
AI-Driven Chemical Design: Transforming the Sustainability of the Pharmaceutical Industry


Abstract
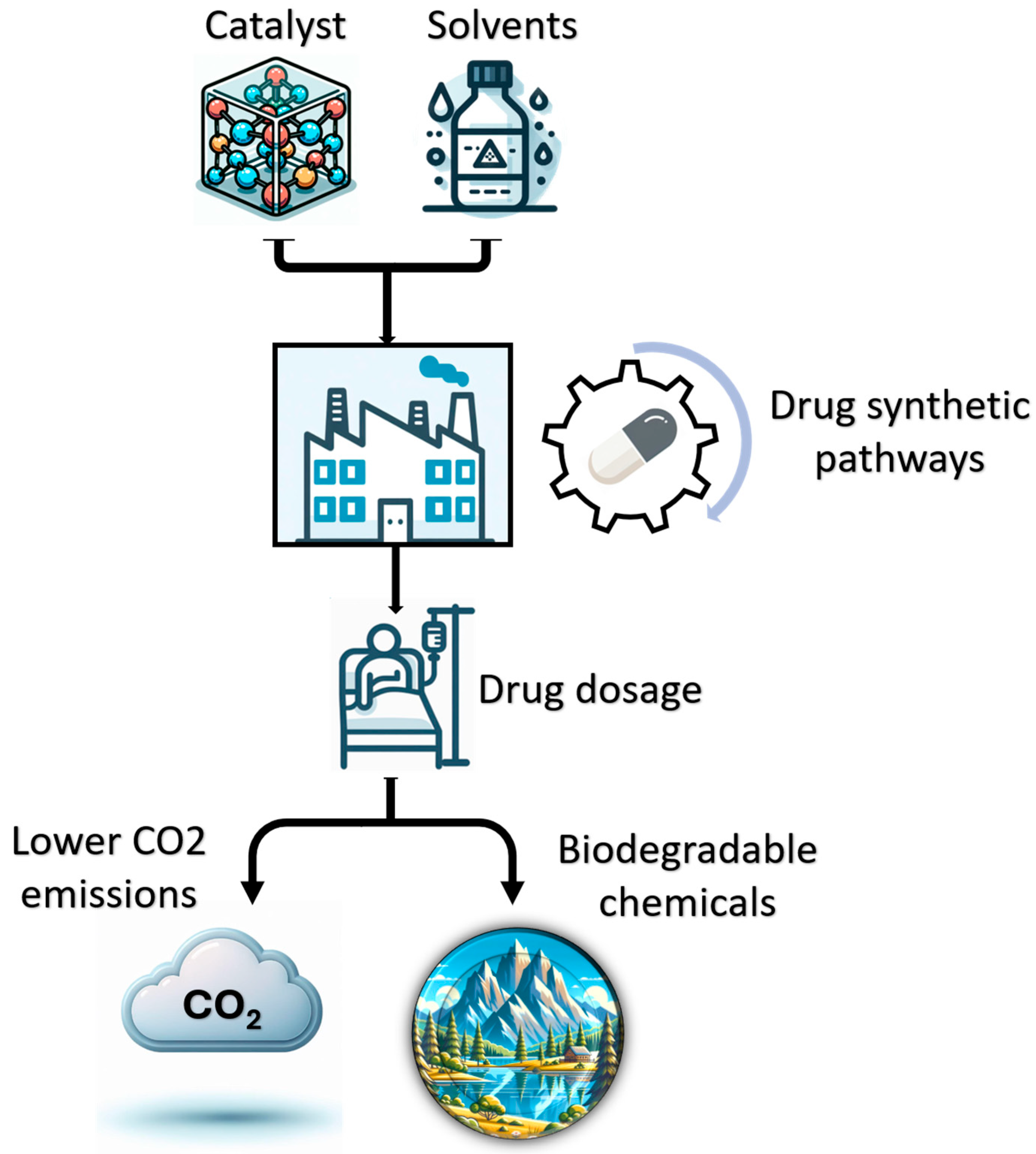
1. Introduction
2. Background Information on AI Models and Key Concepts
- -
- Deep Neural Networks (DNNs) are versatile architectures composed of multiple hidden layers, capable of learning complex, non-linear relationships in data. They are used for both regression (i.e., predicting solubility or melting point) and classification (i.e., toxic, non-toxic substances). DNNs are especially powerful when working with large, high-dimensional datasets such as molecular fingerprints [14].
- -
- Extreme Gradient Boosting (XGBoost) is mainly used with structured data and is widely applied in regression tasks (i.e., predicting pharmacokinetic parameters), as well as in classification problems (i.e., drug–target interaction) [15].
- -
- Support Vector Machines (SVMs) are supervised algorithms that can operate as classifiers or regressors. In drug discovery, they are often used in classification tasks (i.e., active vs. inactive compounds), but they are also applicable in regression settings (i.e., estimating binding affinity or partition coefficients). SVMs perform well in high-dimensional spaces and with small-to-medium-sized datasets [16].
- -
- Random Forests are ensemble models that build multiple decision trees and average their outputs for regression or vote for classification. They are robust to overfitting, handle missing values well, and are commonly used in tasks such as predicting biodegradability, toxicity, or release profiles in drug delivery systems [17].
- -
- Mean Absolute Error (MAE) measures the average magnitude of errors between predicted and true values. It is robust to outliers and provides a straightforward interpretation in the same units as the output variable. It is calculated as in (Equation (1)):where represent the predicted values, represents the actual values, and is the number of samples.
- -
- Root Mean Squared Error (RMSE) is calculated as the square root of the average of the squared differences between the predicted and actual values. This value is more sensitive to outliers than MAE (Equation (2)):
- -
- Mean Relative Error (MRE) expresses the absolute error as a proportion of the true value, averaged across all samples (Equation (3)):
- -
- Mean Absolute Percentage Error (MAPE) represents the average percentage difference between the predicted and actual values (Equation (4)):
- -
- ROC Curves (Receiver Operating Characteristic) are used for binary classification problems. They are graphical representations of the True Positive Rate (Sensitivity) against the False Positive Rate (1 − Specificity) at various threshold settings.
- -
- AUC (Area Under the ROC Curve) summarises the ROC curve into a single number ranging from 0 to 1. It represents the probability that a randomly chosen positive instance is ranked higher than a randomly chosen negative one. Values equal or below 0.5 indicate that the model has no ability to discriminate between categories.
3. Optimisation of Synthetic Routes
- Molecular Complexity: Many pharmaceutical compounds have intricate structures with multiple functional groups. While these complex structures have been associated with higher selectivity [21], it makes their synthesis challenging.
- Chirality and Selectivity: Over half of drugs require specific stereochemistry [24], meaning that reactions must favour the desired enantiomer or diastereomer, which can reduce the effective yield.
- Purification Losses: Rigorous purification steps are needed to meet regulatory purity requirements, leading to additional material loss [25].
3.1. Biocatalyst Design
3.2. Heterogeneous Catalyst Design

4. Synthesis Optimisation
4.1. Speeding Up the Discovery of Drugs

4.2. Solvent Usage
4.3. Synthetic Pathway Optimisation
5. Artificial Intelligence in Drug Waste Reduction
5.1. Reduction in Drug Waste Through Dosage Optimisation
5.2. Design of Biodegradable Drug Delivery Systems
6. Current Status of Molecular Design for Sustainability and Future Perspectives
7. Conclusions
Funding
Conflicts of Interest
References
- Stacciarini, J.H.S. The Global Pharmaceutical Sector: Numbers and dynamics. Caminhos Geogr. 2024, 25. [Google Scholar] [CrossRef]
- Rachet-Jacquet, L.; Rocks, S.; Charlesworth, A. Long-term projections of health care funding, bed capacity and workforce needs in England. Health Policy 2023, 132, 104815. [Google Scholar] [CrossRef] [PubMed]
- Stegemann, S.; van Riet-Nales, D.; de Boer, A. Demographics in the 2020s—Longevity as a challenge for pharmaceutical drug development, prescribing, dispensing, patient care and quality of life. Pharmacology 2020, 86, 1899–1903. [Google Scholar] [CrossRef] [PubMed]
- Benam, K.H.; Gilchrist, S.; Kleensang, A.; Satz, A.B.; Willett, C.; Zhang, Q. Exploring new technologies in biomedical research. Drug Discov. Today 2019, 24, 1242–1247. [Google Scholar] [CrossRef]
- Pichler, P.-P.; Jaccard, I.S.; Weisz, U.; Weisz, H. International comparison of health care carbon footprints. Environ. Res. Lett. 2019, 14, 064004. [Google Scholar] [CrossRef]
- Tennison, I.; Roschnik, S.; Ashby, B.; Boyd, R.; Hamilton, I.; Oreszczyn, T.; Owen, A.; Romanello, M.; Ruyssevelt, P.; Sherman, J.D.; et al. Health care’s response to climate change: A carbon footprint assessment of the NHS in England. Lancet Planet. Health 2021, 5, e84–e92. [Google Scholar] [CrossRef]
- Iyer, J.K. 6 Ways the Pharmaceutical Industry can Reduce Its Climate Impact. 2022. Available online: https://www.weforum.org/stories/2022/11/pharmaceutical-industry-reduce-climate-impact/ (accessed on 14 April 2025).
- Wynendaele, E.; Furman, C.; Wielgomas, B.; Larsson, P.; Hak, E.; Block, T.; Van Calenbergh, S.; Willand, N.; Markuszewski, M.; Odell, L.R.; et al. Sustainability in drug discovery. Med. Drug Discov. 2021, 12, 100107. [Google Scholar] [CrossRef]
- De Spiegeleer, B.; Wynendaele, E. Sustainability of drug discovery, development and use as embedded in European pharmaceutical policies. Curr. Opin. Green Sustain. Chem. 2025, 53, 101028. [Google Scholar] [CrossRef]
- Niemi, L.; Arakawa, N.; Glendell, M.; Gagkas, Z.; Gibb, S.; Anderson, C.; Pfleger, S. Co-developing frameworks towards environmentally directed pharmaceutical prescribing in Scotland—A mixed methods study. Sci. Total Environ. 2024, 955, 176929. [Google Scholar] [CrossRef]
- Mohd Nasir, F.A.; Praveena, S.M.; Aris, A.Z. Public awareness level and occurrence of pharmaceutical residues in drinking water with potential health risk: A study from Kajang (Malaysia). Ecotoxicol. Environ. Saf. 2019, 185, 109681. [Google Scholar] [CrossRef]
- Domingo-Echaburu, S.; Abajo, Z.; Sánchez-Pérez, A.; Elizondo-Alzola, U.; de la Casa-Resino, I.; Lertxundi, U.; Orive, G. Knowledge and attitude about drug pollution in pharmacy students: A questionnaire-based cross sectional study. Curr. Pharm. Teach. Learn. 2023, 15, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Sahrawat, T.R. Role of Artificial Intelligence and Machine Learning in Sustainable Drug Discovery. Braz. Arch. Biol. Technol. 2024, 67, e24240538. [Google Scholar] [CrossRef]
- Askr, H.; Elgeldawi, E.; Ella, H.A.; Elshaier, Y.A.M.M.; Gomaa, M.M. Deep learning in drug discovery: An integrative review and future challenges. Artif. Intell. Rev. 2023, 56, 5975–6037. [Google Scholar] [CrossRef]
- Wiens, M.; Verone-Boyle, A.; Henscheid, N.; Podichetty, J.T.; Burton, J. A Tutorial and Use Case Example of the eXtreme Gradient Boosting (XGBoost) Artificial Intelligence Algorithm for Drug Development Applications. Clin. Transl. Sci. 2025, 18, e70172. [Google Scholar] [CrossRef]
- Heikamp, K.; Bajorath, J. Support vector machines for drug discovery. Expert Opin. Drug Discov. 2014, 9, 93–104. [Google Scholar] [CrossRef]
- Ahn, S.; Lee, S.E.; Kim, M.-H. Random-forest model for drug–target interaction prediction via Kullback–Leibler divergence. J. Cheminform. 2022, 14, 67. [Google Scholar] [CrossRef]
- Ruffolo, J.A.; Madani, A. Designing proteins with language models. Nat. Biotechnol. 2024, 42, 200–202. [Google Scholar] [CrossRef]
- Chen, Z.; Lian, J.Z.; Zhu, H.; Zhang, J.; Zhang, Y.; Xiang, X.; Huang, D.; Tjokro, K.; Barbarossa, V.; Cucurachi, S.; et al. Application of Life Cycle Assessment in the pharmaceutical industry: A critical review. J. Clean. Prod. 2024, 459, 142550. [Google Scholar] [CrossRef]
- Struble, H.G.J.; Coley, C.W.; Green, Y.W.H.; Jensen, K.F. Using Machine Learning to Predict Suitable Conditions for Organic Reactions. ACS Cent. Sci. 2018, 4, 1465–1476. [Google Scholar]
- Méndez-Lucio, O.; Medina-Franco, J.L. The many roles of molecular complexity in drug discovery. Drug Discov. Today 2017, 22, 120–126. [Google Scholar] [CrossRef]
- Bloemendal, V.R.L.J.; Janssen, M.A.C.H.; van Hest, J.C.M.; Rutjes, F.P.J.T. Continuous one-flow multi-step synthesis of active pharmaceutical ingredients. React. Chem. Eng. 2020, 5, 1186–1197. [Google Scholar] [CrossRef]
- Roy, J. Pharmaceutical impurities—A mini-review. AAPS PharmSciTech 2002, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Senkuttuvan, N.; Komarasamy, B.; Krishnamoorthy, R.; Sarkar, S.; Dhanasekarand, S.; Anaikutti, P. The significance of chirality in contemporary drug discovery-a mini review. RSC Adv. 2024, 14, 33429–33448. [Google Scholar] [CrossRef]
- Portoghese, P.S. Revision of Purity Criteria for Tested Compounds. J. Med. Chem. 2009, 52, 1. [Google Scholar] [CrossRef]
- Alcántara, A.R. Special Issue Entitled “10th Anniversary of Catalysts: Recent Advances in the Use of Catalysts for Pharmaceuticals”. Catalysts 2024, 14, 161. [Google Scholar] [CrossRef]
- Verlinden, A.; Boone, L.; De Soete, W.; Dewulf, J. Environmental impacts of drug products: The effect of the selection of production sites in the supply chain. Sustain. Prod. Consum. 2024, 52, 1–11. [Google Scholar] [CrossRef]
- Reetz, M.T.; Qu, G.; Sun, Z. Engineered enzymes for the synthesis of pharmaceuticals and other high-value products. Nat. Synth. 2024, 3, 19–32. [Google Scholar] [CrossRef]
- Alcántara, A.R. Biocatalysis and Pharmaceuticals: A Smart Tool for Sustainable Development. Catalysts 2019, 9, 792. [Google Scholar] [CrossRef]
- Grimaldi, F.; Tran, N.N.; Sarafraz, M.M.; Lettieri, P.; Gonzalez, O.M.M.; Hessel, V. Life Cycle Assessment of an Enzymatic Ibuprofen Production Process with Automatic Recycling and Purification. ACS Sustain. Chem. Eng. 2021, 9, 13135–13150. [Google Scholar] [CrossRef]
- Kumagai, H.; Katayama, T.; Koyanagi, T.; Suzuki, H. Research overview of L-DOPA production using a bacterial enzyme, tyrosine phenol-lyase. Proc. Jpn. Acad. Ser. B 2023, 99, 75–101. [Google Scholar] [CrossRef]
- García-Bofill, M.; Sutton, P.W.; Guillén, M.; Álvaro, G. Enzymatic synthesis of a statin precursor by immobilised alcohol dehydrogenase with NADPH oxidase as cofactor regeneration system. Appl. Catal. A Gen. 2021, 609, 117909. [Google Scholar] [CrossRef]
- Zhao, S.; Tan, M.-Z.; Wang, R.-X.; Ye, F.-T.; Chen, Y.-P.; Luo, X.-M.; Feng, J.-X. Combination of genetic engineering and random mutagenesis for improving production of raw-starch-degrading enzymes in Penicillium oxalicum. Microb. Cell Factories 2022, 21, 272. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.C.; Jin, H.-M.; Kuchner, O.; Arnold, F.H. Strategies for the in vitro evolution of protein function: Enzyme evolution by random recombination of improved sequences. J. Mol. Biol. 1997, 272, 336–347. [Google Scholar] [CrossRef]
- Xie, W.J.; Warshel, A. Harnessing generative AI to decode enzyme catalysis and evolution for enhanced engineering. Natl. Sci. Rev. 2023, 10, nwad331. [Google Scholar] [CrossRef]
- Munsamy, G.; Lindner, S.; Lorenz, P.; Ferruz, N. ZymCTRL: A conditional language model for the controllable generation of artificial enzymes. In Proceedings of the NeurIPS Machine Learning in Structural Biology Workshop, New Orleans, LA, USA, 3 December 2022. [Google Scholar]
- Munsamy, G.; Illanes-Vicioso, R.; Funcillo, S.; Nakou, I.T.; Lindner, S.; Ayres, G.; Sheehan, L.S.; Moss, S.; Eckhard, U.; Ferruz, N. Conditional language models enable the efficient design of proficient enzymes. bioRxiv 2024. [Google Scholar] [CrossRef]
- Madani, A.; Krause, B.; Greene, E.R.; Subramanian, S.; Mohr, B.P.; Holton, J.M.; Olmos, J.L., Jr.; Xiong, C.; Sun, Z.Z.; Socher, R.; et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol. 2023, 41, 1099–1106. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, X.; Zhang, H.; Chu, H.; Shi, C.; Zhang, L.; Bai, J.; Liu, P.; Li, J.; Zhu, X.; et al. Cytochrome P450 Enzyme Design by Constraining the Catalytic Pocket in a Diffusion Model. Research 2024, 7, 0413. [Google Scholar] [CrossRef]
- Hayes, T.; Rao, R.; Akin, H.; Sofroniew, N.J.; Oktay, D.; Lin, V.O.R.I.P.; Verkuil, R.; Tran, V.Q.; Deaton, J.; Wiggert, M.; et al. Simulating 500 million years of evolution with a language model. Science 2025, 387, 850–858. [Google Scholar] [CrossRef]
- Lauko, A.; Pellock, S.J.; Sumida, K.H.; Anishchenko, I.; Juergens, D.; Ahern, W.; Jeung, J.; Shida, A.F.; Hunt, A.; Kalvet, I.; et al. Computational design of serine hydrolases. Science 2025, 388, eadu2454. [Google Scholar] [CrossRef]
- Nielsen, P.H.; Oxenbøll, K.M.; Wenzel, H. Cradle-to-gate environmental assessment of enzyme products produced industrially in denmark by novozymes A/S. Int. J. Life Cycle Assess. 2007, 12, 432–438. [Google Scholar] [CrossRef]
- Kim, S.; Jiménez-González, C.; Dale, B.E. Enzymes for pharmaceutical applications—A cradle-to-gate life cycle assessment. Int. J. Life Cycle Assess. 2009, 14, 392–400. [Google Scholar] [CrossRef]
- Vanella, R.; Küng, C.; Schoepfer, A.A.; Doffini, V.; Nash, J.R.M.A. Understanding activity-stability tradeoffs in biocatalysts by enzyme proximity sequencing. Nat. Commun. 2024, 15, 1807. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, H.C.; Dalby, P.A. Fine-tuning the activity and stability of an evolved enzyme active-site through noncanonical amino-acids. FEBS J. 2021, 288, 1935–1955. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Gao, J.; Ding, Y. DeepPPThermo: A Deep Learning Framework for Predicting Protein Thermostability Combining Protein-Level and Amino Acid-Level Features. J. Comput. Biol. 2023, 31, 147–160. [Google Scholar] [CrossRef]
- Nielsen, G.L.S.R.; Engqvist, M.K.M. Machine Learning Applied to Predicting Microorganism Growth Temperatures and Enzyme Catalytic Optima. ACS Synth. Biol. 2019, 8, 1411–1420. [Google Scholar]
- Bian, J.; Tan, P.; Nie, T.; Hong, L.; Yang, G.-Y. Optimizing enzyme thermostability by combining multiple mutations using protein language model. mLife 2024, 3, 492–504. [Google Scholar] [CrossRef]
- Pak, M.A.; Markhieva, K.A.; Maksimova, E.S.; Kondrashov, F.A.; Ivankov, D.N. Using AlphaFold to predict the impact of single mutations on protein stability and function. PLoS ONE 2023, 18, e0282689. [Google Scholar] [CrossRef]
- Repecka, D.; Jauniskis, V.; Karpus, L.; Rembeza, E.; Rokaitis, I.; Zrimec, J.; Poviloniene, S.; Laurynenas, A.; Viknander, S.; Abuajwa, W.; et al. Expanding functional protein sequence spaces using generative adversarial networks. Nat. Mach. Intell. 2021, 3, 324–333. [Google Scholar] [CrossRef]
- Li, H. AI unveils metal-support interaction principle to optimize catalyst design. Chem Catal. 2025, 5, 101231. [Google Scholar] [CrossRef]
- Marguí, E.; Queralt, I.; Hidalgo, M. Determination of platinum group metal catalyst residues in active pharmaceutical ingredients by means of total reflection X-ray spectrometry. Spectrochim. Acta Part B At. Spectrosc. 2013, 86, 50–54. [Google Scholar] [CrossRef]
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef] [PubMed]
- Ogba, O.M.; Warner, N.C.; O’leary, D.J.; Grubbs, R.H. Recent advances in ruthenium-based olefin metathesis. Chem. Soc. Rev. 2018, 47, 4510–4544. [Google Scholar] [CrossRef] [PubMed]
- Motagamwala, A.H.; Dumesic, J.A. Microkinetic Modeling: A Tool for Rational Catalyst Design. Chem. Rev. 2021, 121, 1049–1076. [Google Scholar] [CrossRef]
- Ahn, S.; Hong, M.; Sundararajan, M.; Ess, D.H.; Baik, M.-H. Design and Optimization of Catalysts Based on Mechanistic Insights Derived from Quantum Chemical Reaction Modeling. Chem. Rev. 2019, 119, 6509–6560. [Google Scholar] [CrossRef]
- Wang, T.; Hu, J.; Ouyang, R.; Wang, Y.; Huang, Y.; Hu, S.; Li, W.-X. Nature of metal-support interaction for metal catalysts on oxide supports. Science 2024, 386, 915–920. [Google Scholar] [CrossRef]
- Deng, B.; Zhong, P.; Jun, K.; Riebesell, J.; Han, K.; Bartel, C.J.; Ceder, G. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nat. Mach. Intell. 2023, 5, 1031–1041. [Google Scholar] [CrossRef]
- Merchant, A.; Batzner, S.; Schoenholz, S.S.; Aykol, M.; Cheon, G.; Cubuk, E.D. Scaling deep learning for materials discovery. Nature 2023, 624, 80–85. [Google Scholar] [CrossRef]
- Chanussot, L.; Das, A.; Goyal, S.; Lavril, T.; Shuaibi, M.; Riviere, M.; Tran, K.; Heras-Domingo, J.; Ho, C.; Hu, W.; et al. Open Catalyst 2020 (OC20) Dataset and Community Challenges. ACS Catal. 2021, 11, 6059–6072. [Google Scholar] [CrossRef]
- Mace, S.; Xu, Y.; Nguyen, B.N. Automated Transition Metal Catalysts Discovery and Optimisation with AI and Machine Learning. ChemCatChem 2024, 16, e202301475. [Google Scholar] [CrossRef]
- Broderick, K.; Lopato, E.; Wander, B.; Bernhard, S.; Kitchin, J.; Ulissi, Z. Identifying limitations in screening high-throughput photocatalytic bimetallic nanoparticles with machine-learned hydrogen adsorptions. Appl. Catal. B Environ. 2023, 320, 121959. [Google Scholar] [CrossRef]
- Choung, S.; Park, W.; Moon, J.; Han, J.W. Rise of machine learning potentials in heterogeneous catalysis: Developments, applications, and prospects. Chem. Eng. J. 2024, 494, 152757. [Google Scholar] [CrossRef]
- Lai, N.S.; Tew, Y.S.; Zhong, X.; Yin, J.; Li, J.; Yan, B.; Wang, X. Artificial Intelligence (AI) Workflow for Catalyst Design and Optimization. Ind. Eng. Chem. Res. 2023, 62, 17835–17848. [Google Scholar] [CrossRef]
- Wang, L.; Chen, X.; Du, Y.; Zhou, Y.; Gao, Y.; Cui, W. CataLM: Empowering catalyst design through large language models. Int. J. Mach. Learn. Cybern. 2025. [Google Scholar] [CrossRef]
- Park, N.H.; Manica, M.; Born, J.; Hedrick, J.L.; Erdmann, T.; Zubarev, D.Y.; Arrechea, N.A.-M.P.L. Artificial intelligence driven design of catalysts and materials for ring opening polymerization using a domain-specific language. Nat. Commun. 2023, 14, 3686. [Google Scholar] [CrossRef]
- Benavides-Hernández, J.; Dumeignil, F. From Characterization to Discovery: Artificial Intelligence, Machine Learning and High-Throughput Experiments for Heterogeneous Catalyst Design. ACS Catal. 2024, 14, 11749–11779. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Jayatunga, M.K.P.; Ayers, M.; Bruens, L.; Jayanth, D.; Meier, C. How successful are AI-discovered drugs in clinical trials? A first analysis and emerging lessons. Drug Discov. Today 2024, 29, 104009. [Google Scholar] [CrossRef]
- Adsheada, F.; Salmanb, R.A.-S.; Aumonierc, S.; Collinsc, M.; Hoodd, K.; McNamarae, C.; Moorea, K.; Smithf, R.; Sydesg, M.R.; Williamson, P.R. A strategy to reduce the carbon footprint of clinical trials. Lancet 2021, 398, 281–282. [Google Scholar] [CrossRef]
- Huang, K.; Chandak, P.; Wang, Q.; Havaldar, S.; Vaid, A.; Leskovec, J.; Nadkarni, G.N.; Glicksberg, B.S.; Zitnik, N.G.M. A foundation model for clinician-centered drug repurposing. Nat. Med. 2024, 30, 3601–3613. [Google Scholar] [CrossRef]
- Gillet, V.J.; Myatt, G.; Zsoldos, Z.; Johnson, A.P. SPROUT, HIPPO and CAESA: Tools for de novo structure generation and estimation of synthetic accessibility. Perspect. Drug Discov. Des. 1995, 3, 34–50. [Google Scholar] [CrossRef]
- Parrot, M.; Tajmouati, H.; da Silva, V.B.R.; Atwood, B.R.; Fourcade, R.; Gaston-Mathé, Y.; Huu, N.D.; Perron, Q. Integrating synthetic accessibility with AI-based generative drug design. J. Cheminform. 2023, 15, 83. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.; Liu, G.; Catacutan, D.B.; Arnold, A.; Zou, J.; Stokes, J.M. Generative AI for designing and validating easily synthesizable and structurally novel antibiotics. Nat. Mach. Intell. 2024, 6, 338–353. [Google Scholar] [CrossRef]
- Available online: https://www.mckinsey.com/industries/life-sciences/our-insights/decarbonizing-api-manufacturing-unpacking-the-cost-and-regulatory-requirements (accessed on 20 February 2025).
- Constable, D.J.C.; Jimenez-Gonzalez, C.; Henderson, R.K. Perspective on Solvent Use in the Pharmaceutical Industry. Org. Process Res. Dev. 2007, 11, 133–137. [Google Scholar] [CrossRef]
- An, M.; Western, L.M.; Say, D.; Chen, L.; Claxton, T.; Ganesan, A.L.; Hossaini, R.; Krummel, P.B.; Manning, A.J.; Mühle, J.; et al. Rapid increase in dichloromethane emissions from China inferred through atmospheric observations. Nat. Commun. 2021, 12, 7279. [Google Scholar] [CrossRef]
- Tsai, W.T. Fate of Chloromethanes in the Atmospheric Environment: Implications for Human Health, Ozone Formation and Depletion, and Global Warming Impacts. Toxics 2017, 5, 23. [Google Scholar] [CrossRef]
- Sels, H.; De Smet, H.; Geuens, J. SUSSOL—Using Artificial Intelligence for Greener Solvent Selection and Substitution. Molecules 2020, 25, 3037. [Google Scholar] [CrossRef]
- Amar, Y.; Schweidtmann, A.M.; Deutsch, P.; Cao, L.; Lapkin, A. Machine learning and molecular descriptors enable rational solvent selection in asymmetric catalysis. Chem. Sci. 2019, 10, 6697–6706. [Google Scholar] [CrossRef]
- Dai, Y.; van Spronsen, J.; Witkamp, G.-J.; Verpoorte, R.; Choi, Y.H. Natural deep eutectic solvents as new potential media for green technology. Anal. Chim. Acta 2013, 766, 61–68. [Google Scholar] [CrossRef]
- Joarder, S.; Bansal, D.; Meena, H.; Kaushik, N.; Tomar, J.; Kumari, K.; Bahadur, I.; Choi, E.H.; Kaushik, N.K.; Singh, P. Bioinspired green deep eutectic solvents: Preparation, catalytic activity, and biocompatibility. J. Mol. Liq. 2023, 376, 121355. [Google Scholar] [CrossRef]
- Available online: https://www.chemistryworld.com/features/solvents-and-sustainability/3008751.article (accessed on 20 February 2025).
- Prabhune, A.; Dey, R. Green and sustainable solvents of the future: Deep eutectic solvents. J. Mol. Liq. 2023, 379, 121676. [Google Scholar] [CrossRef]
- Usmani, Z.; Sharma, M.; Tripathi, M.; Lukk, T.; Karpichev, Y.; Gathergood, N.; Singh, B.N.; Thakur, V.K.; Tabatabaei, M.; Gupta, V.K. Biobased natural deep eutectic system as versatile solvents: Structure, interaction and advanced applications. Sci. Total Environ. 2023, 881, 163002. [Google Scholar] [CrossRef] [PubMed]
- Zaib, Q.; Eckelman, M.J.; Yang, Y.; Kyung, D. Are deep eutectic solvents really green? A life-cycle perspective. Green Chem. 2022, 24, 7924–7930. [Google Scholar] [CrossRef]
- Hayyan, M. Versatile applications of deep eutectic solvents in drug discovery and drug delivery systems: Perspectives and opportunities. Asian J. Pharm. Sci. 2023, 18, 100780. [Google Scholar] [CrossRef] [PubMed]
- Socas-Rodríguez, B.; Torres-Cornejo, M.V.; Álvarez-Rivera, G.; Mendiola, J.A. Deep Eutectic Solvents for the Extraction of Bioactive Compounds from Natural Sources and Agricultural By-Products. Appl. Sci. 2021, 11, 4897. [Google Scholar] [CrossRef]
- Domingues, L.; Duarte, A.R.C.; Jesus, A.R. How Can Deep Eutectic Systems Promote Greener Processes in Medicinal Chemistry and Drug Discovery? Pharmaceuticals 2024, 17, 221. [Google Scholar] [CrossRef]
- Shah, P.A.; Chavda, V.; Hirpara, D.; Sharma, V.S.; Shrivastav, P.S.; Kumar, S. Exploring the potential of deep eutectic solvents in pharmaceuticals: Challenges and opportunities. J. Mol. Liq. 2023, 390, 123171. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Wu, M.; Lifang, H.C.; Qi, C.Z. Deep Deterpenation of Citrus Essential Oils Intensified by In Situ Formation of a Deep Eutectic Solvent in Associative Extraction. Ind. Eng. Chem. Res. 2020, 59, 9223–9232. [Google Scholar] [CrossRef]
- Halder, A.K.; Haghbakhsh, R.; Ferreira, E.S.C.; Duarte, A.R.C.; Cordeiro, M.N.D.S. Machine learning-driven prediction of deep eutectic solvents’ heat capacity for sustainable process design. J. Mol. Liq. 2025, 418, 126707. [Google Scholar] [CrossRef]
- Cysewski, P.; Jeliński, T.; Przybyłek, M.; Mai, A.; Kułak, J. Experimental and Machine-Learning-Assisted Design of Pharmaceutically Acceptable Deep Eutectic Solvents for the Solubility Improvement of Non-Selective COX Inhibitors Ibuprofen and Ketoprofen. Molecules 2024, 29, 2296. [Google Scholar] [CrossRef]
- Abbas, U.L.; Zhang, Y.; Tapia, J.; Md, S.; Chen, J.; Shi, J.; Shao, Q. Machine-Learning-Assisted Design of Deep Eutectic Solvents Based on Uncovered Hydrogen Bond Patterns. Engineering 2024, 39, 74–83. [Google Scholar] [CrossRef]
- Lavrinenko, A.K.; Chernyshov, I.Y.; Pidko, E.A. Machine Learning Approach for the Prediction of Eutectic Temperatures for Metal-Free Deep Eutectic Solvents. ACS Sustain. Chem. Eng. 2023, 11, 15492–15502. [Google Scholar] [CrossRef]
- Chen, M.; Xie, T.; Xu, C. Continuous counter-current centrifugal extraction column with high throughput using a spiral inner cylinder. Chem. Eng. Process. Process Intensif. 2018, 125, 1–7. [Google Scholar] [CrossRef]
- Patel, D.; Suthar, K.J.; Balsora, H.K.; Patel, D.; Panda, S.R.; Bhavsar, N. Estimation of density and viscosity of deep eutectic solvents: Experimental and machine learning approach. Asia-Pac. J. Chem. Eng. 2024, 19, e3151. [Google Scholar] [CrossRef]
- Luu, R.K.; Wysokowski, M.; Buehler, M.J. Generative discovery of de novo chemical designs using diffusion modeling and transformer deep neural networks with application to deep eutectic solvents. Appl. Phys. Lett. 2023, 122, 234103. [Google Scholar] [CrossRef]
- Jin, H.; Jin, Z.; Kim, Y.-G.; Fan, C.; Ghanbari, A. A promising artificial intelligence-based tool to simulate the efficient and sustainable hydrogen sulfide elimination using deep eutectic solvents. Sep. Purif. Technol. 2023, 324, 124472. [Google Scholar] [CrossRef]
- Odegova, V.; Lavrinenko, A.; Rakhmanov, T.; Sysuev, G.; Dmitrenko, A.; Vinogradov, V. DESignSolvents: An open platform for the search and prediction of the physicochemical properties of deep eutectic solvents. Green Chem. 2024, 26, 3958–3967. [Google Scholar] [CrossRef]
- Ayres, L.B.; Gomez, F.J.V.; Silva, M.F.; Garcia, J.R.L.C.D. Predicting the formation of NADES using a transformer-based model. Sci. Rep. 2024, 14, 2715. [Google Scholar] [CrossRef]
- Abdollahzadeh, M.; Khosravi, M.; Masjidi, B.H.K.; Behbahan, A.S.; Bagherzadeh, A.; Shahdost, A.S.F.T. Estimating the density of deep eutectic solvents applying supervised machine learning techniques. Sci. Rep. 2022, 12, 4954. [Google Scholar] [CrossRef]
- Corey, E.J.; Wipke, W.T. Computer-Assisted Design of Complex Organic Syntheses. Science 1969, 166, 178–192. [Google Scholar] [CrossRef]
- Sarkar, R.; De Joarder, D.; Mukhopadhyay, C. Recent advances in the syntheses and reactions of biologically promising β-lactam derivatives. Tetrahedron 2025, 177, 134565. [Google Scholar] [CrossRef]
- Takabatake, T.; Fujiwara, K.; Okamoto, S.; Kishimoto, R.; Kagawa, N.; Toyota, M. Discovery of orthogonal synthesis using artificial intelligence: Pd(OAc)2-catalyzed one-pot synthesis of benzofuran and bicyclo[3.3.1]nonane scaffolds. Tetrahedron Lett. 2020, 61, 152275. [Google Scholar] [CrossRef]
- King-Smith, E.; Berritt, S.; Bernier, L.; Hou, X.; Klug-McLeod, J.L.; Mustakis, J.; Sach, N.W.; Tucker, J.W.; Yang, Q.; Lee, R.M.H.A.A. Probing the chemical ‘reactome’ with high-throughput experimentation data. Nat. Chem. 2024, 16, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, W.; Sremski, W.; Piccini, B.; Palluel, O.; Maillot-Maréchal, E.; Betoulle, S.; Jaffal, A.; Aït-Aïssa, S.; Brion, F.; Thybaud, E.; et al. Adverse effects in wild fish living downstream from pharmaceutical manufacture discharges. Environ. Int. 2011, 37, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Kidd, K.A.; Blanchfield, P.J.; Mills, K.H.; Flick, R.W. Collapse of a fish population after exposure to a synthetic estrogen. Proc. Natl. Acad. Sci. USA 2007, 104, 8897–8901. [Google Scholar] [CrossRef]
- The State of the World’s Antibiotics 2021, a Global Analysis of Antimicrobial Resistance and Its Drivers, the Center for Disease Dynamics, Economics & Policy. 2021. Available online: https://onehealthtrust.org/wp-content/uploads/2021/02/SOWA_01.02.2021_Low-Res.pdf (accessed on 10 October 2021).
- Chung, S.-S.; Brooks, B.W. Identifying household pharmaceutical waste characteristics and population behaviors in one of the most densely populated global cities. Resour. Conserv. Recycl. 2019, 140, 267–277. [Google Scholar] [CrossRef]
- Vieno, N.; Hallgren, P.; Wallberg, P.; Pyhälä, M.; Zandaryaa, S. Baltic Marine Environment Protection Commission. In Pharmaceuticals in the Aquatic Environment of the Baltic Sea Region: A Status Report; CCB Report: Pharmaceutical Pollution in the Baltic Sea Region; UNESCO Publishing: Uppsala, Sweden, 2017; Volume 1. [Google Scholar]
- Daughton, C.G.; Ruhoy, I.S. Lower-dose prescribing: Minimizing “side effects” of pharmaceuticals on society and the environment. Sci. Total Environ. 2013, 443, 324–337. [Google Scholar] [CrossRef]
- Zhu, X.; Huang, W.; Lu, H.; Wang, Z.; Ni, X.; Hu, J.; Deng, S.; Tan, Y.; Li, L.; Zhang, M.; et al. A machine learning approach to personalized dose adjustment of lamotrigine using noninvasive clinical parameters. Sci. Rep. 2021, 11, 5568. [Google Scholar] [CrossRef]
- Iancu, A.; Leb, I.; Prokosch, H.-U.; Rödle, W. Machine learning in medication prescription: A systematic review. Int. J. Med. Inform. 2023, 180, 105241. [Google Scholar] [CrossRef]
- Lai, Y.; Chu, X.; Di, L.; Gao, W.; Guo, Y.; Liu, X.; Lu, C.; Mao, J.; Shen, H.; Tang, H.; et al. Recent advances in the translation of drug metabolism and pharmacokinetics science for drug discovery and development. Acta Pharm. Sin. B 2022, 12, 2751–2777. [Google Scholar] [CrossRef]
- Satheeskumar, R. Enhancing drug discovery with AI: Predictive modeling of pharmacokinetics using Graph Neural Networks and ensemble learning. Intell. Pharm. 2024, 3, 127–140. [Google Scholar] [CrossRef]
- Swanson, K.; Walther, P.; Leitz, J.; Mukherjee, S.; Wu, J.C.; Shivnaraine, R.V.; Zou, J. ADMET-AI: A machine learning ADMET platform for evaluation of large-scale chemical libraries. Bioinformatics 2024, 40, btae416. [Google Scholar] [CrossRef] [PubMed]
- Farhud, D.D.; Zokaei, S. Ethical Issues of Artificial Intelligence in Medicine and Healthcare. Iran J. Public Health 2021, 50, i–v. [Google Scholar] [CrossRef] [PubMed]
- Sulbaek Andersen, M.P.; Nielsen, O.J.; Sherman, J.D. Assessing the potential climate impact of anaesthetic gases. Lancet Planet. Health 2023, 7, e622–e629. [Google Scholar] [CrossRef]
- White, S.M.; Shelton, C.L. Abandoning inhalational anaesthesia. Sci. Rep. 2020, 75, 451–454. [Google Scholar] [CrossRef]
- Miyaguchi, N.; Takeuchi, K.; Kashima, H.; Morita, M. Predicting anesthetic infusion events using machine learning. Sci. Rep. 2021, 11, 23648. [Google Scholar] [CrossRef]
- Hu, Y.-J.; Ku, T.-H.; Jan, R.-H.; Wang, K.; Tseng, Y.-C. Decision tree-based learning to predict patient controlled analgesia consumption and readjustment. BMC Med. Inform. Decis. Mak. 2012, 12, 131. [Google Scholar] [CrossRef]
- Ortolani, O.; Conti, A.; Di Filippo, A.; Adembri, C.; Moraldi, E.; Evangelisti, A.; Maggini, M.; Roberts, S.J. EEG signal processing in anaesthesia. Use of a neural network technique for monitoring depth of anaesthesia. Br. J. Anaesth. 2002, 88, 644–648. [Google Scholar] [CrossRef]
- Anderson, W.A.; Rao, A. Anesthetic Gases: Environmental Impacts and Mitigation Strategies for Fluranes and Nitrous Oxide. Environments 2024, 11, 275. [Google Scholar] [CrossRef]
- Schamberg, G.; Badgeley, M.; Meschede-Krasa, B.; Kwon, O.; Brown, E.N. Continuous action deep reinforcement learning for propofol dosing during general anesthesia. Artif. Intell. Med. 2022, 123, 102227. [Google Scholar] [CrossRef] [PubMed]
- Nyirenda, J.; Mwanza, A.; Lengwe, C. Assessing the biodegradability of common pharmaceutical products (PPs) on the Zambian market. Heliyon 2020, 6, e05286. [Google Scholar] [CrossRef]
- Barra Caracciolo, A.; Topp, E.; Grenni, P. Pharmaceuticals in the environment: Biodegradation and effects on natural microbial communities. A review. J. Pharm. Biomed. Anal. 2015, 106, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.L.; Boxall, A.B.A.; Kolpin, D.W.; Leung, K.M.Y.; Lai, R.W.S.; Galbán-Malagón, C.; Adell, A.D.; Mondon, J.; Metian, M.; Marchant, R.A.; et al. Pharmaceutical pollution of the world’s rivers. Proc. Natl. Acad. Sci. USA 2022, 119, e2113947119. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.; Muhammad, F.; Akhtar, B. Biodegradable nanoparticles as drug delivery devices. J. Drug Deliv. Sci. Technol. 2021, 64, 102638. [Google Scholar] [CrossRef]
- Sun, S.; Cui, Y.; Yuan, B.; Dou, M.; Wang, G.; Xu, H.; Wang, J.; Yin, W.; Wu, D.; Peng, C. Drug delivery systems based on polyethylene glycol hydrogels for enhanced bone regeneration. Front. Bioeng. Biotechnol. 2023, 11, 1117647. [Google Scholar] [CrossRef]
- Sanchez Armengol, E.; Alexander, U.; Laffleur, F. PEGylated drug delivery systems in the pharmaceutical field: Past, present and future perspective. Drug Dev. Ind. Pharm. 2022, 48, 129–139. [Google Scholar] [CrossRef]
- Vlachopoulos, A.; Karlioti, G.; Balla, E.; Daniilidis, V.; Kalamas, T.; Stefanidou, M.; Bikiaris, N.D.; Christodoulou, E.; Koumentakou, I.; Karavas, E.; et al. Poly(Lactic Acid)-Based Microparticles for Drug Delivery Applications: An Overview of Recent Advances. Pharmaceutics 2022, 14, 359. [Google Scholar] [CrossRef]
- Tyler, B.; Gullotti, D.; Mangraviti, A.; Utsuki, T.; Brem, H. Polylactic acid (PLA) controlled delivery carriers for biomedical applications. Adv. Drug Deliv. Rev. 2016, 107, 163–175. [Google Scholar] [CrossRef]
- Hines, D.J.; Kaplan, D.L. Poly(lactic-co-glycolic) acid-controlled-release systems: Experimental and modeling insights. Crit. Rev. Ther. Drug Carr. Syst. 2013, 30, 257–276. [Google Scholar] [CrossRef]
- El Allaoui, B.; Benzeid, H.; Zari, N.; el kacem Qaiss, A.; Bouhfid, R. Functional cellulose-based beads for drug delivery: Preparation, functionalization, and applications. J. Drug Deliv. Sci. Technol. 2023, 88, 104899. [Google Scholar] [CrossRef]
- Sivamaruthi, B.S.; Nallasamy, P.K.; Suganthy, N.; Kesika, P.; Chaiyasut, C. Pharmaceutical and biomedical applications of starch-based drug delivery system: A review. J. Drug Deliv. Sci. Technol. 2022, 77, 103890. [Google Scholar] [CrossRef]
- Boztepe, C.; Künkül, A.; Yüceer, M. Application of artificial intelligence in modeling of the doxorubicin release behavior of pH and temperature responsive poly(NIPAAm-co-AAc)-PEG IPN hydrogel. J. Drug Deliv. Sci. Technol. 2020, 57, 101603. [Google Scholar] [CrossRef]
- Aghajanpour, S.; Amiriara, H.; Esfandyari-Manesh, M.; Ebrahimnejad, P.; Jeelani, H.; Henschel, A.; Singh, H.; Dinarvand, R.; Hassan, S. Utilizing machine learning for predicting drug release from polymeric drug delivery systems. Comput. Biol. Med. 2025, 188, 109756. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, R.; da Costa, C.A.; de Figueiredo, R.M.; Zehetmeyer, G.; Schmith, J. A method to predict the percentage of biodegradation in polymeric materials. Comput. Electr. Eng. 2024, 118, 109473. [Google Scholar] [CrossRef]
- Chen, T.; Pang, Z.; He, S.; Li, Y.; Shrestha, S.; Little, J.M.; Yang, H.; Chung, T.-C.; Sun, J.; Whitley, H.C.; et al. Machine intelligence-accelerated discovery of all-natural plastic substitutes. Nat. Nanotechnol. 2024, 19, 782–791. [Google Scholar] [CrossRef]
- Alshahrani, S.M.; Alotaibi, H.F.; Alqarni, M. Modeling and validation of drug release kinetics using hybrid method for prediction of drug efficiency and novel formulations. Front. Chem. 2024, 12, 1395359. [Google Scholar] [CrossRef]
- AL-Rajabi, M.M.; Alzyod, S.; Patel, A.; Teow, Y.H. A hybrid machine learning framework for predicting drug-release profiles, kinetics, and mechanisms of temperature-responsive hydrogels. Polym. Bull. 2025, 82, 2911–2932. [Google Scholar] [CrossRef]
- WHO Calls for Transformative Action Towards a Greener Future in Pharmaceutical Manufacturing and Distribution. 23 December 2024. Available online: https://www.who.int/news/item/23-12-2024-who-calls-for-transformative-action-towards-a-greener-future-in-pharmaceutical-manufacturing-and-distribution (accessed on 16 May 2025).
- Available online: https://acsgcipr.org/ (accessed on 16 May 2025).
- Benison, C.H.; Payne, P.R. Manufacturing mass intensity: 15 Years of Process Mass Intensity and development of the metric into plant cleaning and beyond. Curr. Res. Green Sustain. Chem. 2022, 5, 100229. [Google Scholar] [CrossRef]
- Available online: https://open-reaction-database.org/browse (accessed on 16 May 2025).
- Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 16 May 2025).
- Available online: https://alphafold.ebi.ac.uk/ (accessed on 16 May 2025).
- Available online: http://www.swissadme.ch/ (accessed on 16 May 2025).
- Available online: https://deepchem.io/ (accessed on 16 May 2025).
- Available online: https://github.com/salesforce/progen (accessed on 16 May 2025).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Model Prediction | Algorithm | Performance | Refs. |
|---|---|---|---|---|
| 280 million protein sequences from >19,000 families | Protein function | 1.2-billion-parameter neural network | AUC = 0.85 (experimentally validated) | [38] |
| BRENDA DATASET (37,624,812 sequences) | Protein activity (model made available as Open Source) | Language model | - | [36,37] |
| 3.15 billion protein sequences, 236 million protein structures, and 539 million proteins with function annotations | Protein activity | Language model | SS3 > 80% pTM > 0.8 pLDDT > 0.8 RMSD < 1.5 Å (experimentally validated) | [40] |
| Protein–small molecule complexes in PDB | Enzyme structure | Deep Neural Network | RMSD < 1.5 Å (experimentally validated) | [41] |
| 2950 thermophilic protein sequences | Enzyme thermostability | Deep Neural Network and bi-long short-term memory | ACC (%) = 94.34 PR (%) = 93.97 REC (%) = 94.81 REC (%) = 94.36 MCC (%) = 88.73 AUROC (%) = 98.68 | [46] |
| Dataset from 21,498 microorganisms | Enzyme optimal temperature (model made available as Open Source) | Random Forest regressor | R2 = 0.94 RMSE = 4.46 | [47] |
| Optimal growth temperatures for 96 million host bacterial strains | Enzyme thermostability | Language model | Experimentally validated | [48] |
| 16,706 unique sequences | Catalytic activity | Generative Adversarial Network | Experimentally validated | [50] |
| Dataset | Model Prediction | Algorithm | Performance | Ref. |
|---|---|---|---|---|
| 603 articles | Catalyst synthesis optimisation | Large language model and Bayesian optimisation | Experimentally validated | [64] |
| 22,000 articles from Web of Science | Catalyst synthesis optimisation | Language model | Average predicted faraday efficiency = 64.15% | [65] |
| 549 monomer–catalyst pairs for catalyst model | Monomer conversion, dispersity, and average molecular weight | Regression transformer | Pearson correlation = 0.59 (Experimentally validated) | [66] |
| Dataset | Model Prediction | Algorithm | Performance | Ref. |
|---|---|---|---|---|
| 186 (simulated systems) + 34 (experimentally verified) | DES formation | Support Vector Machine | Average ROC-AUC score = 0.8 (experimentally validated) | [94] |
| 237 experimentally validated DESs | Melting temperature | Support Vector Regression | R2 = 0.74, RMSE = 22.5 | [95] |
| 530 DESs | Heat capacity | Neural Network Multilayer Perceptron | AARD = 4% | [92] |
| 402 different DES compositions | Diffusion model with self-/cross-attention | R2 = 0.93 (experimentally validated) | [98] | |
| 435 records | Hydrogen sulphide (H2S) elimination capacity | Cascade neural network | MAE = 0.02 MSE = 0.0031 AARE = 3.03 R2 = 0.99943 (experimentally validated) | [99] |
| - | Melting temperature, density, viscosity (model made available as Open Source) | CatBoost | R2 = 0.6–0.91 AARD = 3.14–19.05% (experimentally validated) | [100] |
| - | Solubility | Non-linear Support Vector Regression | Experimentally validated | [93] |
| 1000 labelled mixtures of DESs and NADESs | DES formation | Transformer-based neural network model | F1-Score = 0.82 (experimentally validated) | [101] |
| 1239 records | DES density | Least-squares Support Vector Regression | R2 = 0.99798 MAPE = 0.26% | [102] |
| Dataset | Model Prediction | Algorithm | Performance | Ref. |
|---|---|---|---|---|
| 1141 therapeutic drug-monitoring measurements from 347 patients | Dose-adjusted concentrations of lamotrigine | Extra-trees regression algorithm | MAE = 8.7 μg mL−1 g−1 day Mean Relative Error (%) = 3% (experimentally validated) | [113] |
| 1099 patients, each described with 280 attributes | Postoperative analgesic requirements | Decision trees | Prediction accuracies of total analgesic consumption of 80.9% (experimentally validated) | [122] |
| 10,000 bioactive compounds from ChEMBL database | Clearance, volume distribution, half-life, bioavailability | Stacking ensemble model | R2 = 0.92 MAE = 0.062 | [116] |
| Simulated pharmacokinetic and pharmacodynamic data | Anaesthetic dosing | Reinforcement learning algorithm | Median episode median performance error of 1.1% ± 0.5 (experimentally validated with retrospective data) | [125] |
| 210 case data | Increase in flow rate of remifentanil | Long short-term memory | Specificity = 0.73 Sensitivity = 0.66 ROC-AUC = 0.75 | [121] |
| 13 processed EEG parameters from 200 patients | Anaesthesia scores | Reinforcement learning algorithm | R2 = 0.94 (experimentally validated) | [123] |
| Dataset | Model Prediction | Algorithm | Performance | Ref. |
|---|---|---|---|---|
| 540 experimental data points | Release kinetics | Support Vector Machine | R2 = 0.998 RMSE = 0.3701 MSE = 0.137 MAPE = 0.944 (experimentally validated) | [137] |
| 76 biodegradation curves | Biodegradation kinetics (%) | Long short-term memory neural network | MAPE = 7.6–16.99% MAE = 0.018–0.027 MSE = 0.0004–0.001 RMSE = 6.71–11.33 (experimentally validated with validation dataset) | [139] |
| Data from 133 experiments | Polymer properties (Young’s modulus, flammability) | Artificial neural network | MRE = 21% (experimentally validated with molecular dynamic simulations) | [140] |
| 15,000 data samples | Drug release kinetics | Decision Tree Regression | R2 = 0.94652 RMSE = 6.04 × 10−5 MAE = 4.83 × 10−5 | [141] |
| 500 datapoints | Drug release kinetics | Random Forest | R2 = 0.99 MAPE = 0.002 (experimentally validated) | [142] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Gonzalez, A. AI-Driven Chemical Design: Transforming the Sustainability of the Pharmaceutical Industry. Future Pharmacol. 2025, 5, 24. https://doi.org/10.3390/futurepharmacol5020024
Ruiz-Gonzalez A. AI-Driven Chemical Design: Transforming the Sustainability of the Pharmaceutical Industry. Future Pharmacology. 2025; 5(2):24. https://doi.org/10.3390/futurepharmacol5020024
Chicago/Turabian StyleRuiz-Gonzalez, Antonio. 2025. "AI-Driven Chemical Design: Transforming the Sustainability of the Pharmaceutical Industry" Future Pharmacology 5, no. 2: 24. https://doi.org/10.3390/futurepharmacol5020024
APA StyleRuiz-Gonzalez, A. (2025). AI-Driven Chemical Design: Transforming the Sustainability of the Pharmaceutical Industry. Future Pharmacology, 5(2), 24. https://doi.org/10.3390/futurepharmacol5020024