
Green Tea Extract Solid Dispersion Pellets with Enhanced Permeability for Hyperlipidemia
 ,
, 
Abstract
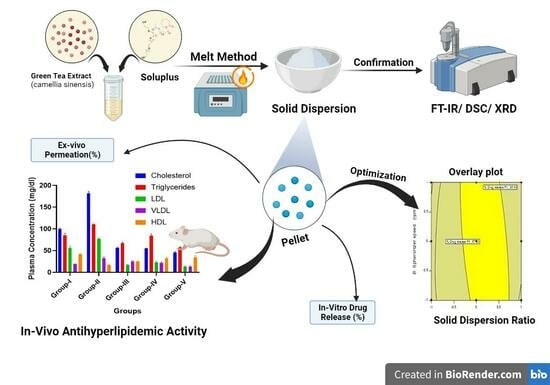
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
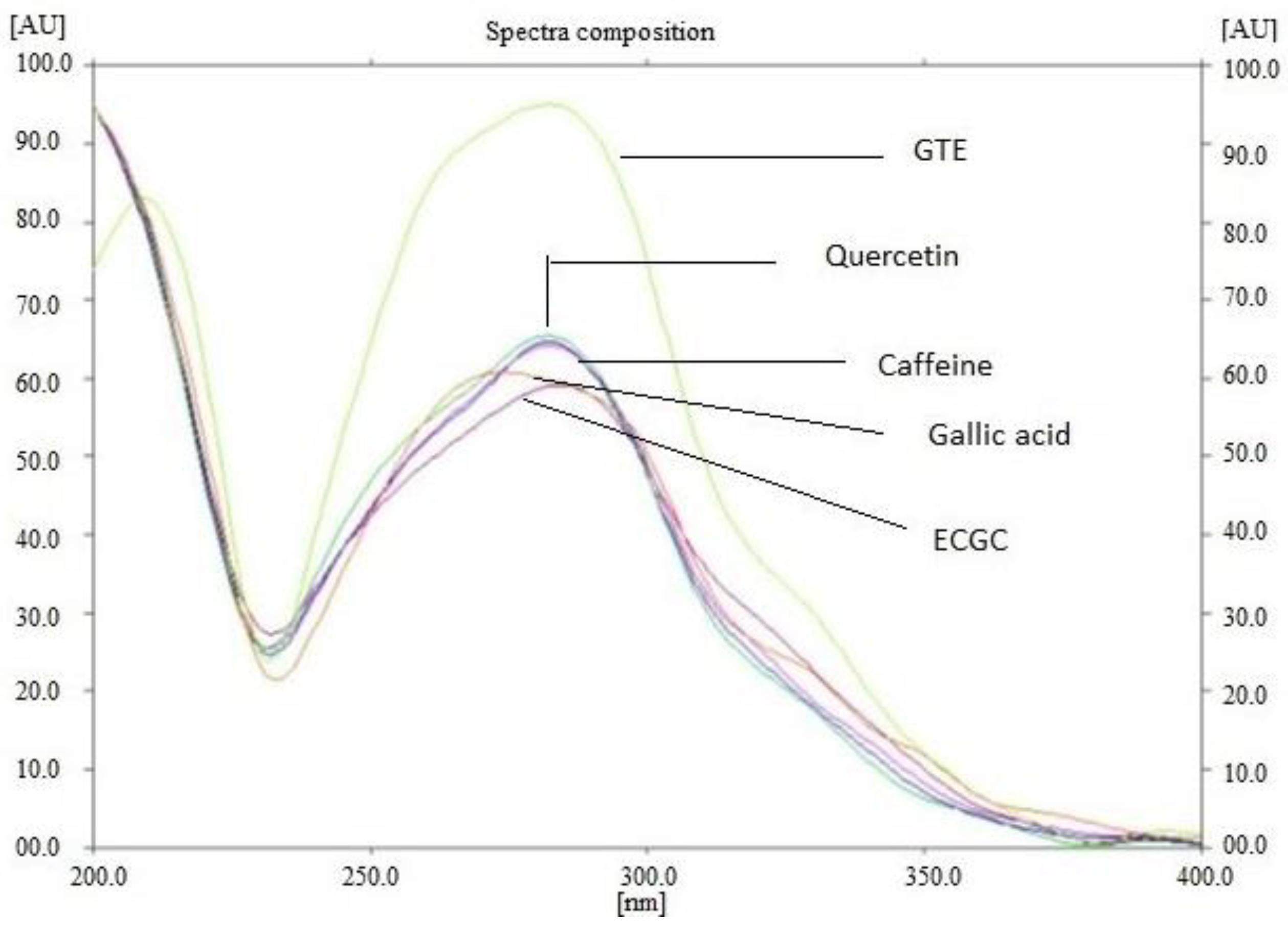
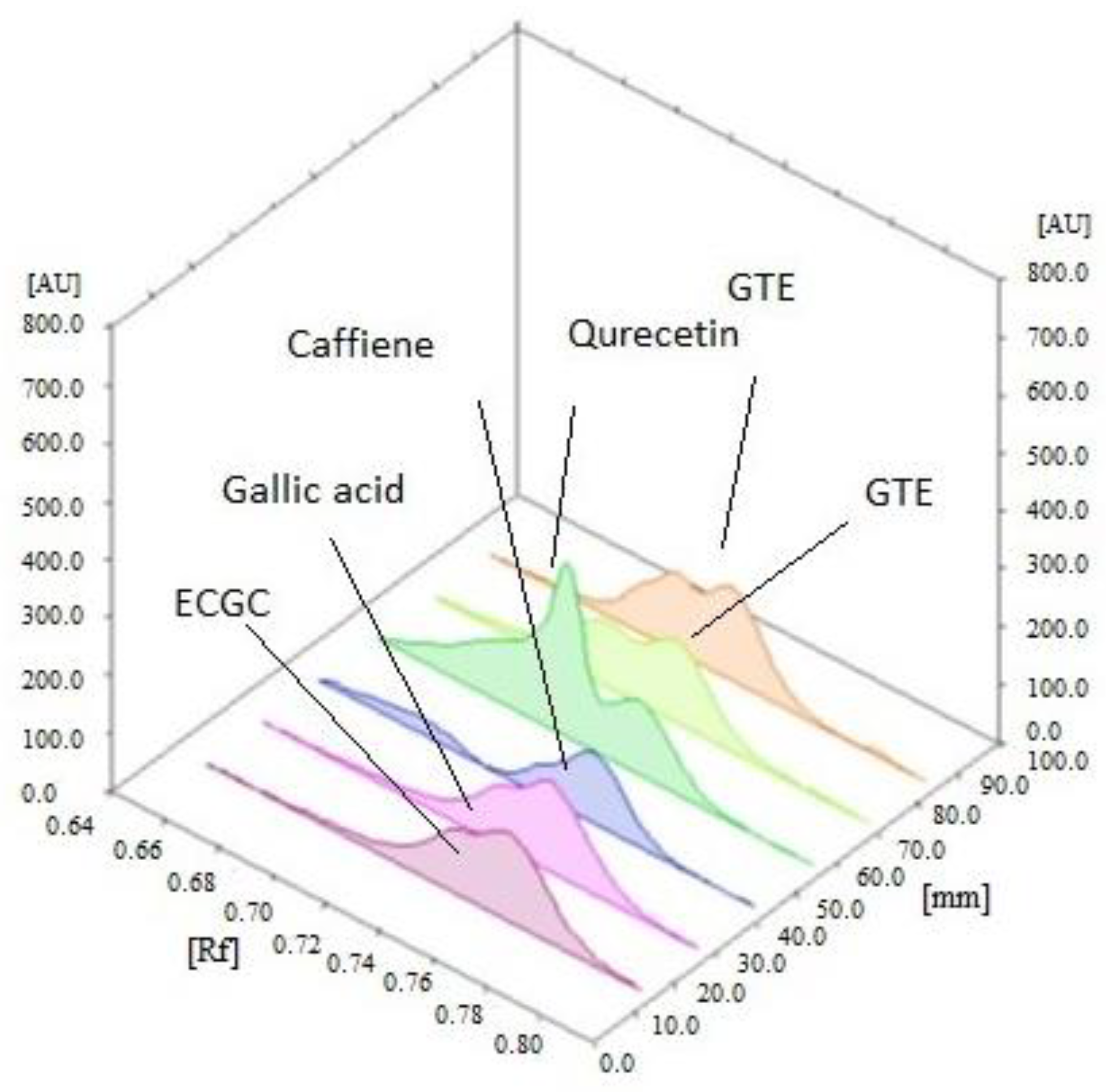
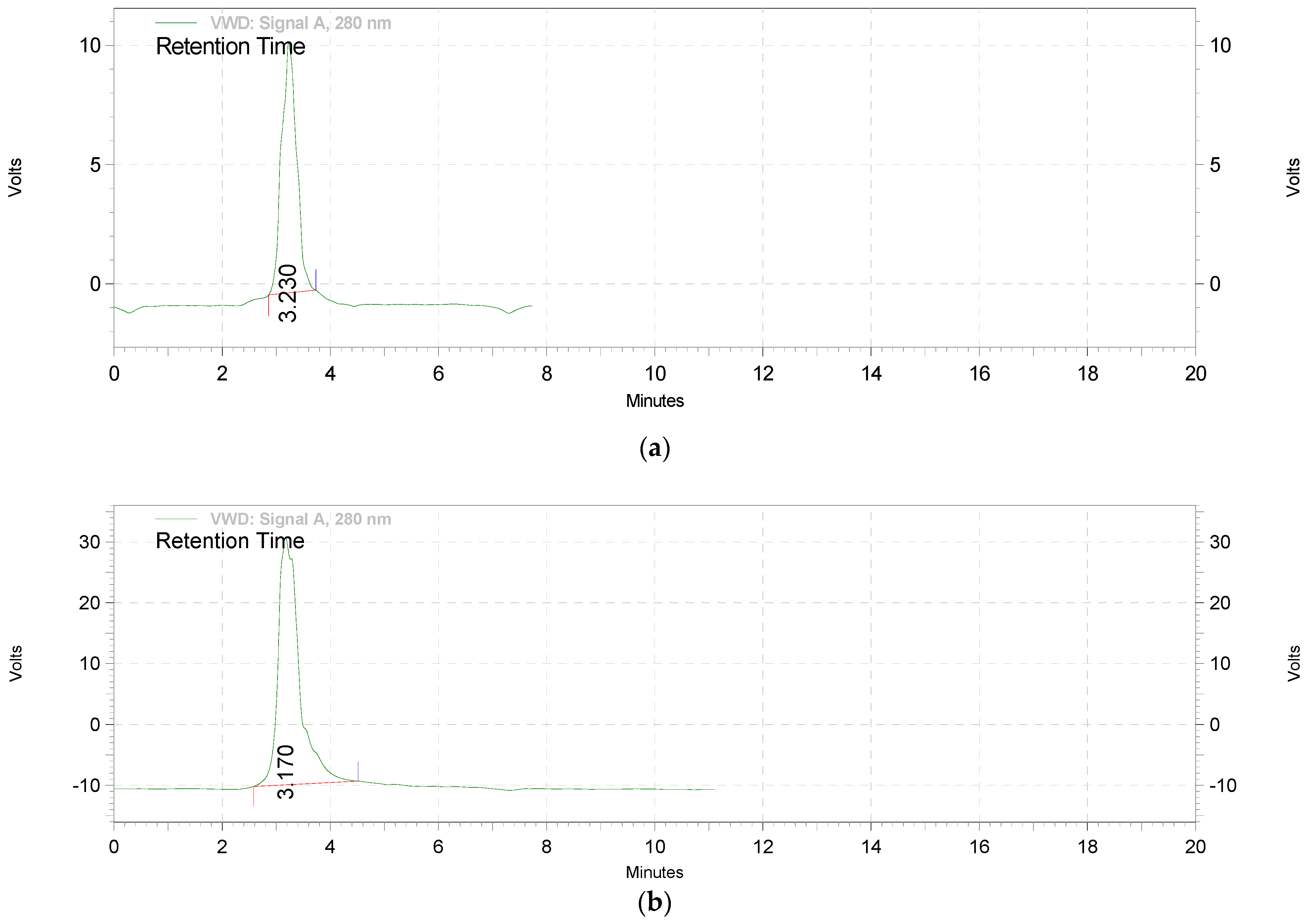
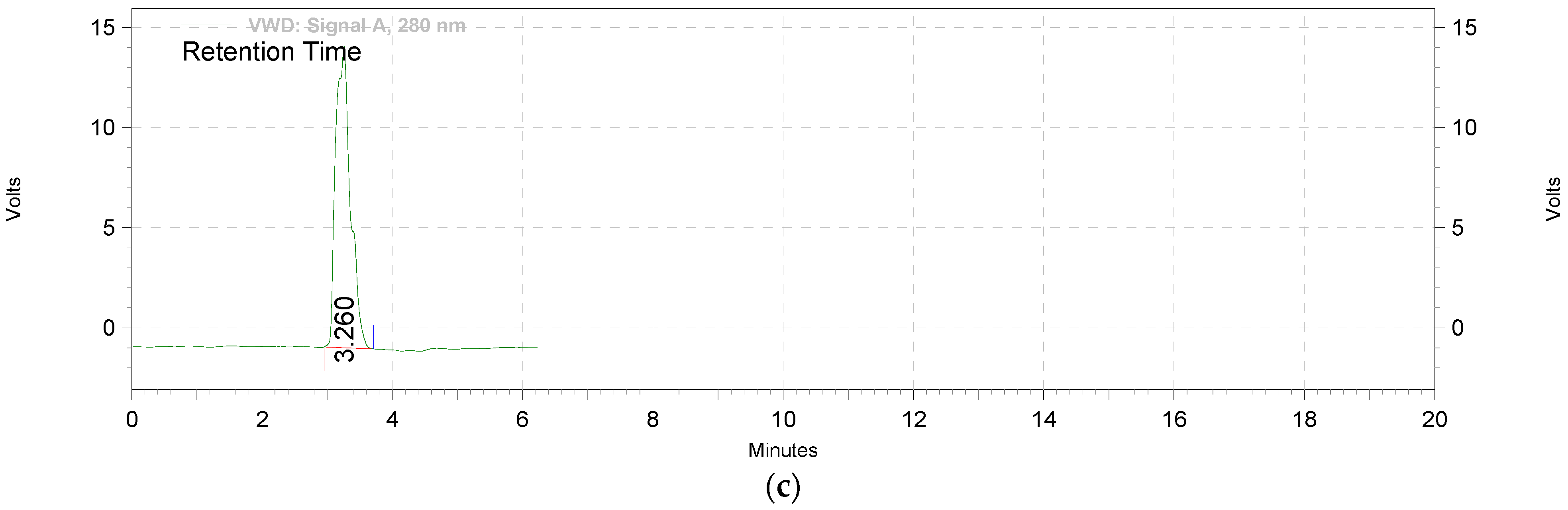
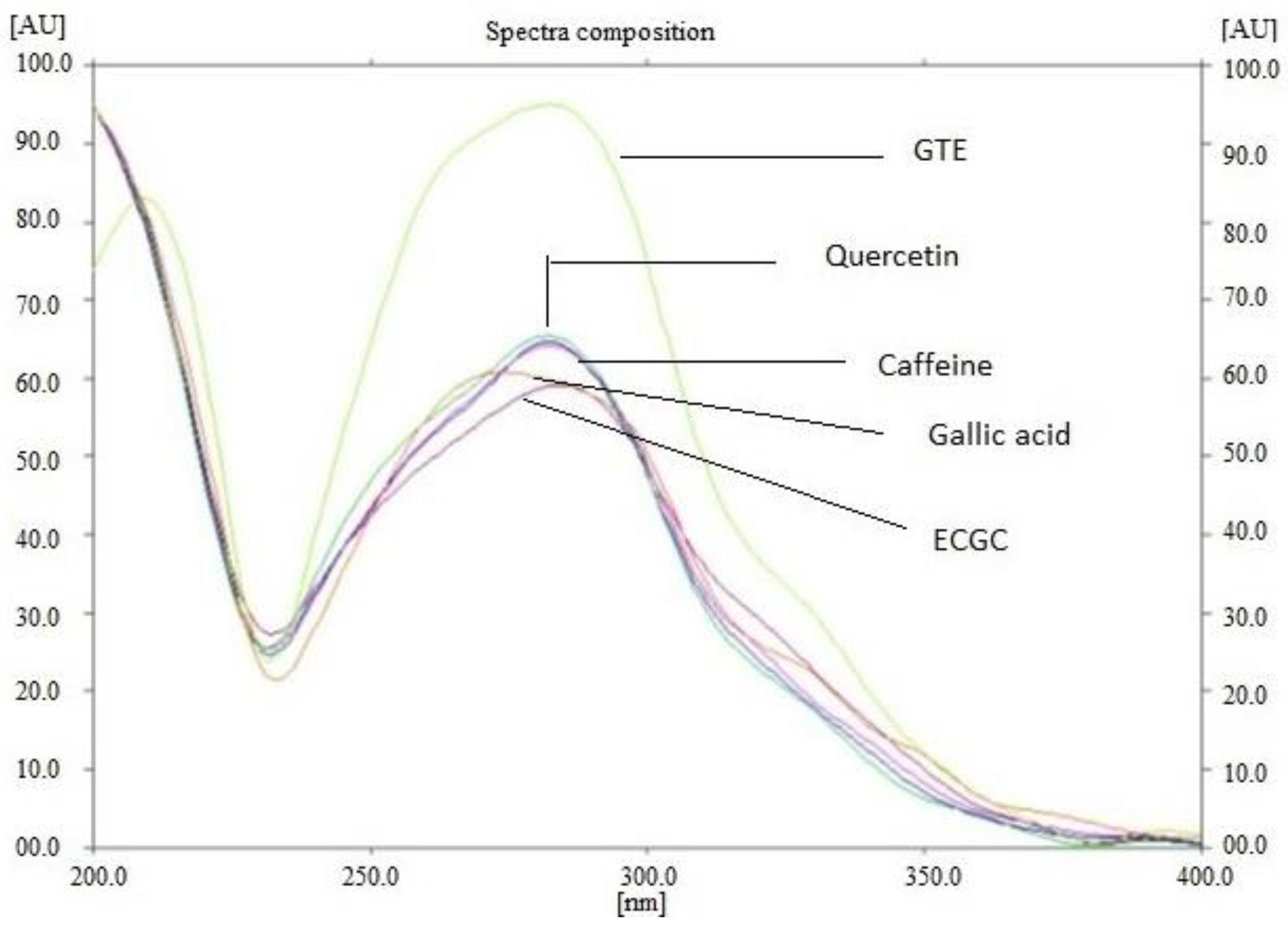
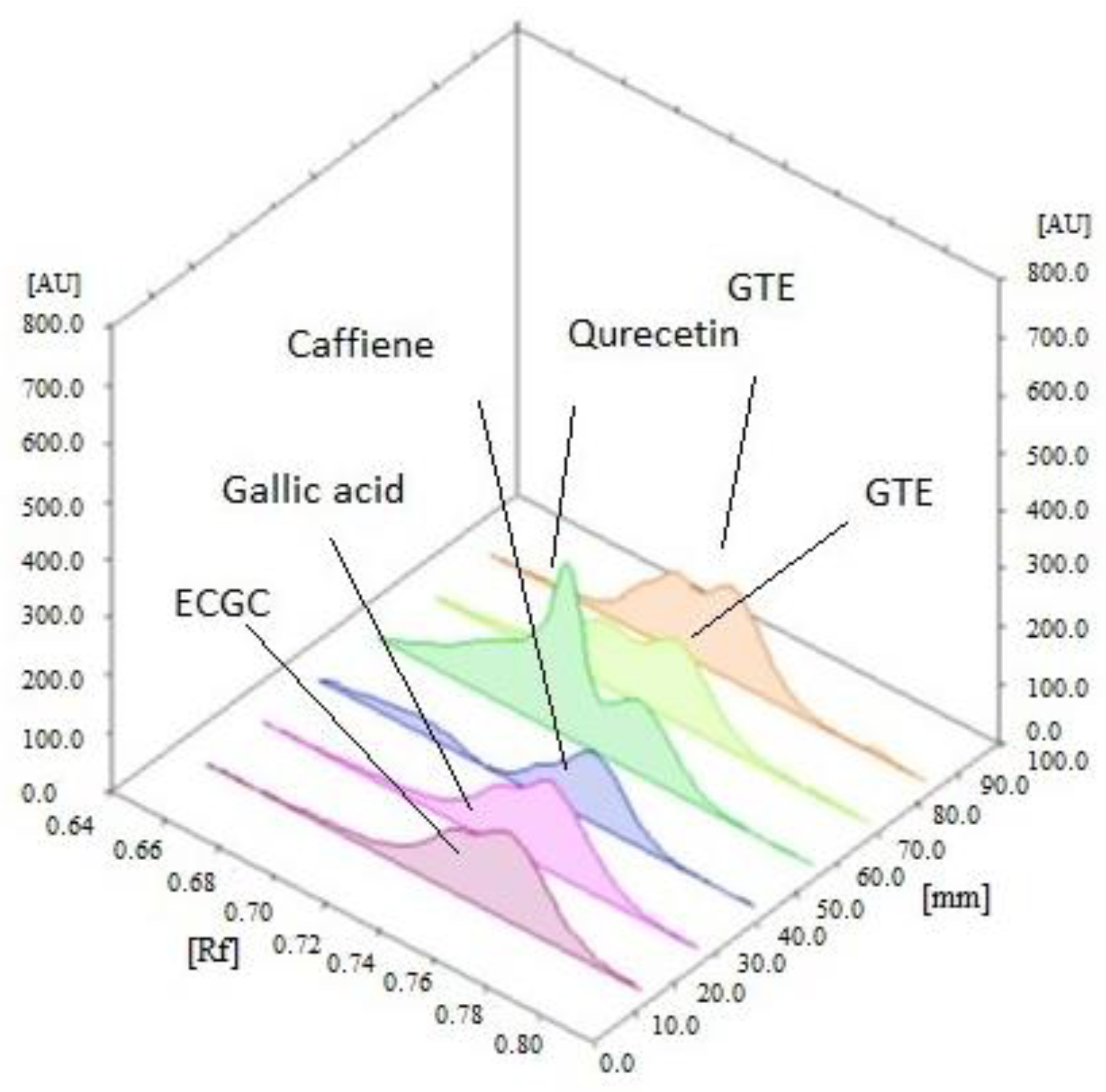
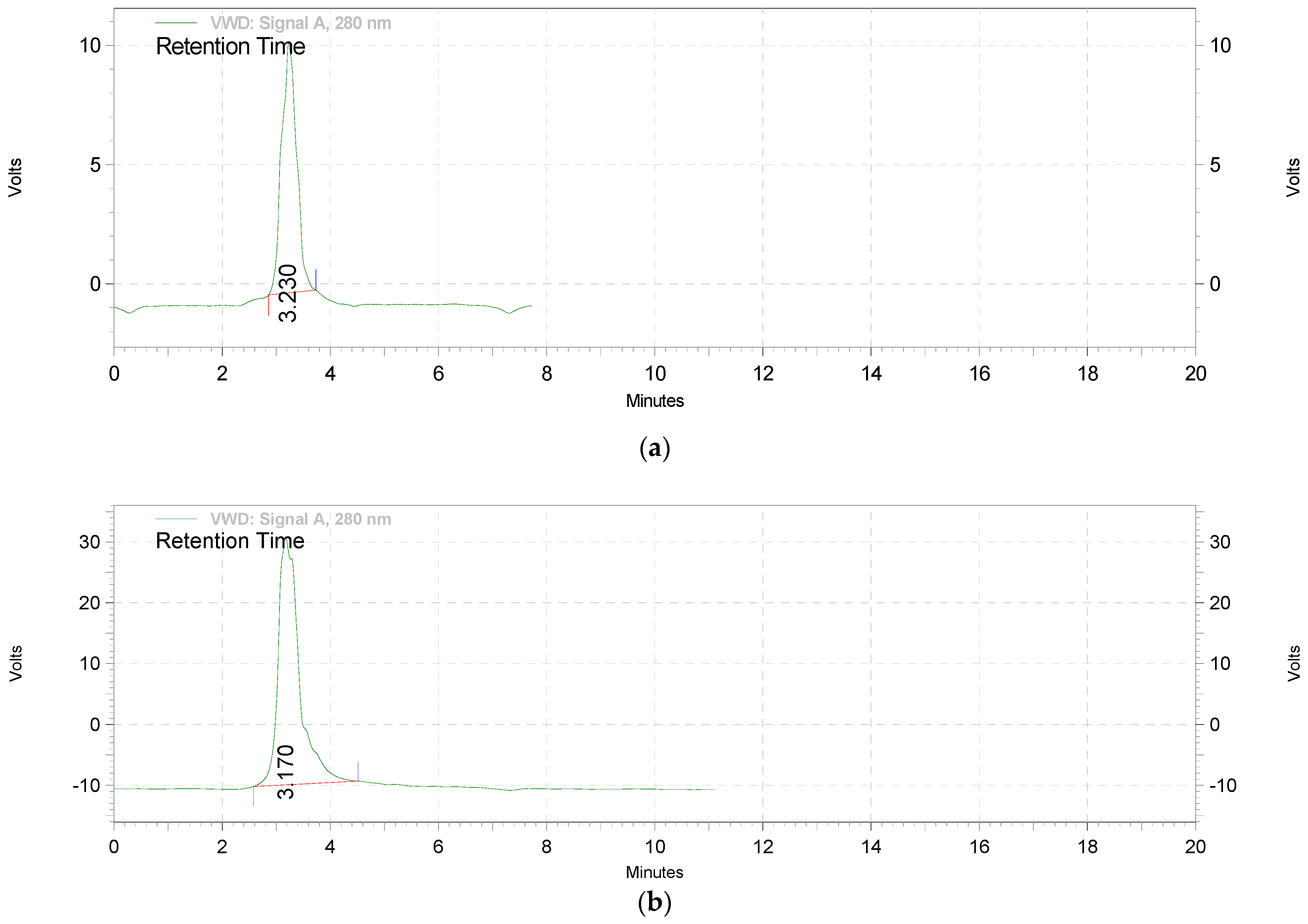
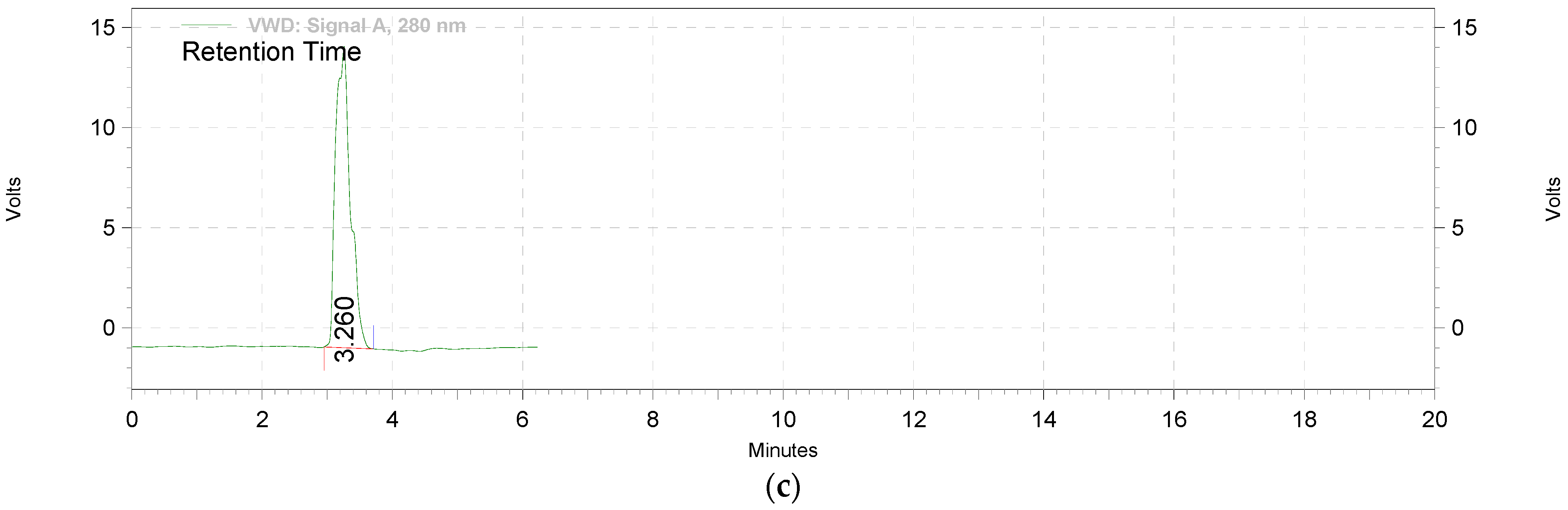
2.2.1. Extraction and Standardization of Green Tea Extract by HPTLC
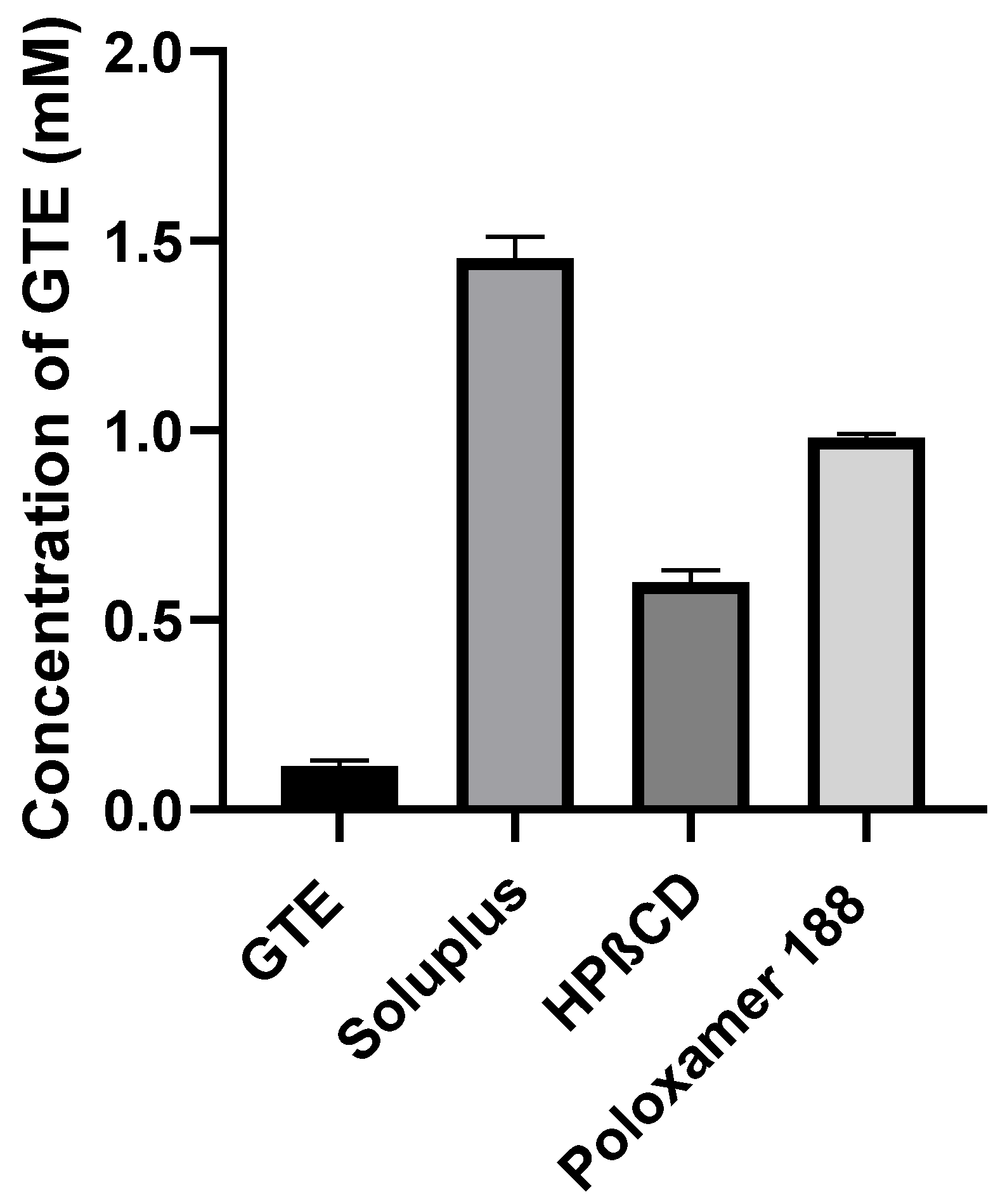
2.2.2. Phase Solubility Analysis
2.2.3. Preparation of GTE Solid Dispersion (GTE-SD)
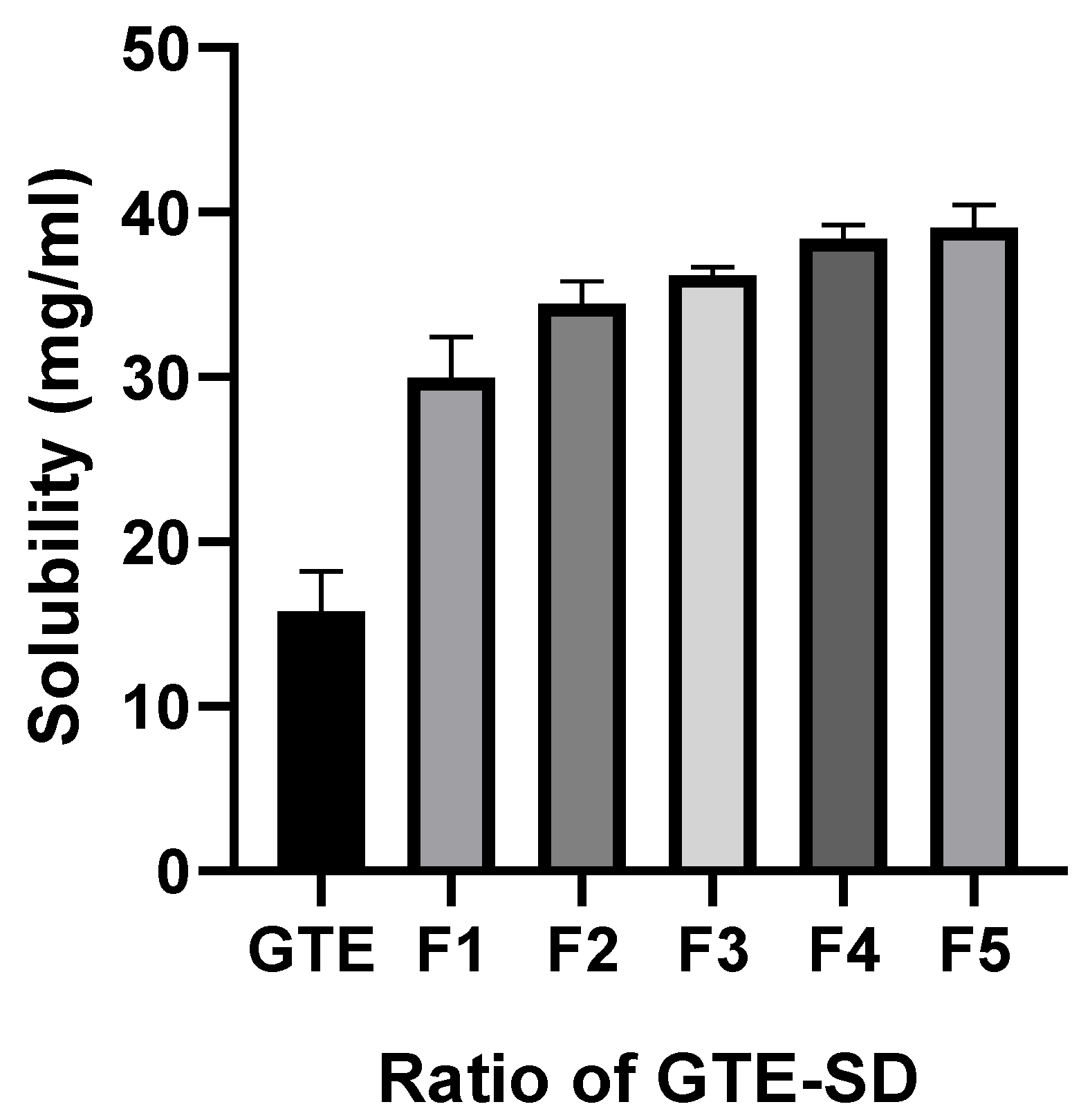
2.2.4. Saturation Solubility of GTE and GTE-
2.2.5. Characterization of GTE-SD
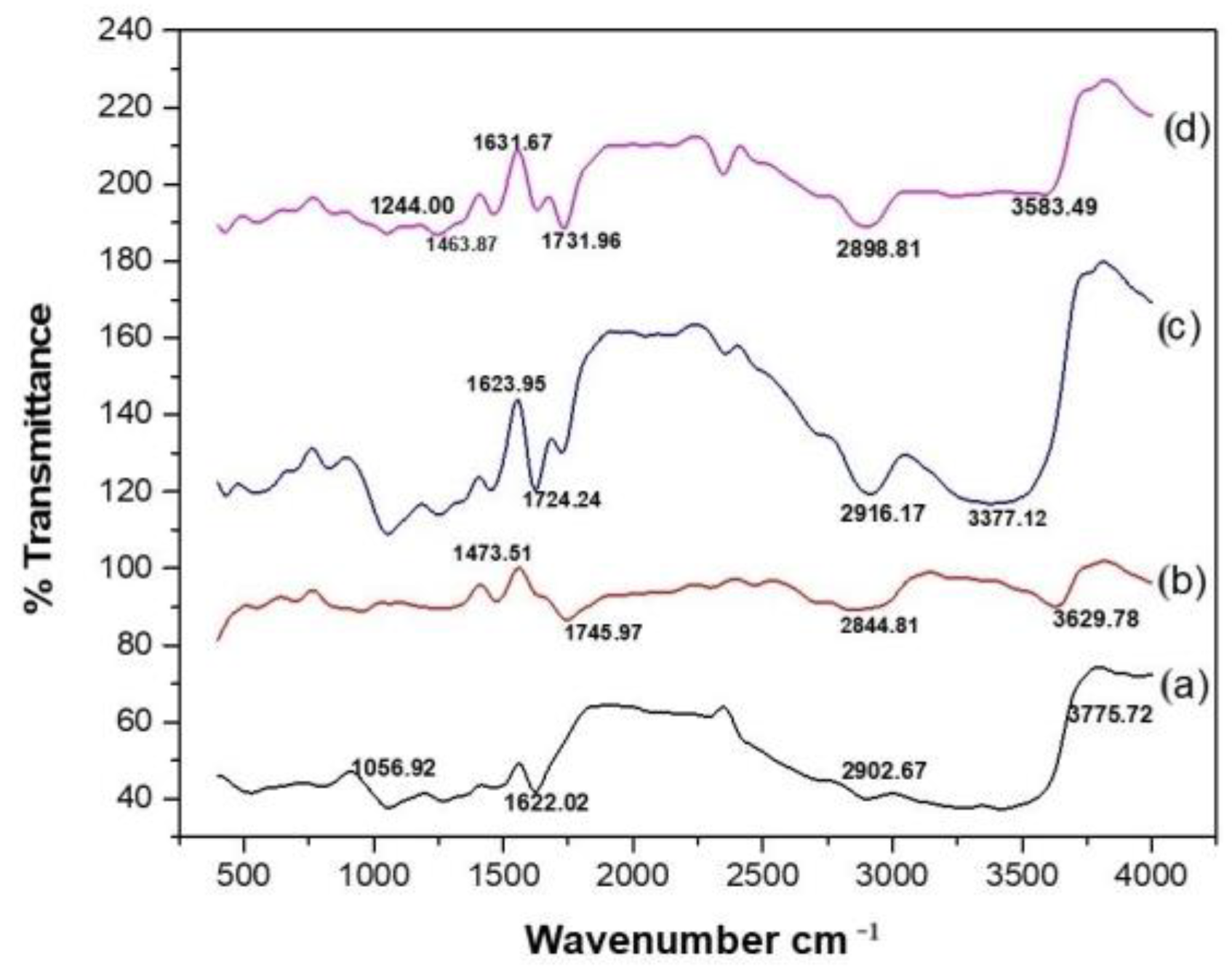
Fourier-Transform Infrared Spectroscopy
Differential Scanning Calorimetry Study
X-ray Diffraction Study
Characterization of GTE-SD for Self-Micellizing Property
Ex Vivo Permeation of GTE-SD
2.2.6. The Formulation of GTE-SD Pellets
2.2.7. Experimental Design
2.2.8. Characterization of GTE-SD Pellets
Scanning Electron Microscopy
Micromeritic Properties
Drug Content
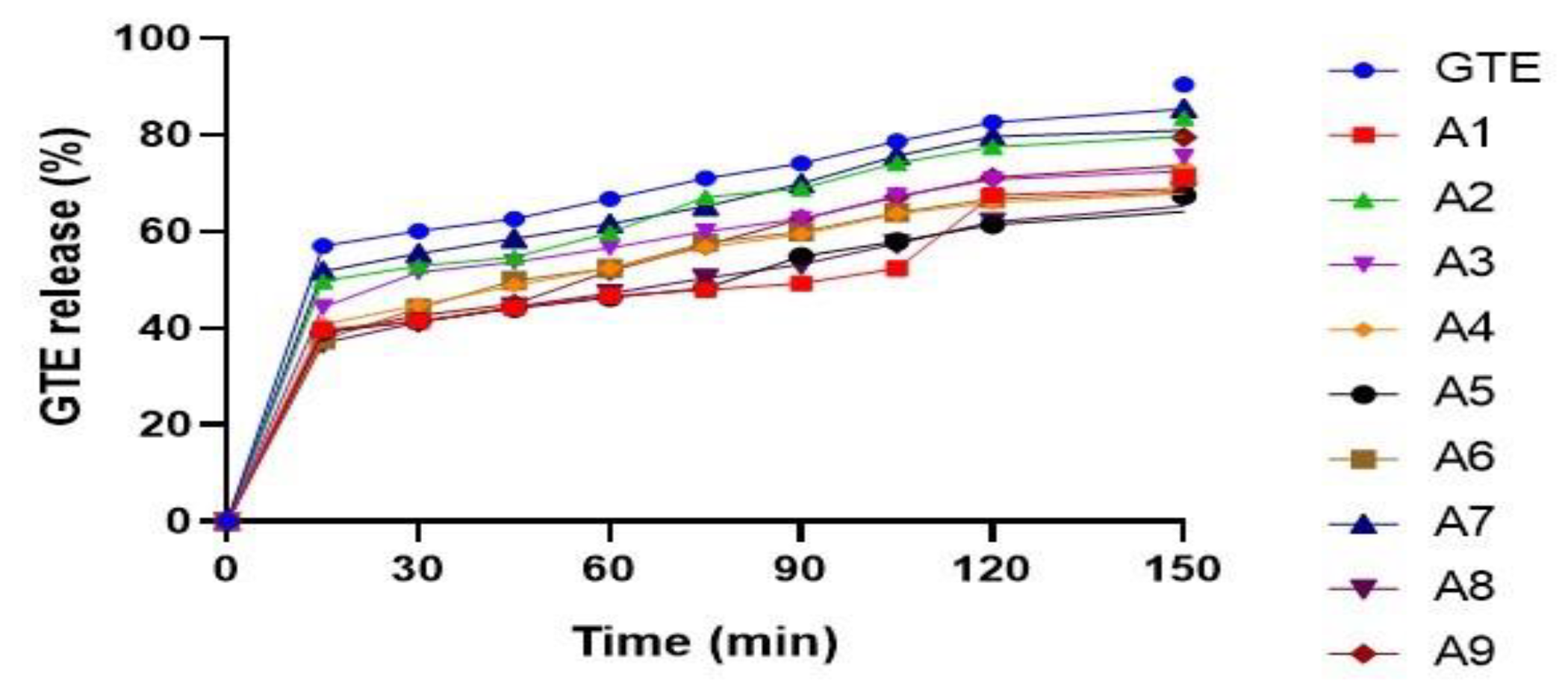
In Vitro Dissolution Study
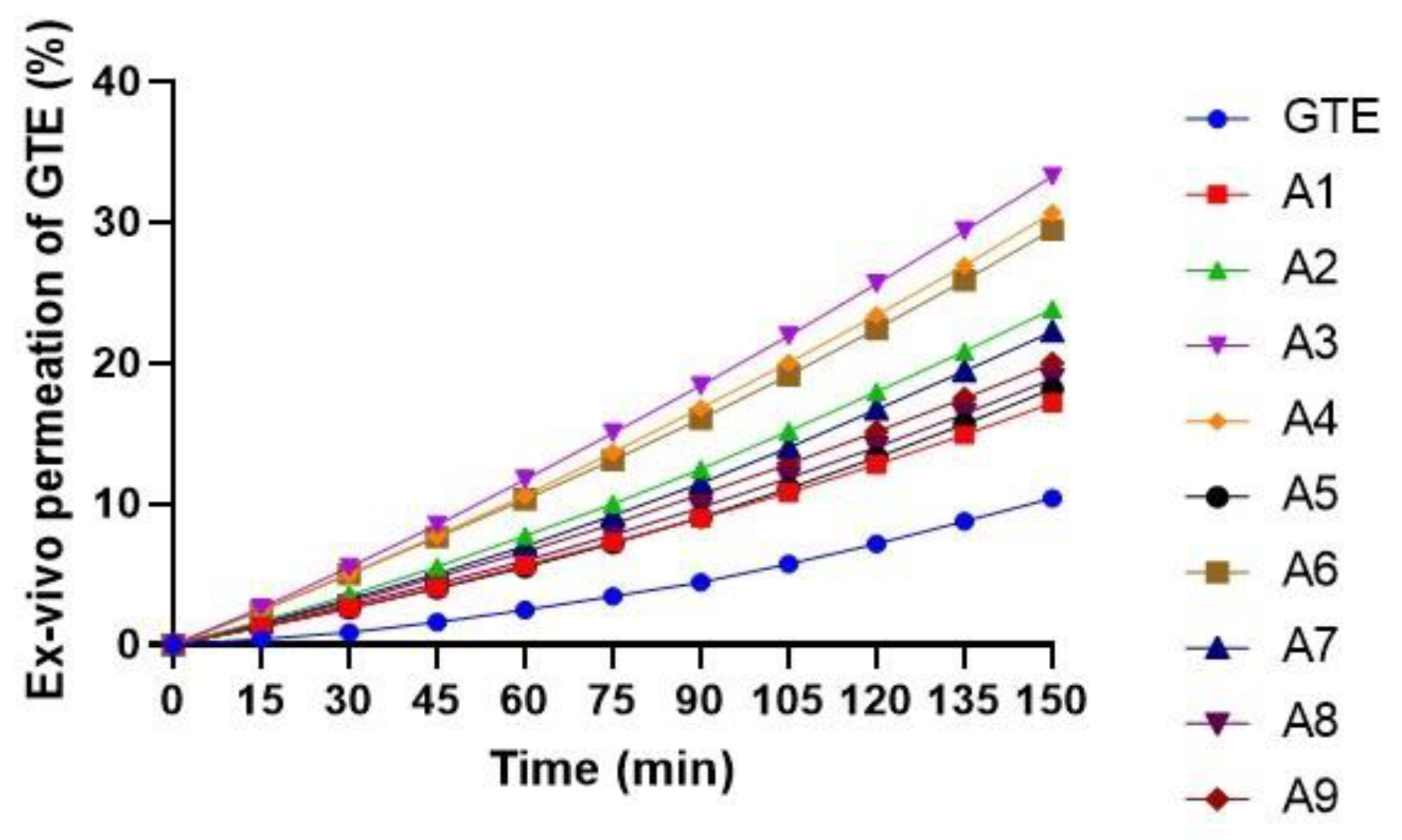
Ex Vivo Permeability Study
In Vivo Study
2.2.9. Statistical Analysis for In Vivo Studies
3. Results and Discussion
3.1. The Extraction and Standardization of Green Tea Extract
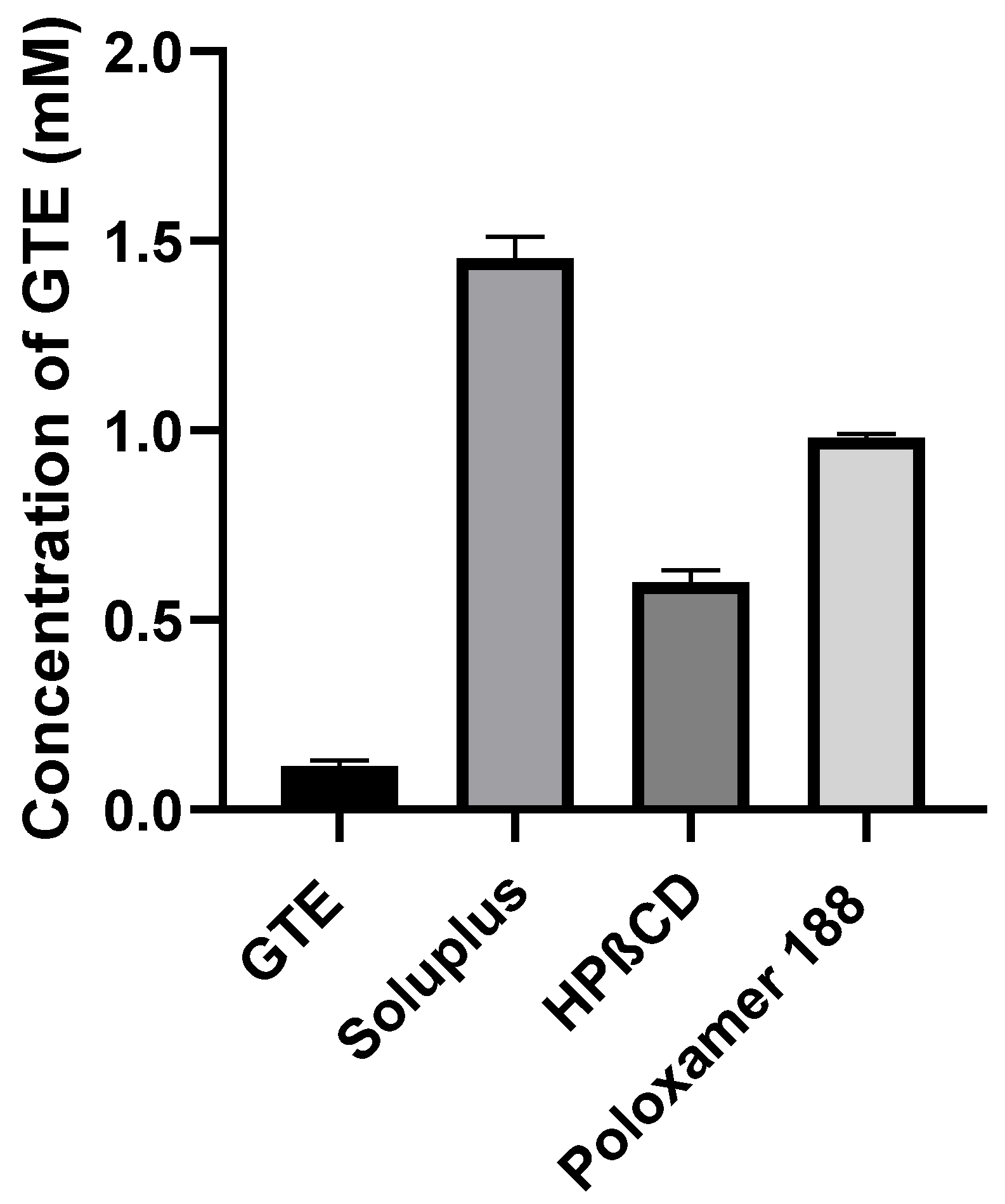
3.2. Phase Solubility Analysis
3.3. Formulation and Evaluation of GTE-SDs
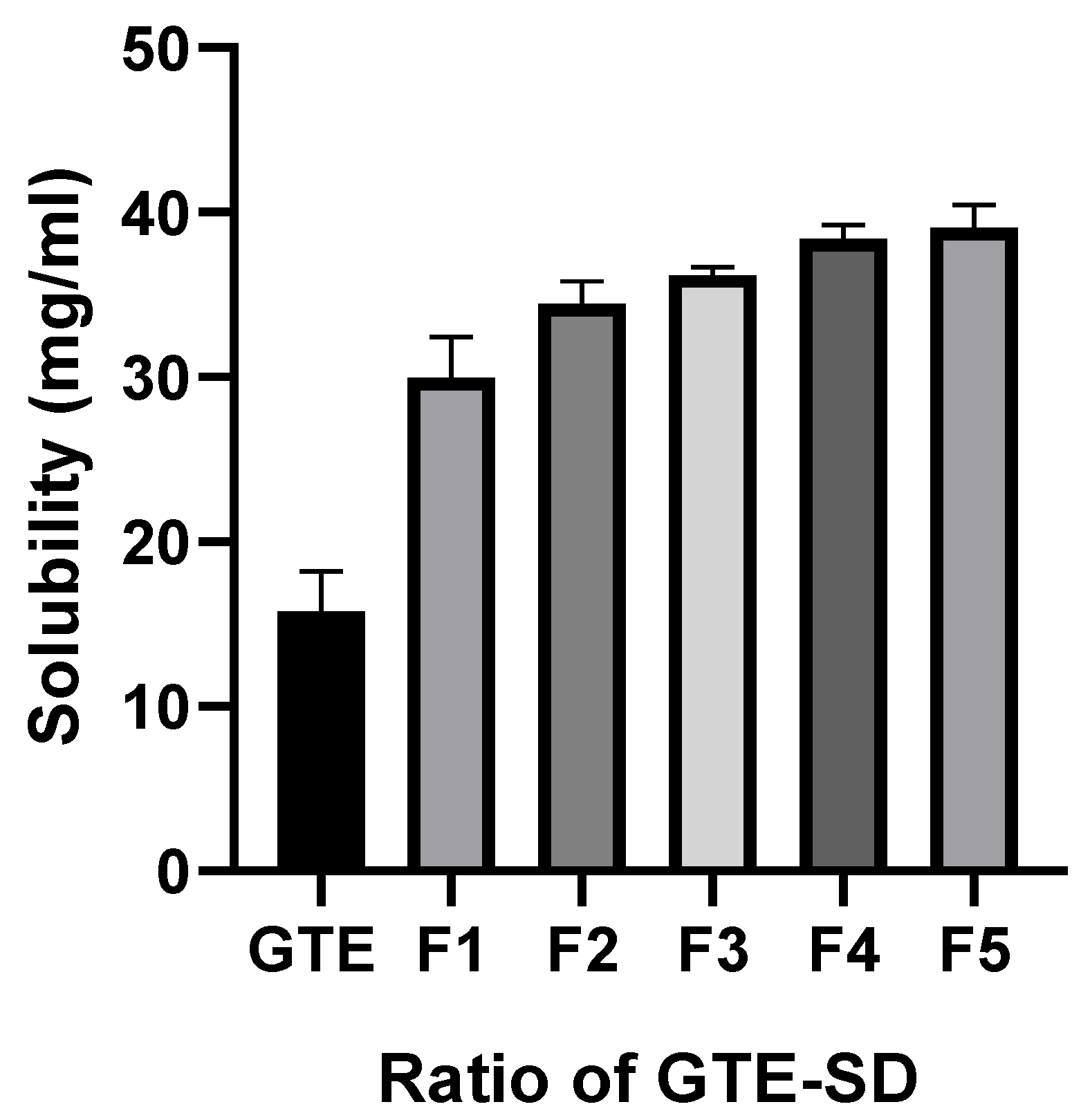
3.3.1. Saturation Solubility
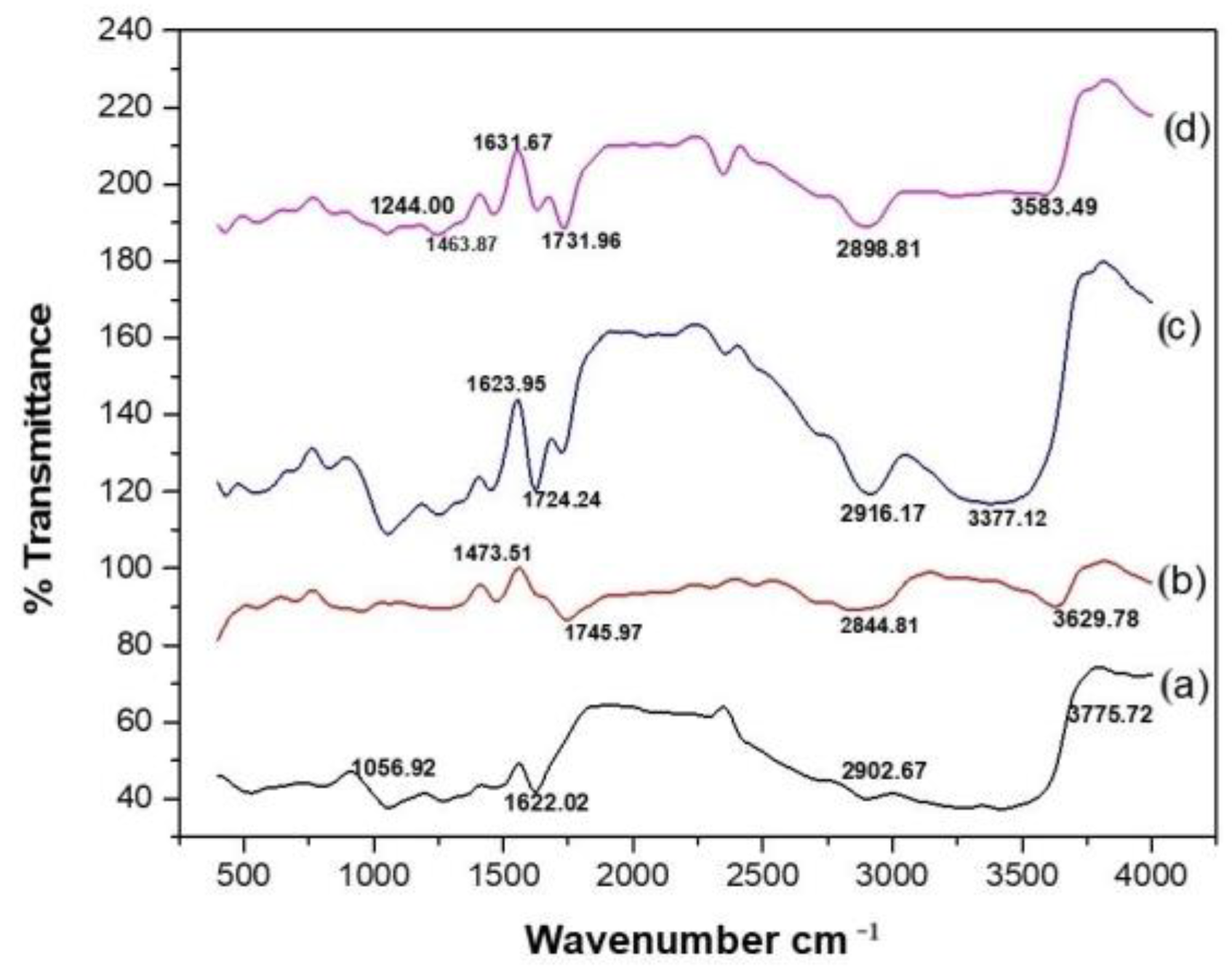
3.3.2. Fourier-Transform Infrared Spectroscopy
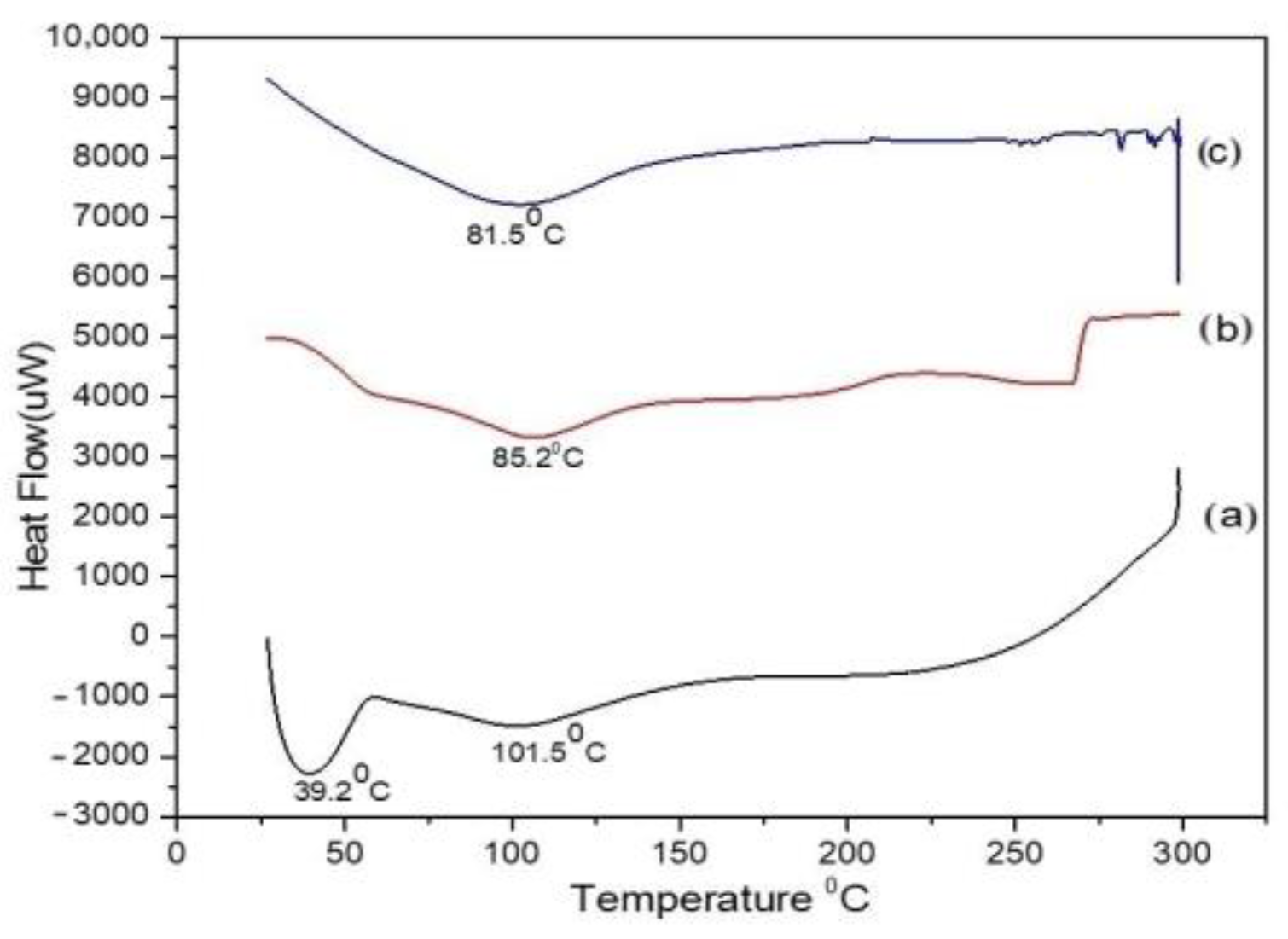
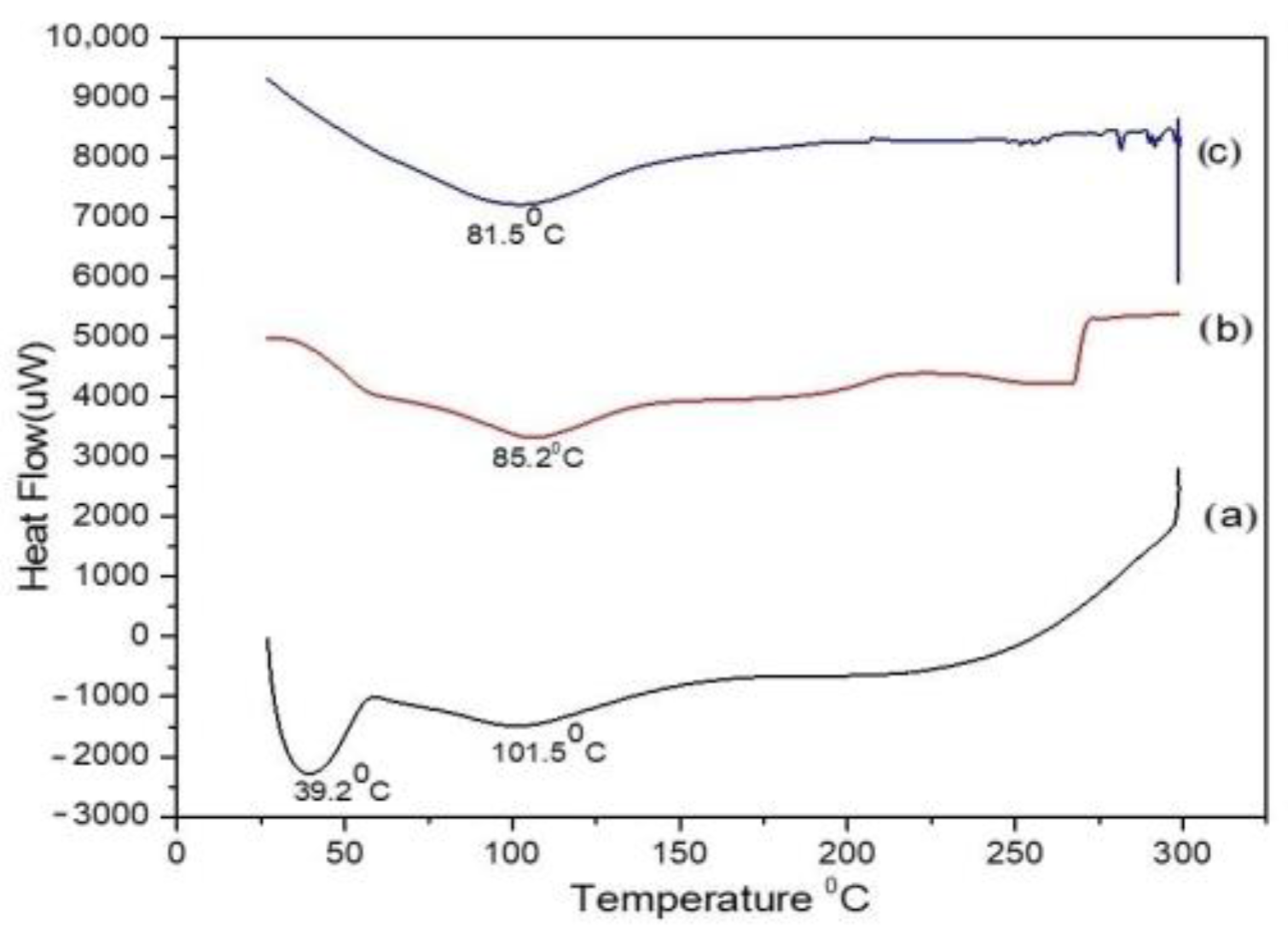
3.3.3. Differential Scanning Calorimetry
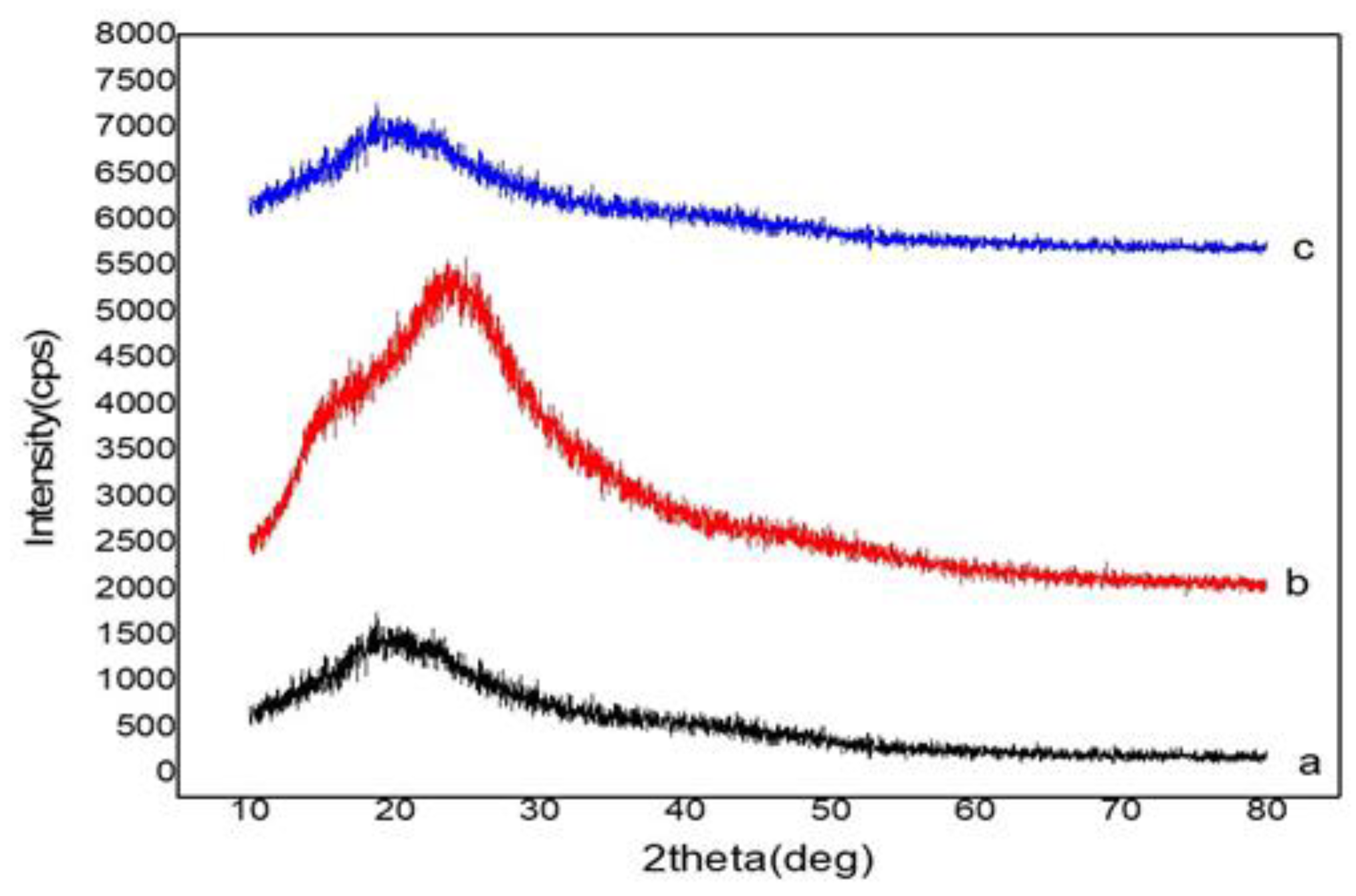
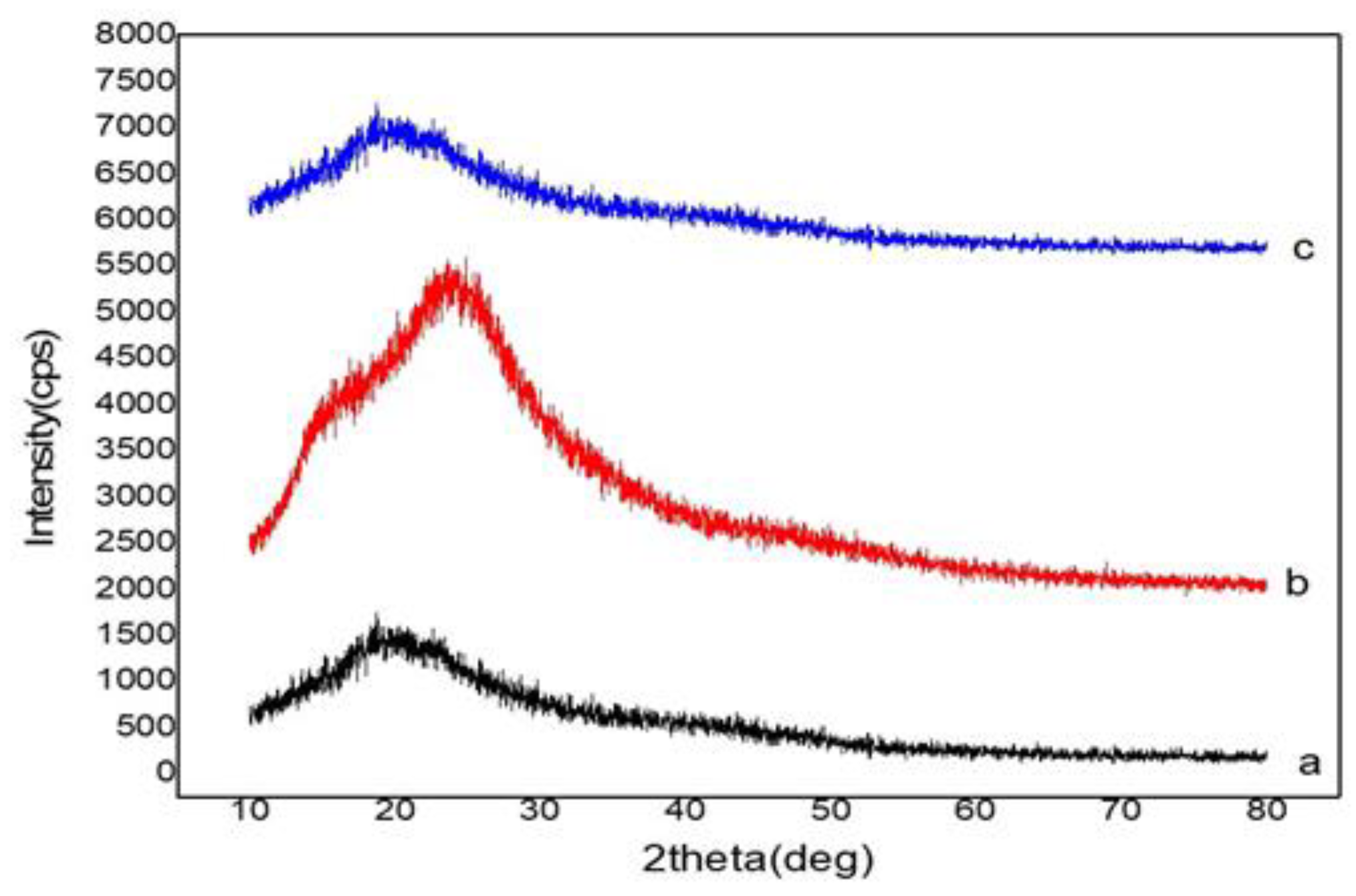
3.3.4. X-ray Diffraction
3.3.5. Characterization of the Self-Micellizing Property and Drug Content of GTE-SD
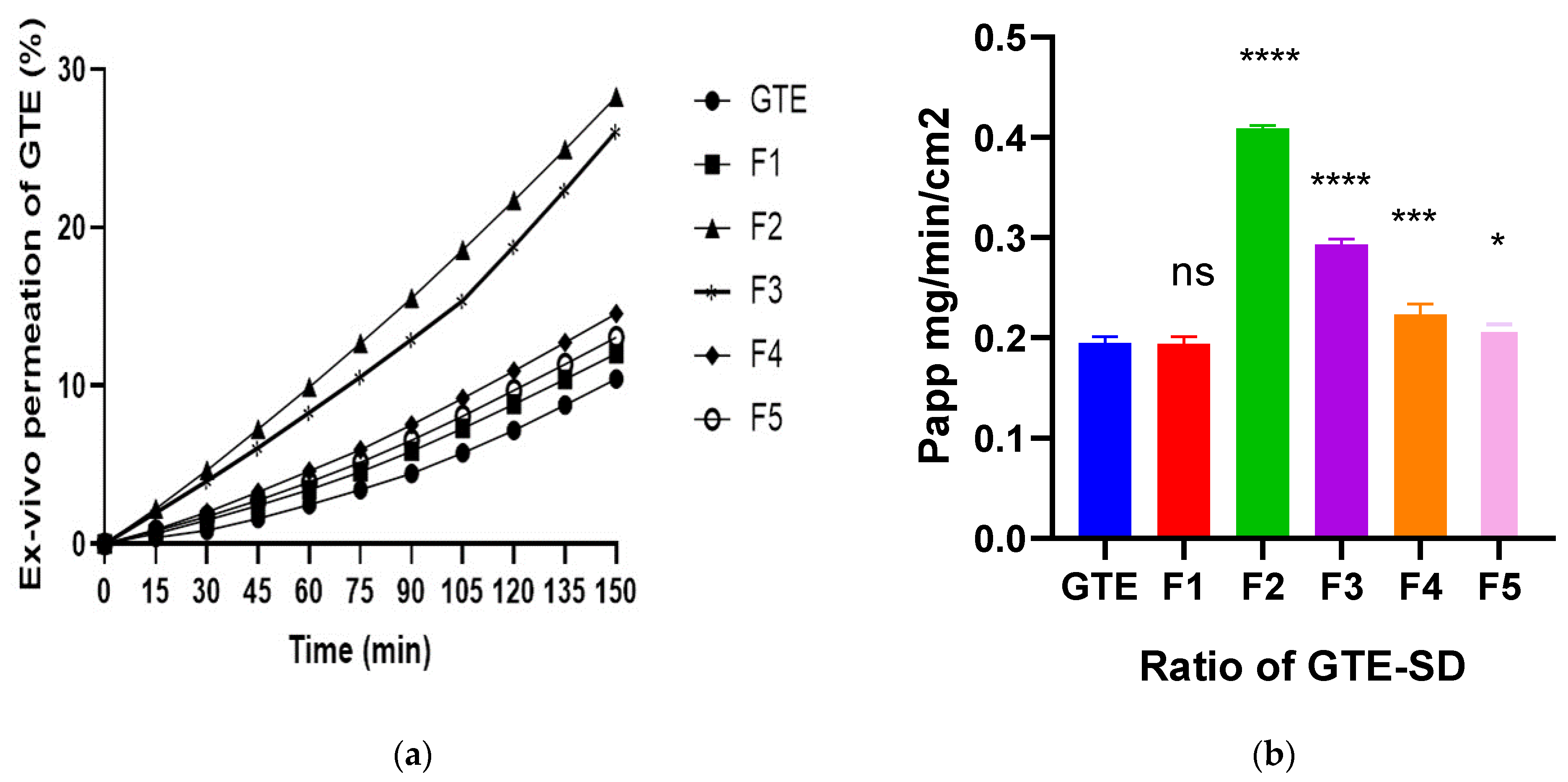
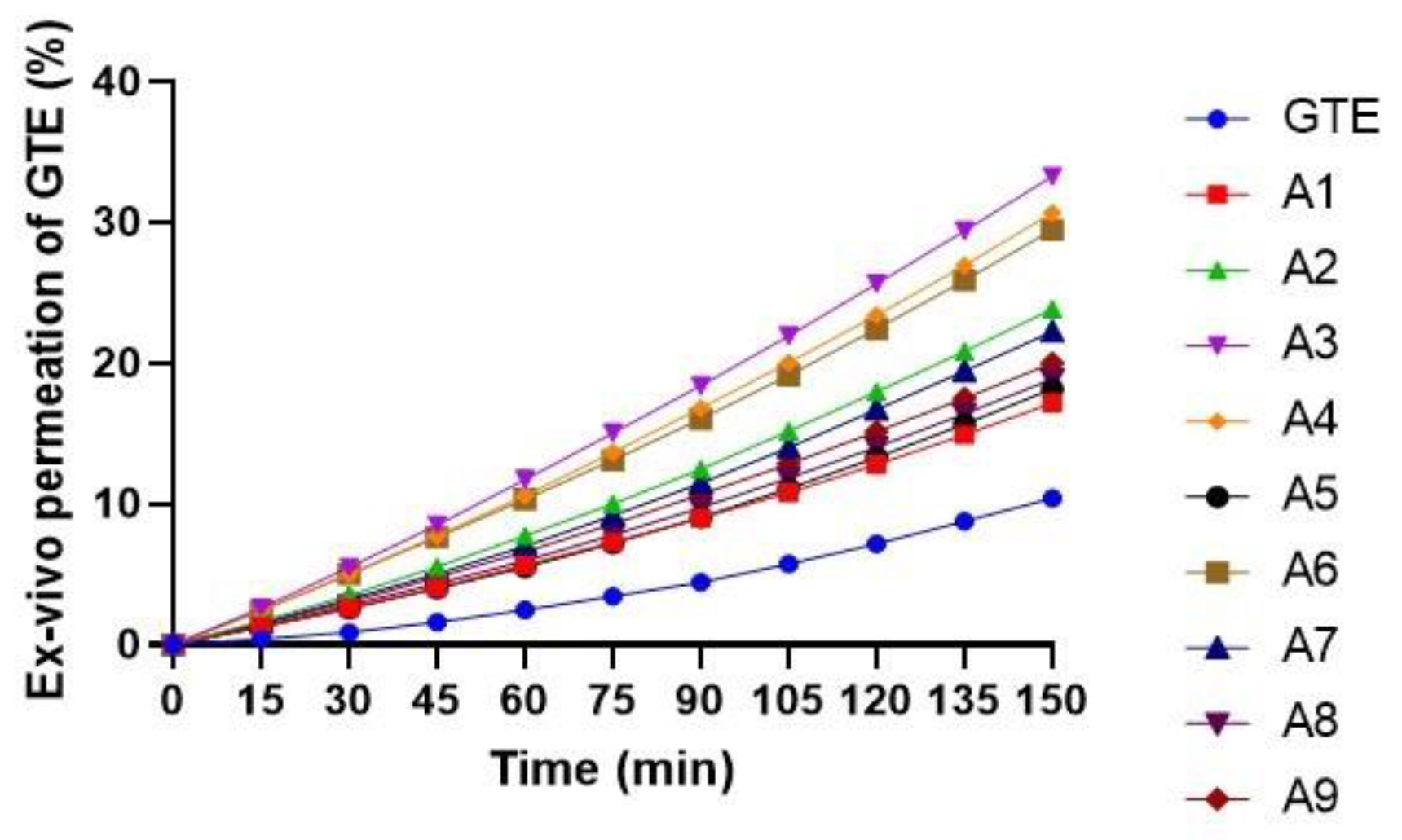
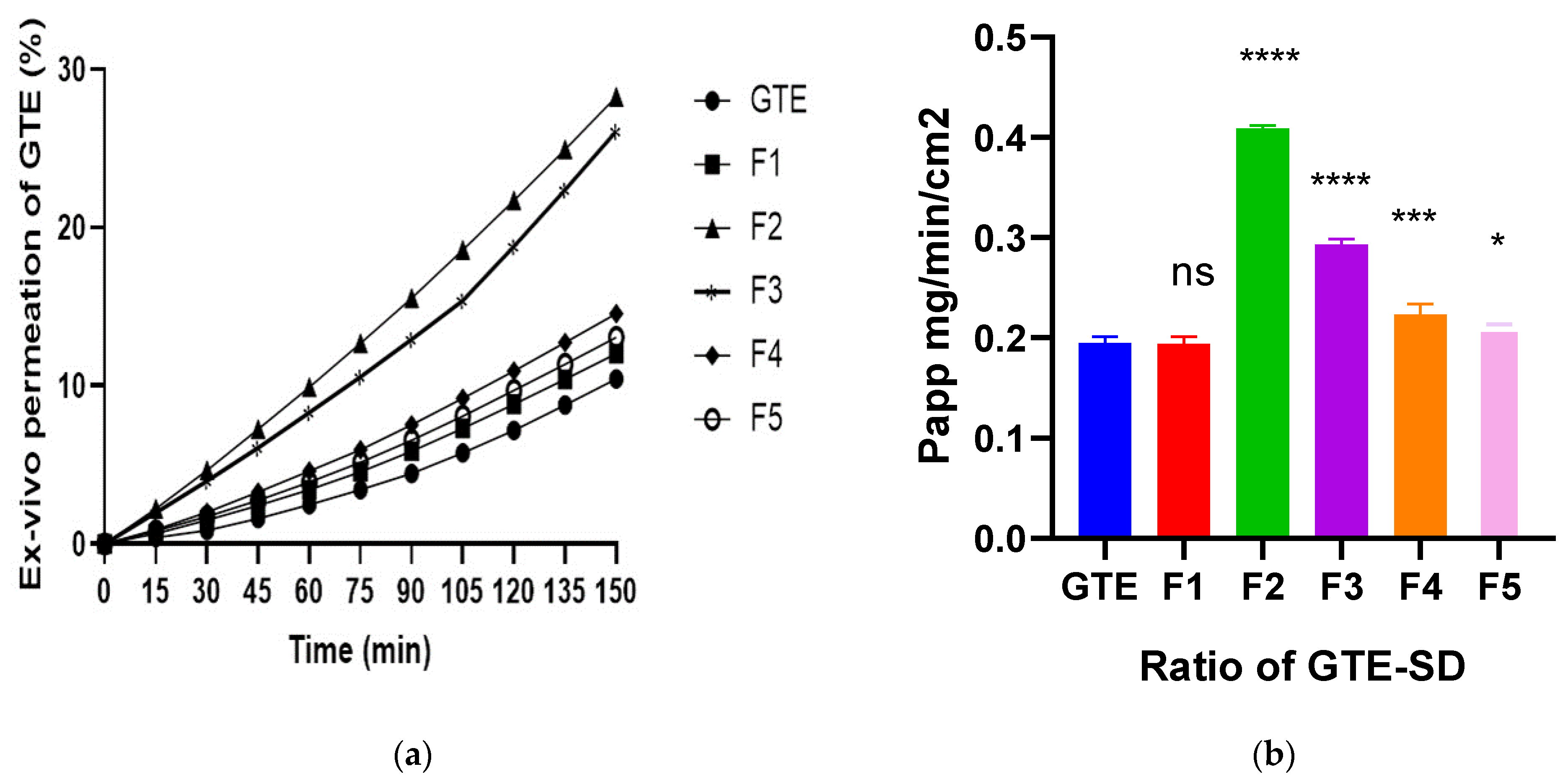
3.3.6. Ex Vivo Permeation of EGCG from GTE-SD
3.4. Characterization of GTE-SD Pellets
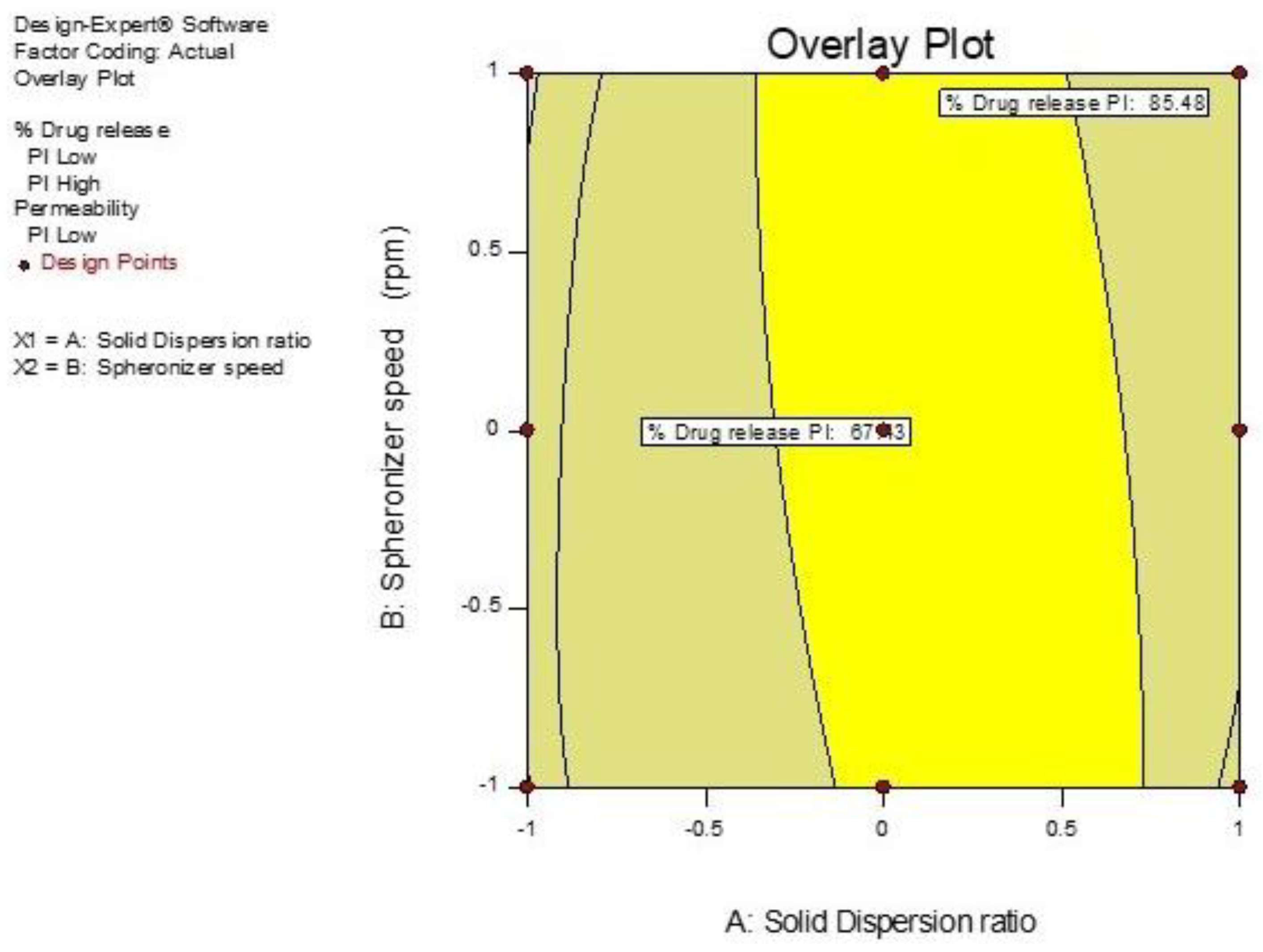
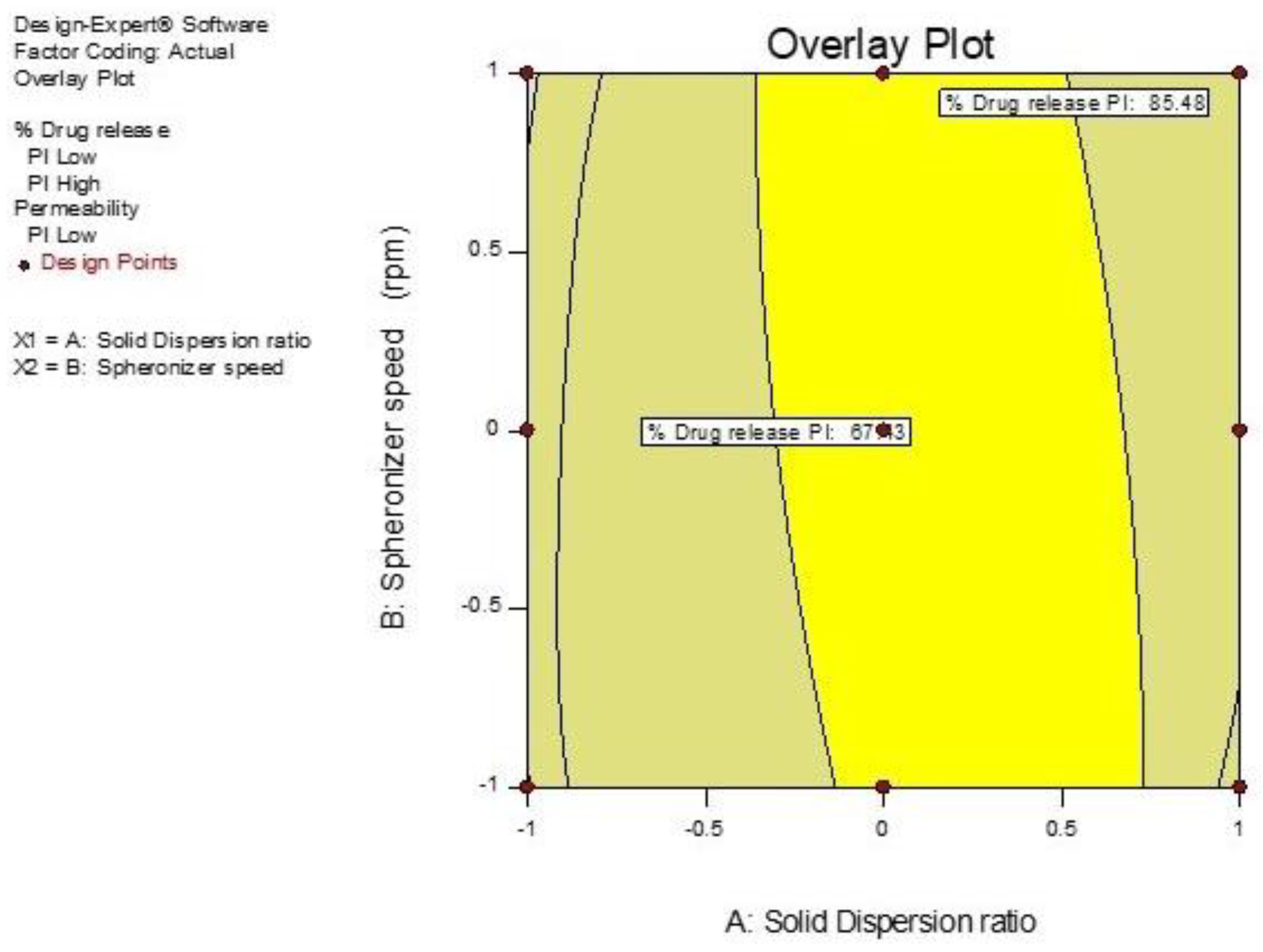
3.5. Optimization of GTE-SD Pellets
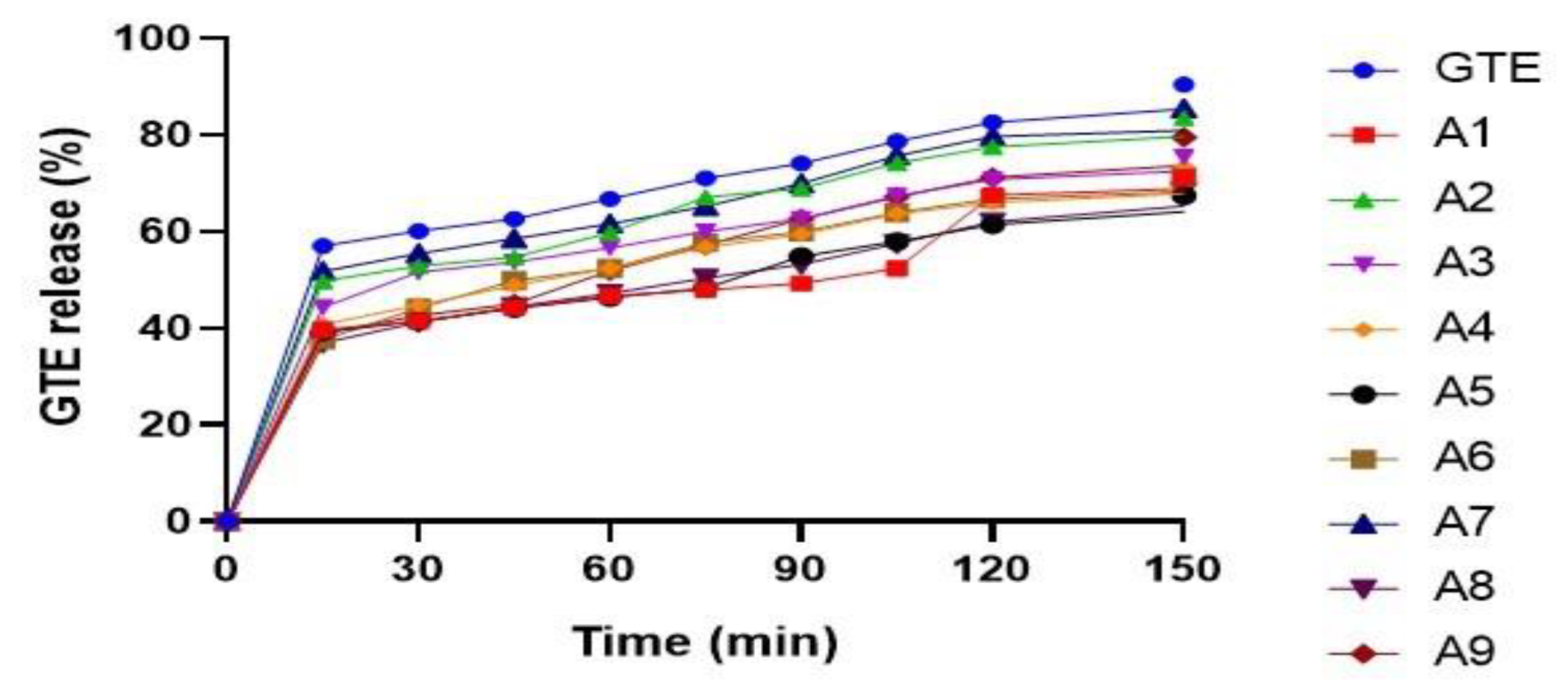
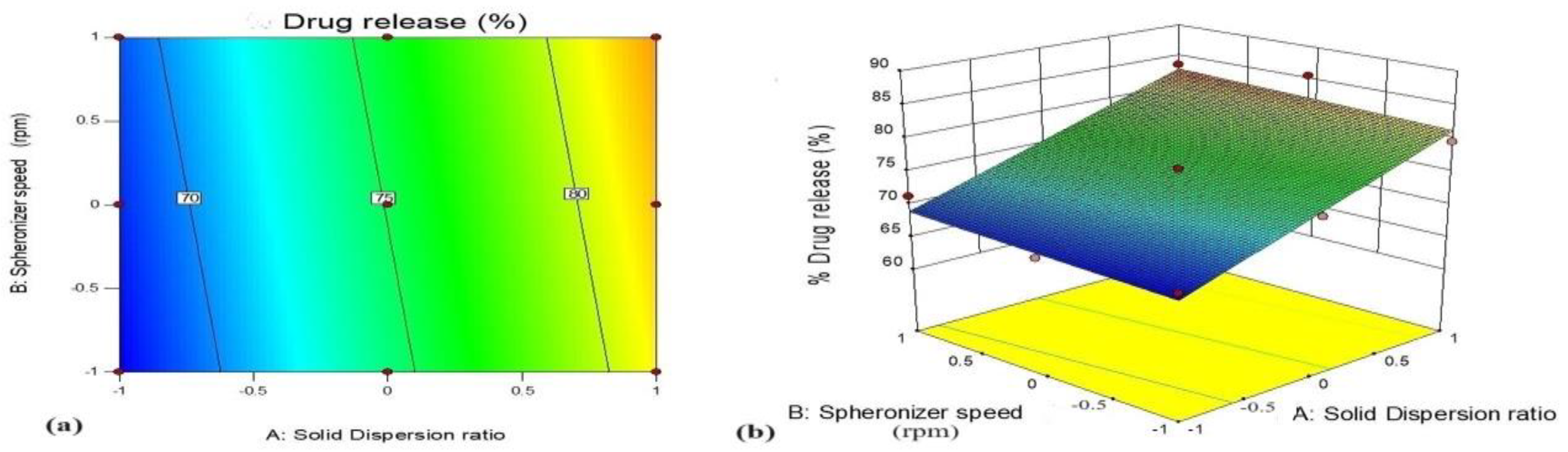
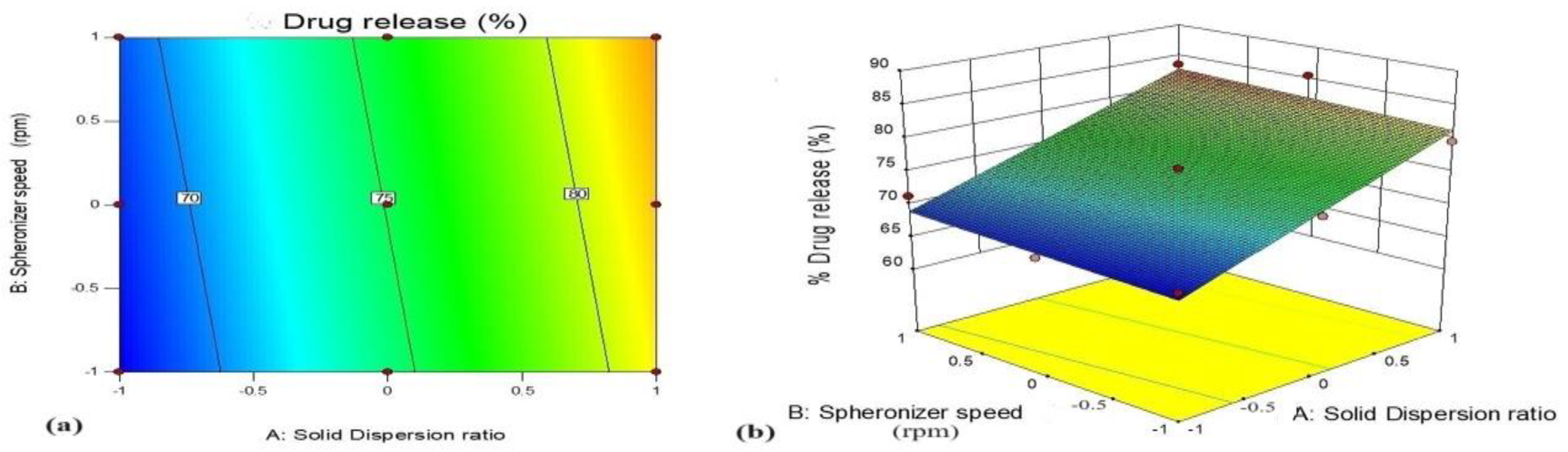
3.5.1. Effect of Variables X1 (GTE-SD Ratio) and X2 (Spheronizer Speed) on % Drug Release
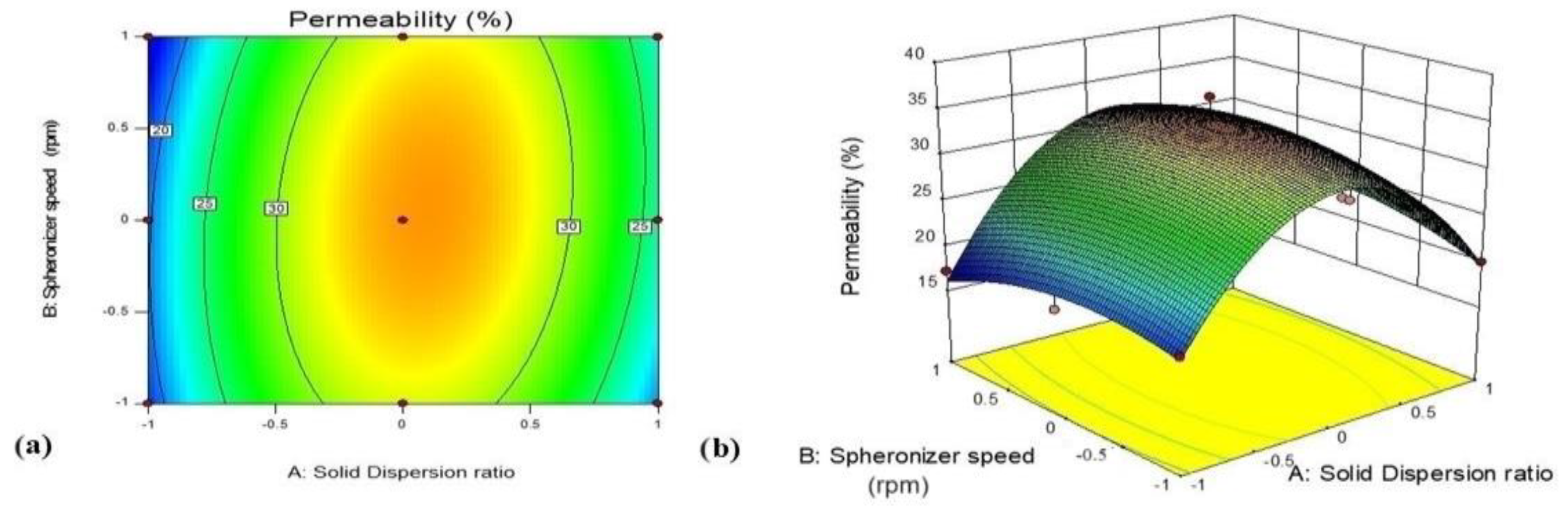
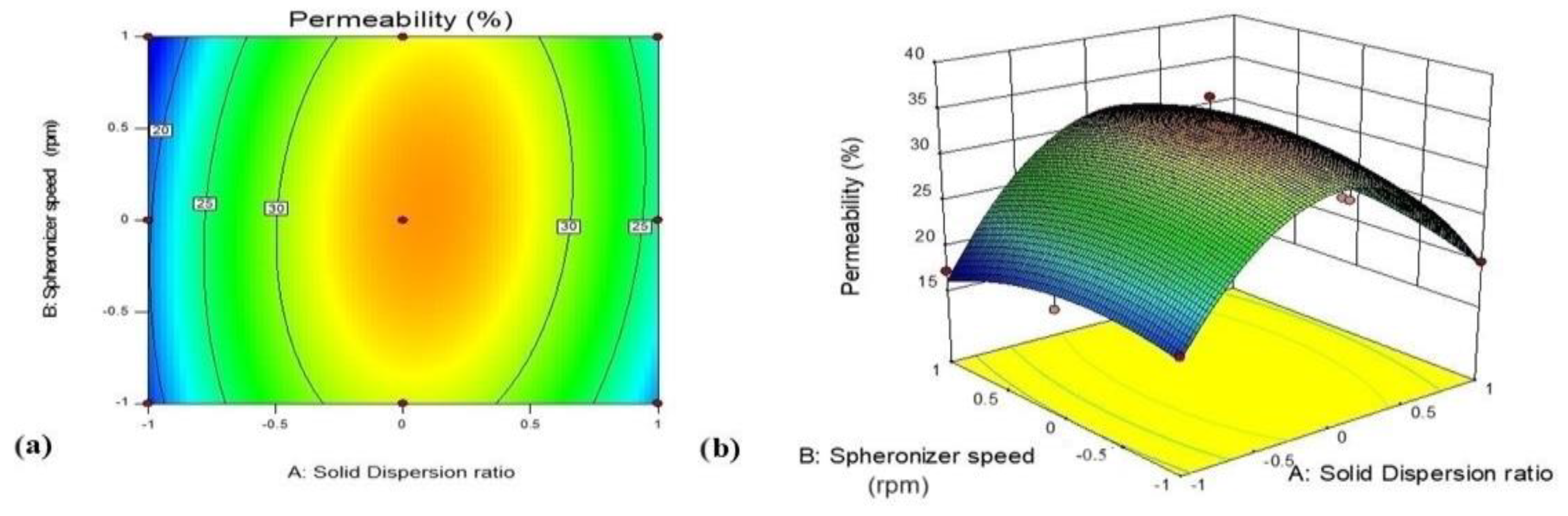
3.5.2. Effect of Variables X1 (GTE-SD Ratio) and X2 (Spheronizer Speed) on Ex Vivo Permeation (%)
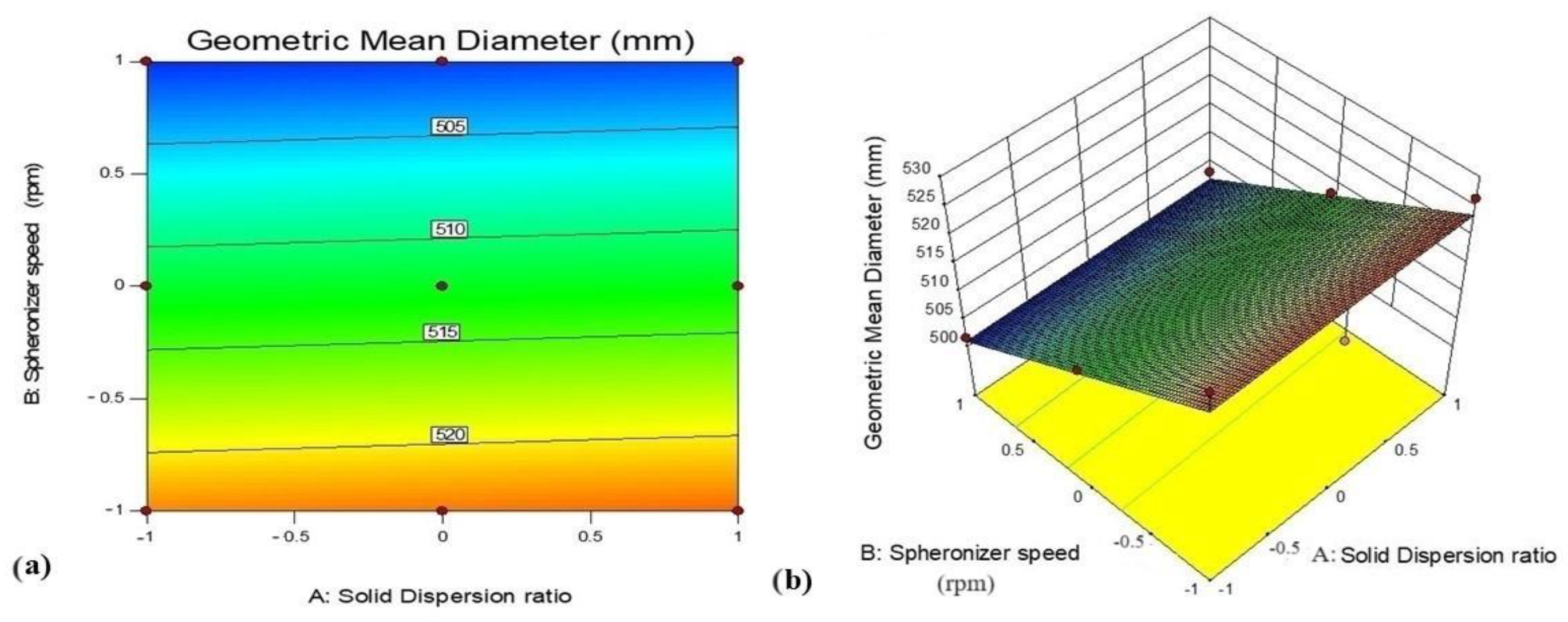
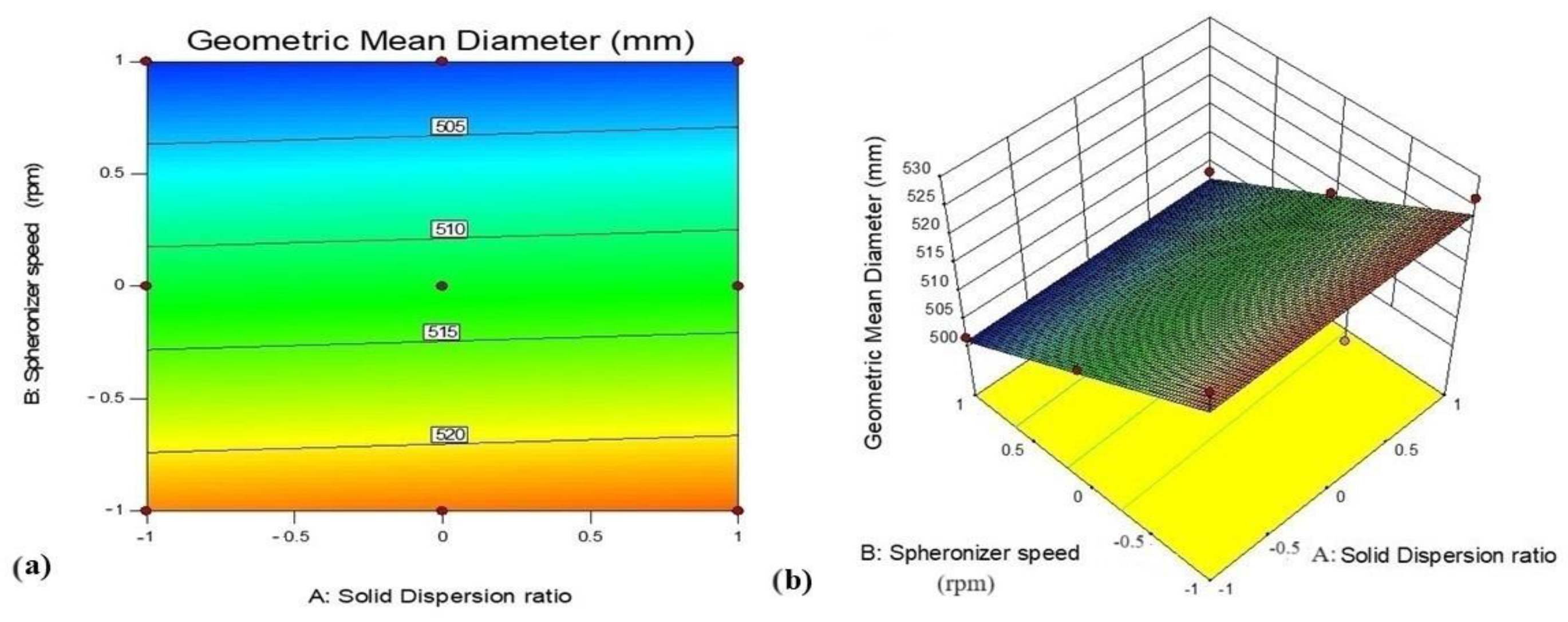
3.5.3. Effect of Variables X1 (GTE-SD Ratio) and X2 (Spheronizer Speed) on Geometric Mean Diameter
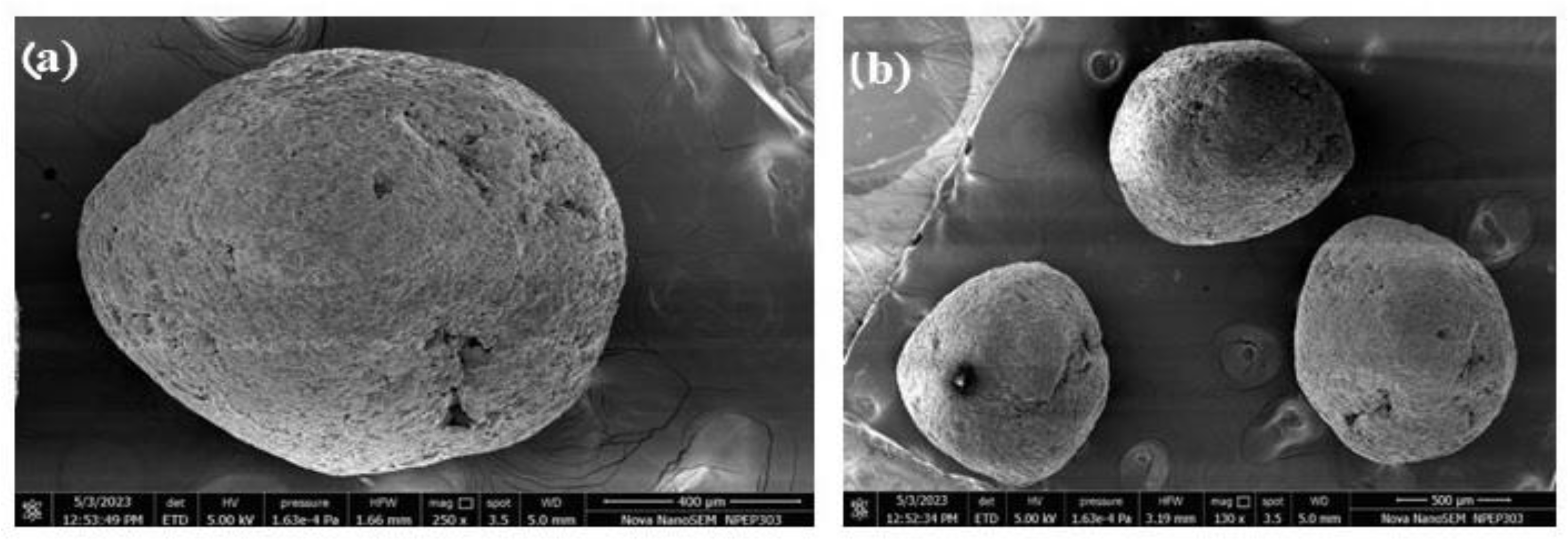
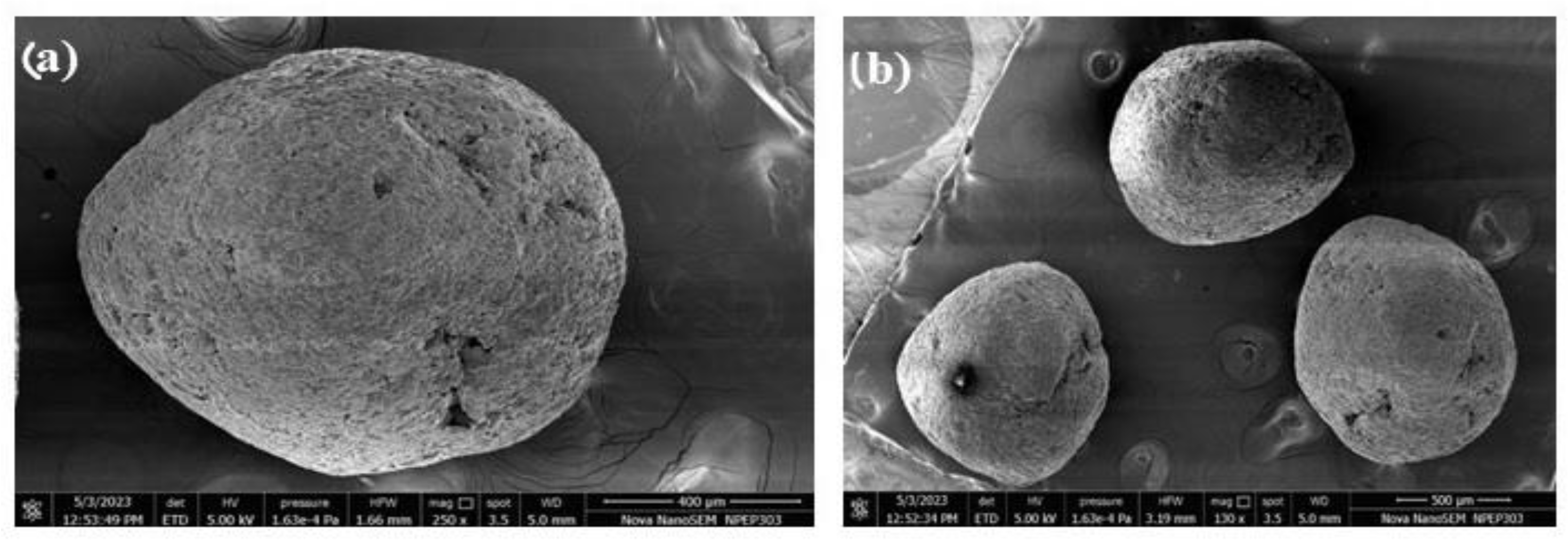
3.5.4. Scanning Electron Microscopy
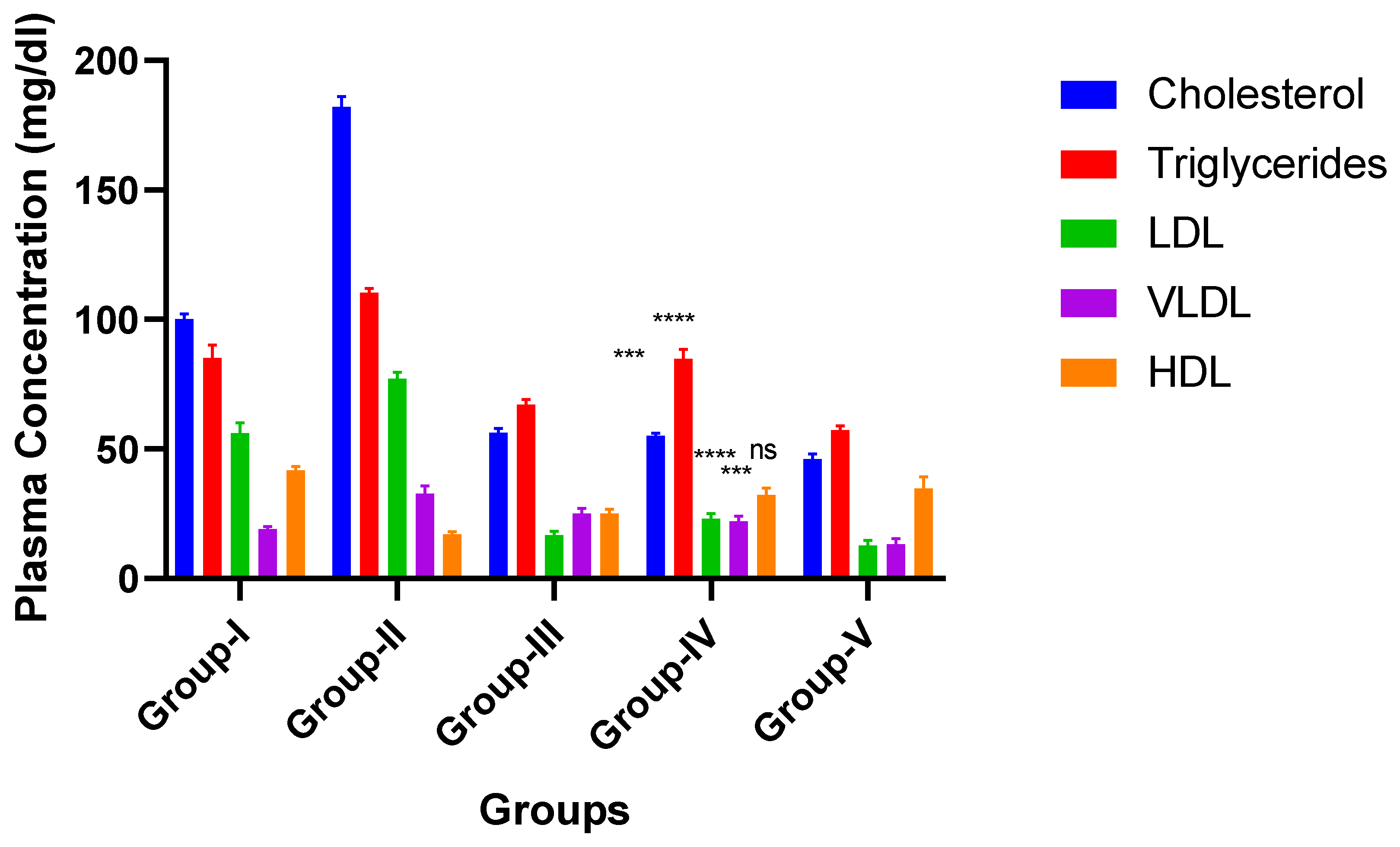
3.5.5. In Vivo Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, Y.; Siddiqui, W.J. Cholesterol Levels. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK542294/ (accessed on 3 March 2023).
- Fredrickson, D.S. An international classification of hyperlipidemias and hyperlipoproteinemias. Ann. Intern. Med. 1971, 75, 471–472. [Google Scholar] [CrossRef]
- Yao, Y.S.; Li, T.D.; Zeng, Z.H. Mechanisms underlying direct actions of hyperlipidemia on myocardium: An updated review. Lipids Health Dis. 2020, 19, 23. [Google Scholar] [CrossRef]
- Nelson, R.H. Hyperlipidemia as a risk factor for cardiovascular disease. Prim. Care 2013, 40, 195–211. [Google Scholar] [CrossRef]
- Hassani, H.; Huang, X.; MacFeely, S.; Entezarian, M.R. Big data and the United Nations sustainable development goals (UN SDGs) at a glance. Big Data Cogn. Comput. 2021, 5, 28. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart disease and stroke statistics update: A report from the American Heart Association. Circulation 2018, 137, 67–492. [Google Scholar] [CrossRef]
- Golomb, B.A.; Evans, M.A. Statin adverse effects: A review of the literature and evidence for a mitochondrial mechanism. Am. J. Cardiovasc. Drugs 2008, 8, 373–418. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, X.; Hu, M.; Zhang, D.; Yuan, S.; Li, P.; Feng, L. Medicinal and edible plants in the treatment of dyslipidemia: Advances and prospects. Chin. Med. 2022, 17, 113. [Google Scholar] [CrossRef]
- Tuhin, R.H.; Begum, M.; Rahman, M.S.; Karim, R.; Begum, T.; Ahmed, S.U.; Mostofa, R.; Hossain, A.; Abdel-Daim, M.; Begum, R. Wound healing effect of Euphorbia hirtalinn. (Euphorbiaceae) in alloxan induced diabetic rats. BMC Complement. Altern. Med. 2017, 17, 423. [Google Scholar] [CrossRef]
- Rudrapal, M.; Khairnar, S.J.; Khan, J.; Dukhyil, A.B.; Ansari, M.A.; Alomary, M.N.; Alshabrmi, F.M.; Palai, S.; Deb, P.K.; Devi, R. Dietary polyphenols and their role in oxidative stress-induced human diseases: Insights into protective effects, antioxidant potentials and mechanism(s) of action. Front. Pharmacol. 2022, 13, 283. [Google Scholar] [CrossRef]
- Gupta, A.U.; Jagadis, K. Reactive Oxygen and Nitrogen Species Signaling and Communication in Plants; Part of Signaling and Communication in Plants; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Fujiki, H.; Suganuma, M.; Imai, K.; Nakachi, K. Green tea: Cancer preventive beverage and/or drug. Cancer Lett. 2002, 188, 9–13. [Google Scholar] [CrossRef]
- Zhao, T.; Li, C.; Wang, S.; Song, X. Green tea (Camellia sinensis): A review of its phytochemistry, pharmacology, and toxicology. Molecules 2022, 27, 3909. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Yang, K.; Li, S.; Dai, M.; Chen, G. Effect of green tea consumption on blood lipids: A systematic review and meta-analysis of randomized controlled trials. Nutr. J. 2020, 19, 48. [Google Scholar] [CrossRef] [PubMed]
- Suzuki-Sugihara, N.; Kishimoto, Y.; Saita, E.; Taguchi, C.; Kobayashi, M.; Ichitani, M.; Ukawa, Y.; Sagesaka, Y.M.; Suzuki, E.; Kondo, K. Green tea catechins prevent low-density lipoprotein oxidation via their accumulation in low-density lipoprotein particles in humans. Nutr. Res. 2016, 36, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.I.; Noh, S.K. Green tea as inhibitor of the intestinal absorption of lipids: Potential mechanism for its lipid-lowering effect. J. NutrBiochem. 2007, 18, 179–183. [Google Scholar] [CrossRef]
- Cai, Z.Y.; Li, X.M.; Liang, J.P.; Xiang, L.P.; Wang, K.R.; Shi, Y.L.; Yang, R.; Shi, M.; Ye, J.H.; Lu, J.L.; et al. Bioavailability of tea catechins and its improvement. Molecules 2018, 23, 2346. [Google Scholar] [CrossRef]
- Krook, M.A.; Hagerman, A.E. Stability of polyphenols epigallocatechingallate and pentagalloyl glucose in a simulated digestive system. Food Res. Int. 2012, 49, 112–116. [Google Scholar] [CrossRef]
- Yin, Z.; Zheng, T.; Ho, C.T.; Huang, Q.; Wu, Q.; Zhang, M. Improving the stability and bioavailability of tea polyphenols by encapsulations: A review. Food Sci. Hum. Wellness 2022, 11, 537–556. [Google Scholar] [CrossRef]
- Cano, A.; Ettcheto, M.; Chang, J.H.; Barroso, E.; Espina, M.; Kühne, B.A.; Barenys, M.; Auladell, C.; Folch, J.; Souto, E.B.; et al. Dual-drug loaded nanoparticles of epigallocatechin-3-gallate (EGCG)/ascorbic acid enhance therapeutic efficacy of EGCG in a APPswe/PS1dE9 alzheimer’s disease mice model. J. Control. Release 2019, 301, 62–75. [Google Scholar] [CrossRef]
- Furniturewalla, A.; Barve, K. Approaches to overcome bioavailability inconsistencies of epigallocatechingallate, a powerful anti-oxidant in green tea. Food Chem. Adv. 2022, 1, 100037. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Duong, V.A.; Maeng, H.J. Pharmaceutical formulations with P-glycoprotein inhibitory effect as promising approaches for enhancing oral drug absorption and bioavailability. Pharmaceutics 2021, 13, 1103. [Google Scholar] [CrossRef]
- Linn, M.; Collnot, E.M.; Djuric, D.; Hempel, K.; Fabian, E.; Kolter, K.; Lehr, C.M. Soluplus® as an effective absorption enhancer of poorly soluble drugs in vitro and in vivo. Eur. J. Pharm. Sci. 2012, 45, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhou, D.; Chen, Y. Preparation and antioxidant activity of green tea extract enriched in epigallocatechin (EGC) and epigallocatechingallate (EGCG). J. Agric. Food Chem. 2009, 57, 1349–1353. [Google Scholar] [CrossRef]
- Pandit, A.P.; Joshi, S.R.; Dalal, P.S.; Patole, V.C. Curcumin as a permeability enhancer enhanced the antihyperlipidemic activity of dietary green tea extract. BMC Complement. Altern. Med. 2019, 19, 129. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, C.; Del PilarBuera, M.; Mazzobre, M.F. Phase solubility studies of terpineol with β-cyclodextrins and stability of the freeze-dried inclusion complex. Procedia Food Sci. 2011, 1, 355–362. [Google Scholar] [CrossRef]
- Dhirendra, K.; Lewis, S.; Udupa, N.; Atin, K. Solid dispersions: A review. Pak. J. Pharm. Sci. 2009, 22, 234–246. [Google Scholar] [PubMed]
- Fule, R.; Amin, P. Development and evaluation of lafutidine solid dispersion via hot melt extrusion: Investigating drug-polymer miscibility with advanced characterisation. Asian J. Pharm. Sci. 2014, 9, 92–106. [Google Scholar] [CrossRef]
- Manea, A.M.; Vasile, B.S.; Meghea, A. Antioxidant and antimicrobial activities of green tea extract loaded into nanostructured lipid carriers. C. R. Chim. 2014, 17, 331–341. [Google Scholar] [CrossRef]
- Senthilkumar, S.R.; Sivakumar, T. Green tea (Camellia sinensis) mediated synthesis of zinc oxide (ZnO) nanoparticles and studies on their antimicrobial activities. Int. J. Pharm. Pharm. Sci. 2014, 6, 461–465. [Google Scholar]
- Fousteris, E.; Tarantili, P.A.; Karavas, E.; Bikiaris, D. Poly (vinyl pyrrolidone)–poloxamer-188 solid dispersions prepared by hot melt extrusion: Thermal properties and release behavior. J. Therm. Anal. Calorim. 2013, 113, 1037–1047. [Google Scholar] [CrossRef]
- Dhome, A.G.; Deshkar, S.S.; Shirolkar, S.V. Gliclazide solid lipid nanoparticles: Formulation, optimization and in vitro characterization. Pharm. Reason. 2018, 1, 8–16. [Google Scholar]
- Najmuddin, M.; Ahmed, A.; Shelar, S.; Patel, V.; Khan, T. Floating microspheres of ketoprofen: Formulation and evaluation. Int. J. Pharm. Pharm. Sci. 2010, 2, 83–87. [Google Scholar]
- Shelar, S.; Shirolkar, S.; Kale, N. Formulation optimization of promethazine theoclate immediate release pellets by using extrusion-spheronization technique. Int. J. Appl. Pharm. 2018, 10, 30–35. [Google Scholar]
- Singh, G.; Pai, R.S.; Devi, V.K. Optimization of pellets containing solid dispersion prepared by extrusion/spheronization using central composite design and desirability function. J. Young Pharm. 2012, 4, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Pharmacopeia US. United States Pharmacopeia and National Formulary (USP 37–NF 32); Rockville, M.D., Ed.; US Pharmacopeia: North Bethesda, MD, USA, 2014. [Google Scholar]
- Ravella, V.N.; Nadendla, R.R.; Kesari, N.C. Design and evaluation of sustained release pellets of aceclofenac. J. Pharm. Res. 2013, 6, 525–531. [Google Scholar] [CrossRef]
- Sareen, S.; Mathew, G.; Joseph, L. Improvement in solubility of poor water-soluble drugs by solid dispersion. Int. J. Pharm. Investig. 2012, 2, 12. [Google Scholar] [CrossRef]
- Dixit, P.; Jain, D.K.; Dumbwani, J. Standardization of an ex vivo method for determination of intestinal permeability of drugs using everted rat intestine apparatus. J. Pharmacol. Toxicol. Methods 2012, 65, 13–17. [Google Scholar] [CrossRef]
- Karaca, T.; Bayiroglu, F.; Cemek, M.; Comba, B.; Ahmet, A.Y.; Karaboğa, İ. Effects of green tea extract and lactobacillus casei strain shirota on levels of serum minerals, cholesterol, triglycerides, glucose and lactate in rats fed on high-carbohydrate and high-lipid diets. KafkasTıpBilim. Derg. 2013, 1, 1–7. [Google Scholar] [CrossRef]
- Thirumalai, T.; Tamilselvan, N.; David, E. Hypolipidemic activity of Piper betel in high fat diet induced hyperlipidemic rat. J. Acute Dis. 2014, 3, 131–135. [Google Scholar] [CrossRef]
- Lee, S.M.; Kim, C.W.; Kim, J.K.; Shin, H.J.; Baik, J.H. GCG-rich tea catechins are effective in lowering cholesterol and triglyceride concentrations in hyperlipidemic rats. Lipids 2008, 43, 419–429. [Google Scholar] [CrossRef]
- Mokra, D.; Adamcakova, J.; Mokry, J. Green tea polyphenol (-)-epigallocatechin-3-gallate (EGCG): A time for a new player in the treatment of respiratory diseases? Antioxidants 2022, 11, 1566. [Google Scholar] [CrossRef]
- Naharros-Molinero, A.; Caballo-González, M.Á.; de la Mata, F.J.; García-Gallego, S. Direct and reverse pluronic micelles: Design and characterization of promising drug delivery nanosystems. Pharmaceutics 2022, 14, 2628. [Google Scholar] [CrossRef] [PubMed]
- Nandi, U.; Ajiboye, A.L.; Patel, P.; Douroumis, D.; Trivedi, V. Preparation of solid dispersions of simvastatin and soluplus using a single-step organic solvent-free supercritical fluid process for the drug solubility and dissolution rate enhancement. Pharmaceuticals 2021, 14, 846. [Google Scholar] [CrossRef] [PubMed]
- Dian, L.; Yu, E.; Chen, X.; Wen, X.; Zhang, Z.; Qin, L.; Wang, Q.; Li, G.; Wu, C. Enhancing oral bioavailability of quercetin using novel soluplus polymeric micelles. Nanoscale Res. Lett. 2014, 9, 684. [Google Scholar] [CrossRef] [PubMed]
- Kshirsagar, S.; Pandit, A.P. Curcumin pellets of carboxymethylated tamarind seed polysaccharide for the treatment of inflammatory bowel disease. Drug Deliv. Lett. 2018, 8, 29–40. [Google Scholar] [CrossRef]
- Shamma, R.N.; Basha, M. Soluplus®: A novel polymeric solubilizer for optimization of carvedilol solid dispersions: Formulation design and effect of method of preparation. Powder Technol. 2013, 237, 406–414. [Google Scholar] [CrossRef]
- Broesder, A.; Bircan, S.Y.; De Waard, A.B.; Eissens, A.C.; Frijlink, H.W.; Hinrichs, W.L. Formulation and in vitro evaluation of pellets containing sulfasalazine and caffeine to verify ileo-colonic drug delivery. Pharmaceutics 2021, 13, 1985. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No | Ingredients | Quantity (%w/w) |
|---|---|---|
| 1 | GTE-SD powder | 30 |
| 2 | Microcrystalline cellulose | 30 |
| 3 | Lactose | 30 |
| 4 | Sodium croscarmellose | 5 |
| 5 | Polyvinyl pyrrolidone K 30 | 5 |
| 6 | IPA and distilled Water | q.s. 1 |
| Run | Batches | X1 (GTE-SD Ratio) | X2 (Spheronizer Speed (rpm)) |
|---|---|---|---|
| 1 | A1 | −1(1:4) | 1(1300) |
| 2 | A2 | 1(1:8) | 1(1300) |
| 3 | A3 | 0(1:6) | 0(1200) |
| 4 | A4 | 0(1:6) | −1(1100) |
| 5 | A5 | −1(1:4) | 0(1200) |
| 6 | A6 | 0(1:6) | 1(1300) |
| 7 | A7 | 1(1:8) | 0(1200) |
| 8 | A8 | −1(1:4) | −1(1100) |
| 9 | A9 | 1(1:8) | −1(1100) |
| SD Ratio | Mean Particle Size (nm) | PDI | Drug Content (%) |
|---|---|---|---|
| F1 | 219.1 | 1.191 | 92.85 ± 0.715 |
| 1:4F2 | 211.9 | 1.628 | 95.47 ± 1.09 |
| 1:6F3 | 198.0 | 0.548 | 96.18 ± 0.409 |
| 1:8F4 | 402.3 | 1.259 | 97.14 ± 1.43 |
| 1:10F5 | 403.5 | 4.425 | 98.8 ± 0.409 |
| Factor 1 | Factor 2 | Response 1 | Response 2 | Response 3 | ||
|---|---|---|---|---|---|---|
| Run | Batches | X1: Solid Dispersion Ratio | X2: Spheronizer Speed (rpm) | Y1 (Drug Release (%)) | Y2 (Drug Permeation (%)) | Y3 (Geometric Mean Diameter (µm)) |
| 1 | A1 | −1(1:4) | 1 (1300) | 71.31 | 17.18 | 501.6 |
| 2 | A2 | 1(1:8) | 1 (1300) | 83.55 | 23.84 | 503.1 |
| 3 | A3 | 0(1:6) | 0 (1200) | 75.45 | 33.27 | 510.4 |
| 4 | A4 | 0(1:6) | −1 (1100) | 73.46 | 30.67 | 517.7 |
| 5 | A5 | −1(1:4) | 0 (1200) | 67.43 | 18.18 | 512.4 |
| 6 | A6 | 0(1:6) | 1 (1300) | 71.35 | 29.46 | 500 |
| 7 | A7 | 1(1:8) | 0 (1200) | 85.48 | 22.24 | 513.2 |
| 8 | A8 | −1(1:4) | −1 (1100) | 68.35 | 18.89 | 526.2 |
| 9 | A9 | 1(1:8) | −1 (1100) | 79.58 | 20.03 | 528.2 |
| Batches | Bulk Density | Tapped Density | Hausner’s Ratio | Carr’s Index (%) | Angle of Repose (°) | Drug Content (%) |
|---|---|---|---|---|---|---|
| A1 | 0.883 ± 0.04 | 1.11 ± 0.07 | 1.25 ± 0.05 | 20.45 ± 2.74 | 22.56 ± 0.152 | 82.85 ± 1.43 |
| A2 | 0.912 ± 0.02 | 1.28 ± 0.06 | 1.40 ± 0.05 | 28.76 ± 2.43 | 30.11 ± 0.203 | 90.47 ± 1.09 |
| A3 | 0.837 ± 0.07 | 1.15 ± 0.08 | 1.37 ± 0.05 | 27.25 ± 2.90 | 22.33 ± 0.325 | 83.56 ± 0.71 |
| A4 | 0.849 ± 0.05 | 1.11 ± 0.01 | 1.30 ± 0.06 | 23.20 ± 1.21 | 29.98 ± 0.091 | 83.80 ± 1.09 |
| A5 | 0.837 ± 0.07 | 1.12 ± 0.12 | 1.33 ± 0.03 | 25.11 ± 2.09 | 27.29 ± 0.323 | 83.33 ± 1.09 |
| A6 | 0.883 ± 0.04 | 1.20 ± 0.08 | 1.36 ± 0.14 | 26.78 ± 2.77 | 28.32 ± 0.187 | 89.04 ± 1.09 |
| A7 | 0.837 ± 0.04 | 1.08 ± 0.07 | 1.28 ± 0.02 | 22.14 ± 1.77 | 35.21 ± 0.281 | 87.85 ± 0.71 |
| A8 | 0.845 ± 0.01 | 1.16 ± 0.04 | 1.37 ± 0.05 | 27.1 ± 2.51 | 25.54 ± 0.352 | 82.37 ± 0.82 |
| A9 | 0.787 ± 0.01 | 1.15 ± 0.05 | 1.46 ± 0.12 | 31.46 ± 2.26 | 29.94 ± 0.892 | 86.18 ± 1.08 |
| Batch | Factor | Responses | Predicted Value | Experimental Value |
|---|---|---|---|---|
| A3 | X1 GTE-SD ratio (1:6) | % Drug release | 79.23 | 75.45 |
| X2 spheronizer speed (1200) | % Drug permeation | 29.92 | 33.27 | |
| Geometric mean diameter | 501.64 | 510.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patole, V.; Gaikwad, P.; Kharat, S.; Jadhav, P.; Deshkar, S.; Giram, P. Green Tea Extract Solid Dispersion Pellets with Enhanced Permeability for Hyperlipidemia. Future Pharmacol. 2023, 3, 708-730. https://doi.org/10.3390/futurepharmacol3040044
Patole V, Gaikwad P, Kharat S, Jadhav P, Deshkar S, Giram P. Green Tea Extract Solid Dispersion Pellets with Enhanced Permeability for Hyperlipidemia. Future Pharmacology. 2023; 3(4):708-730. https://doi.org/10.3390/futurepharmacol3040044
Chicago/Turabian StylePatole, Vinita, Pranita Gaikwad, Shashikant Kharat, Pranali Jadhav, Sanjeevani Deshkar, and Prabhanjan Giram. 2023. "Green Tea Extract Solid Dispersion Pellets with Enhanced Permeability for Hyperlipidemia" Future Pharmacology 3, no. 4: 708-730. https://doi.org/10.3390/futurepharmacol3040044
APA StylePatole, V., Gaikwad, P., Kharat, S., Jadhav, P., Deshkar, S., & Giram, P. (2023). Green Tea Extract Solid Dispersion Pellets with Enhanced Permeability for Hyperlipidemia. Future Pharmacology, 3(4), 708-730. https://doi.org/10.3390/futurepharmacol3040044