulinemia and Macrosomia in the Fetus of the Diabetic Mother. Diabetes Care 1994, 17, 640–648. [Google Scholar] [CrossRef]
- Tam, W.H.; Ma, R.C.W.; Ozaki, R.; Li, A.M.; Chan, M.H.M.; Yuen, L.Y.; Lao, T.T.H.; Yang, X.; Ho, C.S.; Tutino, G.E.; et al. In Utero Exposure to Maternal Hyperglycemia Increases Childhood Cardiometabolic Risk in Offspring. Diabetes Care 2017, 40, 679–686. [Google Scholar] [CrossRef]
- Lee, S.C.; Pu, Y.B.; Chow, C.C.; Yeung, V.T.; Ko, G.T.; So, W.Y.; Li, J.K.; Chan, W.B.; Ma, R.C.; Critchley, J.A.; et al. Diabetes in Hong Kong Chinese: Evidence for familial clustering and parental effects. Diabetes Care 2000, 23, 1365–1368. [Google Scholar] [CrossRef]
- Boudina, S.; Graham, T.E. Mitochondrial function/dysfunction in white adipose tissue. Exp. Physiol. 2014, 99, 1168–1178. [Google Scholar] [CrossRef]
- Arruda, A.P.A.; Pers, B.M.; Parlakgül, G.; Güney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum–mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef]
- Hansen, M.; Lund, M.T.; Gregers, E.; Kraunsøe, R.; Van Hall, G.; Helge, J.W.; Dela, F. Adipose tissue mitochondrial respiration and lipolysis before and after a weight loss by diet and RYGB. Obesity 2015, 23, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M.; Neufer, P.D. Lipid-Induced Mitochondrial Stress and Insulin Action in Muscle. Cell Metab. 2012, 15, 595–605. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, C.D.; Jennings, E.Q.; Harris, P.S.; Galligan, J.J.; Fritz, K.S. Biochemical Mechanisms of Sirtuin-Directed Protein Acylation in Hepatic Pathologies of Mitochondrial Dysfunction. Cells 2022, 11, 2045. [Google Scholar] [CrossRef]
- Waddell, J.; Banerjee, A.; Kristian, T. Acetylation in Mitochondria Dynamics and Neurodegeneration. Cells 2021, 10, 3031. [Google Scholar] [CrossRef] [PubMed]
- Mone, P.; Morgante, M.; Pansini, A.; Jankauskas, S.S.; Rizzo, M.; Lombardi, A.; Frullone, S.; Santulli, G. Effects of insulin resistance on mitochondrial (dys)function. Atherosclerosis 2021, 341, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Deguchi, Y.; Nozaki, Y.; Higami, Y. Contribution of PGC-1α to Obesity- and Caloric Restriction-Related Physiological Changes in White Adipose Tissue. Int. J. Mol. Sci. 2021, 22, 6025. [Google Scholar] [CrossRef]
- Schwärzler, J.; Mayr, L.; Radlinger, B.; Grabherr, F.; Philipp, M.; Texler, B.; Grander, C.; Ritsch, A.; Hunjadi, M.; Enrich, B.; et al. Adipocyte GPX4 protects against inflammation, hepatic insulin resistance and metabolic dysregulation. Int. J. Obes. 2022, 46, 951–959. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef]
- Canto, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Infect. Dis. 2009, 20, 98–105. [Google Scholar] [CrossRef]
- Banks, A.S.; Kon, N.; Knight, C.; Matsumoto, M.; Gutiérrez-Juárez, R.; Rossetti, L.; Gu, W.; Accili, D. SirT1 Gain of Function Increases Energy Efficiency and Prevents Diabetes in Mice. Cell Metab. 2008, 8, 333–341. [Google Scholar] [CrossRef]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. SirT1 Regulates Adipose Tissue Inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef]
- Shoar, Z.; Goldenthal, M.J.; De Luca, F.; Suarez, E. Mitochondrial DNA content and function, childhood obesity, and insulin resistance. Endocr. Res. 2015, 41, 49–56. [Google Scholar] [CrossRef]
- Nishida, K.; Otsu, K. Inflammation and metabolic cardiomyopathy. Cardiovasc. Res. 2017, 113, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.A.; Guo, S. Insulin receptor substrate signaling controls cardiac energy metabolism and heart failure. J. Endocrinol. 2017, 233, R131–R143. [Google Scholar] [CrossRef]
- Kras, K.A.; Langlais, P.R.; Hoffman, N.; Roust, L.R.; Benjamin, T.R.; De Filippis, E.A.; Dinu, V.; Katsanos, C.S. Obesity modifies the stoichiometry of mitochondrial proteins in a way that is distinct to the subcellular localization of the mitochondria in skeletal muscle. Metabolism 2018, 89, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Michalsky, M.P.; Inge, T.H.; Jenkins, T.M.; Xie, C.; Courcoulas, A.; Helmrath, M.; Brandt, M.L.; Harmon, C.M.; Chen, M.; Dixon, J.B.; et al. Cardiovascular Risk Factors After Adolescent Bariatric Surgery. Pediatrics 2018, 141, e20172485. [Google Scholar] [CrossRef] [PubMed]
- Schauer, P.R.; Kashyap, S.R.; Wolski, K.; Brethauer, S.A.; Kirwan, J.P.; Pothier, C.E.; Thomas, S.; Abood, B.; Nissen, S.E.; Bhatt, D.L. Bariatric Surgery versus Intensive Medical Therapy in Obese Patients with Diabetes. N. Engl. J. Med. 2012, 366, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H. Changes in kidney function markers after bariatric surgery in morbidly obese patients. Kidney Res. Clin. Pract. 2020, 39, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Madsen, L.R.; Baggesen, L.M.; Richelsen, B.; Thomsen, R.W. Effect of Roux-en-Y gastric bypass surgery on diabetes remission and complications in individuals with type 2 diabetes: A Danish population-based matched cohort study. Diabetologia 2019, 62, 611–620. [Google Scholar] [CrossRef]
- Bankoglu, E.E.; Seyfried, F.; Rotzinger, L.; Nordbeck, A.; Corteville, C.; Jurowich, C.; Germer, C.T.; Otto, C.; Stopper, H. Impact of weight loss induced by gastric bypass or caloric restriction on oxidative stress and genomic damage in obese Zucker rats. Free. Radic. Biol. Med. 2016, 94, 208–217. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).