
SARS-CoV-2 Mpro Inhibitors: Achieved Diversity, Developing Resistance and Future Strategies


Abstract
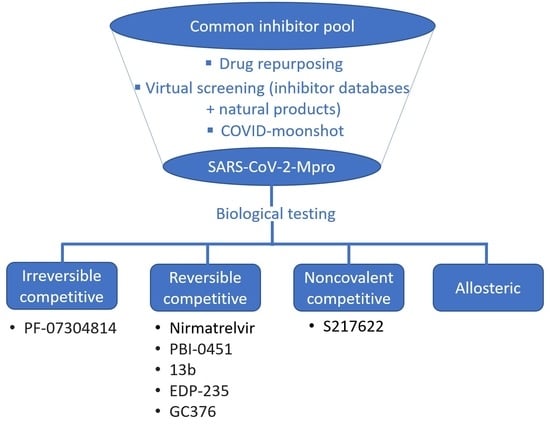
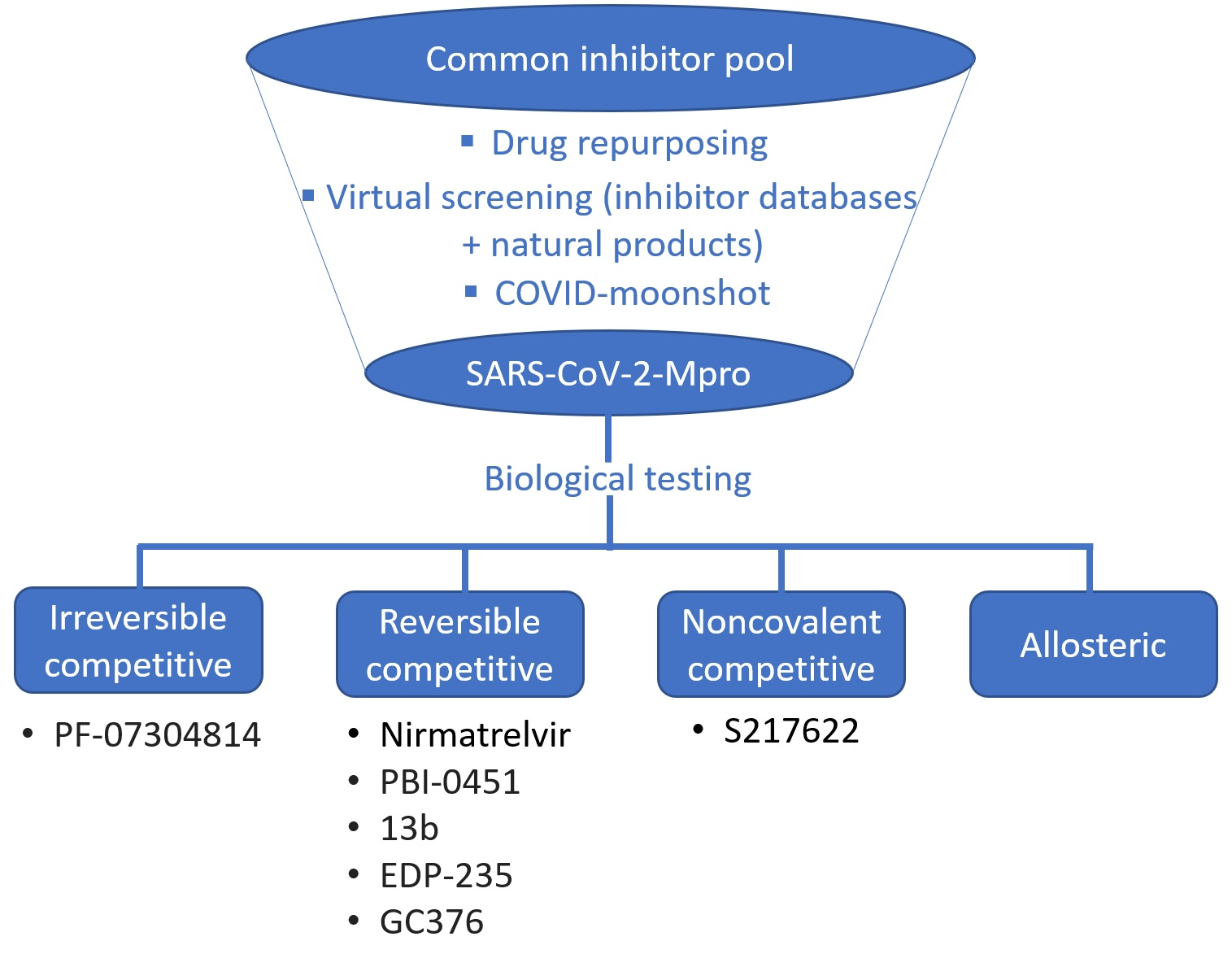
1. Introduction
- The smaller, spatially more defined active site of Mpro, allowing the synthesis of smaller and stiffer inhibitors, whereas efficient binding to PLpro usually relies on tight distal inhibitors [8].
- Dimerization of Mpro for activity and close proximity of active site and dimerization interface, suggesting inhibitor design that additionally prevents dimerization [9].
- The lower mutation rates of Mpro (nsp5) compared to PLpro (nps3), mitigating the risk of mutation-mediated drug resistance [4].
- Less off-target effects expected, since Mpro’s glutamine (Gln) cleavage site recognition is unique and has not been not observed for any human protease. PLpro inhibitors might interfere with human ubiquitin binding motives [10].
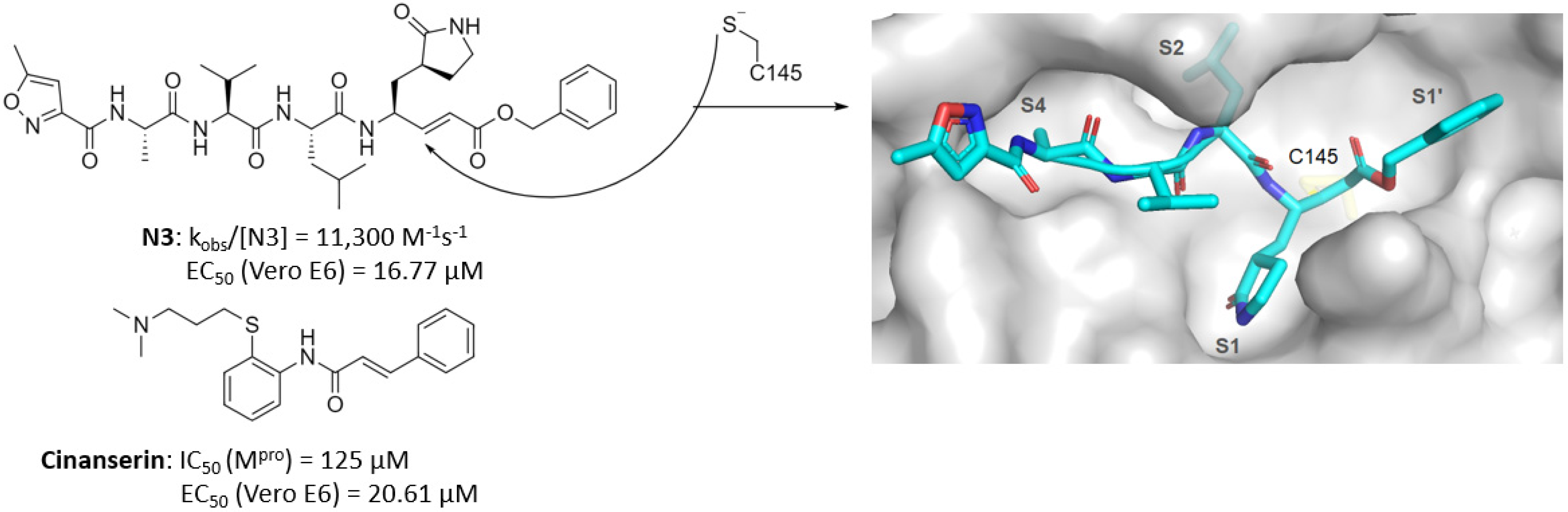
2. Competitive Inhibitor Design Strategies
2.1. Predominantly Irreversible Warheads


| Warhead | Drug Example a | Efficacy | Potency | Toxicology | Pharmacology | Lit. | ||
|---|---|---|---|---|---|---|---|---|
| Ki/IC50 (µM) | EC50 (µM) | CC50 (µM) | t0.5 (h) | Cmax (ng/mL) | Clear. (mL/min/kg) | |||
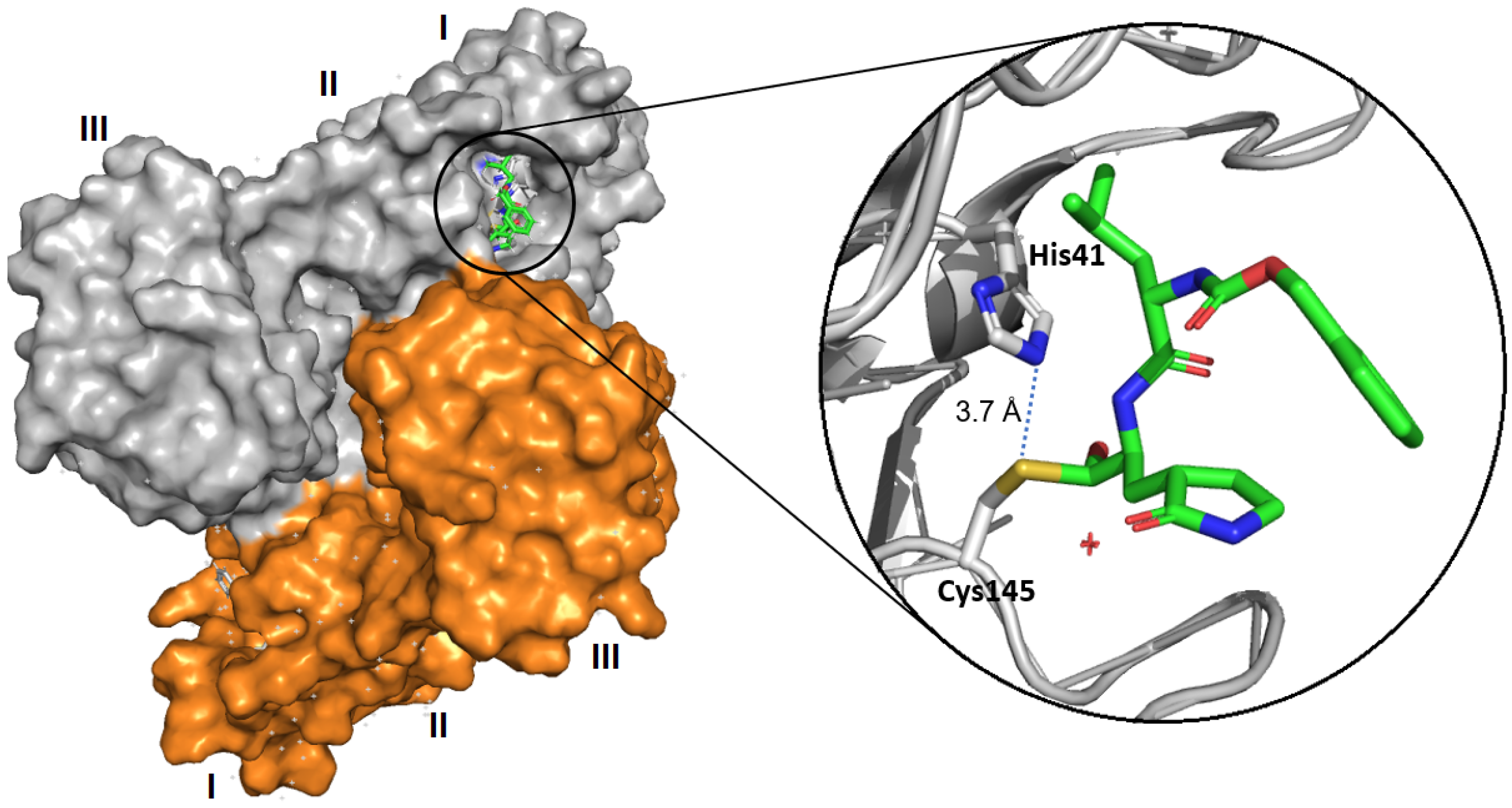
| Michael acceptor | N3, Cinanserin, 1 | - 1: 0.15 (IC50) | N3: 16.77 1: 2.88 | N3: >130 1: >200 | - | - | - | [14] [16] |
| Acrylamide | LON-WEI-adc59df6-47 | 2.9 (IC50) 38.4 (Ki) | - | - | - | - | - | [19] |
| Carbamoyl | Carmofur | 1.82 | 24 | 133 | - | - | - | [24] |
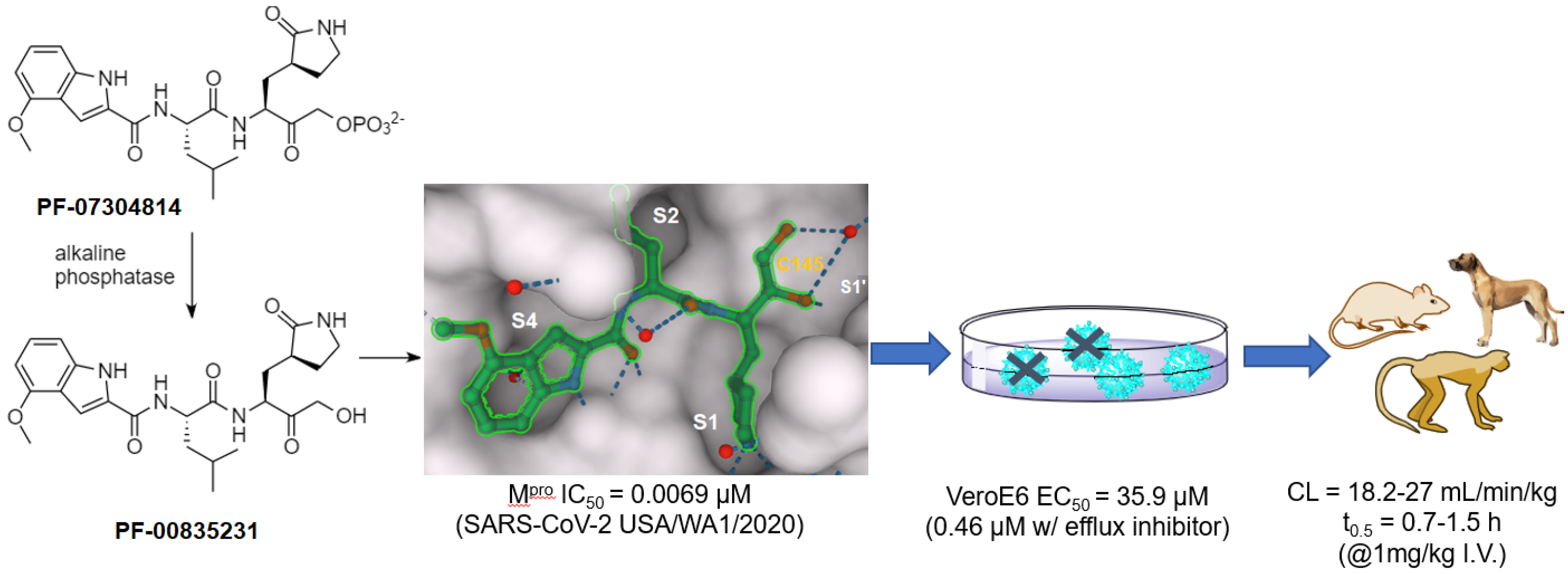
| Hydroxymethyl ketone | PF-00835231 | 0.0069 (IC50) | 0.46 b | >50 (VeroE6) | 0.72 (rat) | 1250 (rat) | 27 (rat) | [26] |
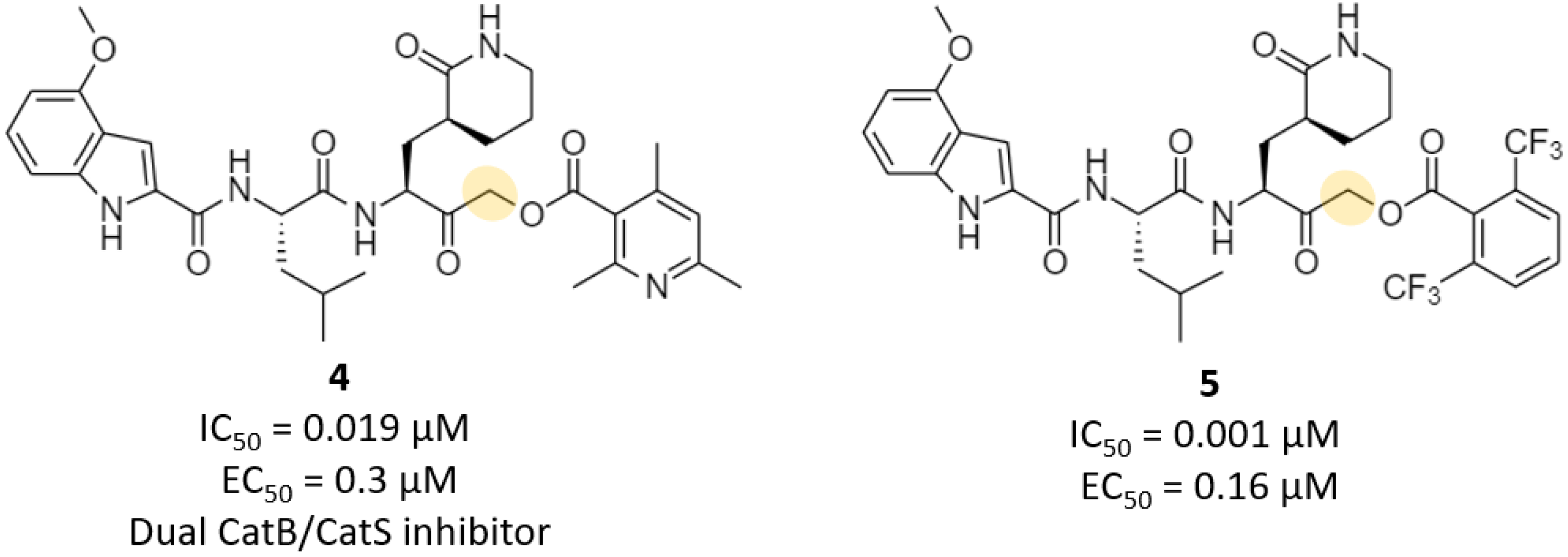
| Acyloxymethyl ketone | 4, 5 | 0.001 | 0.16 | >200 | >4 (mouseplasma) | - | - | [27] |
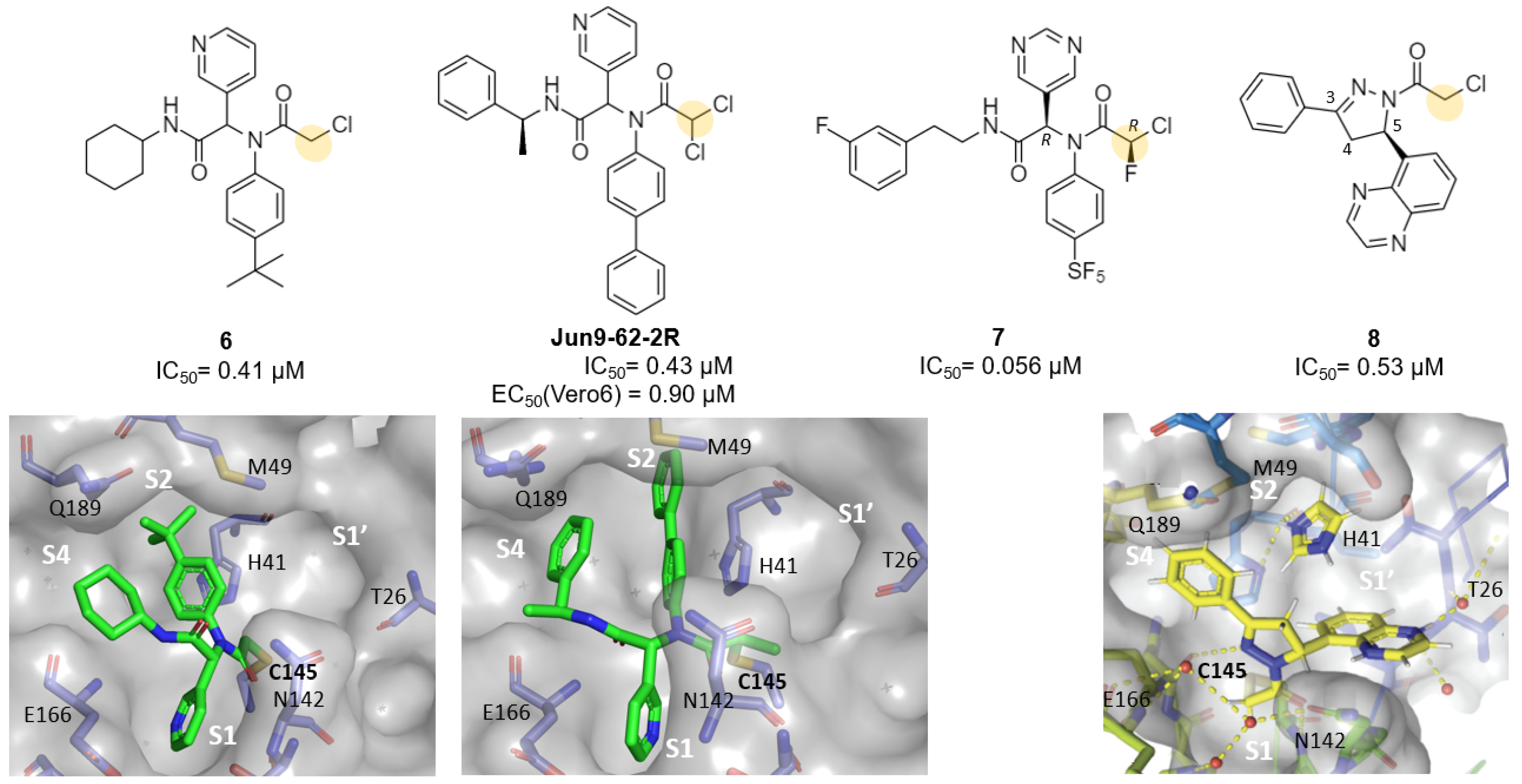
| Chloroacetamides | 6, Jun9-62-2R, Jun9-88-2R | 6a: 0.4 (IC50) Jun9-62-2R: 0.43 (IC50) Jun9-88-2R: 0.08 (IC50) | 6a: -Jun9-62-2R: 0.9 Jun9-88-2R: 0.58 | 6a: -Jun9-62-2R: >100 Jun9-88-2R: 5.48 | - | - | - | [31] |
| Chlorofluoro- acetamides | 7 | 0.056 (IC50) 1.34 | - | - | - | - | - | [34] |
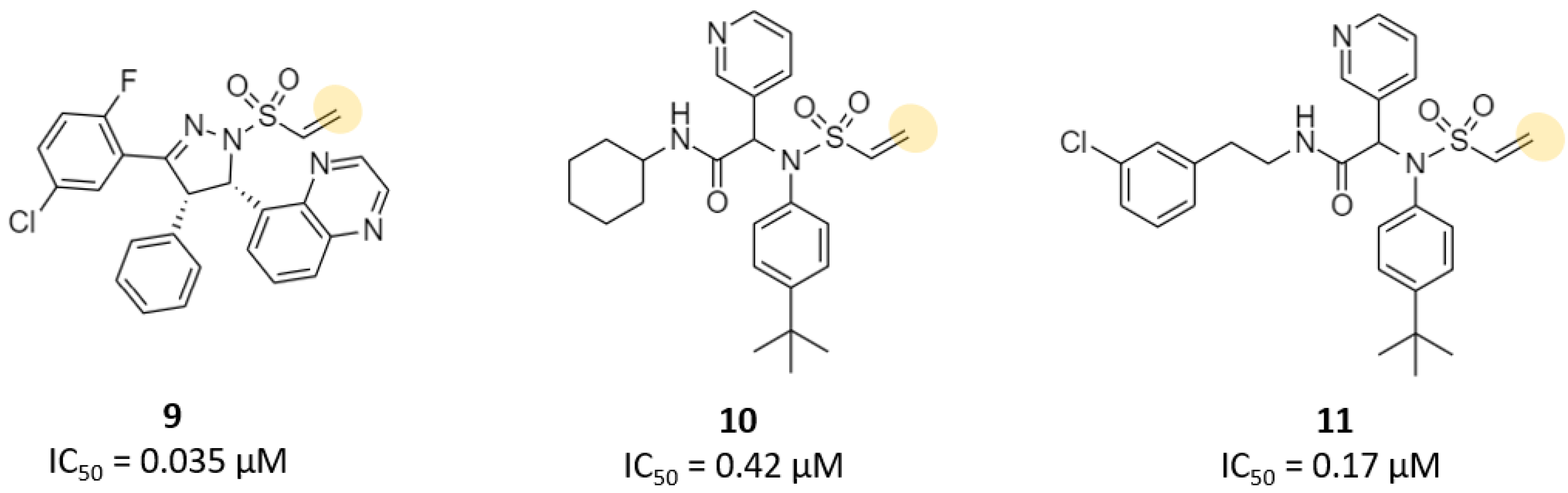
| Vinylsulfonamide | 11 | 2.3 (Ki) 0.17 (IC50) | - | - | - | - | - | [31] |
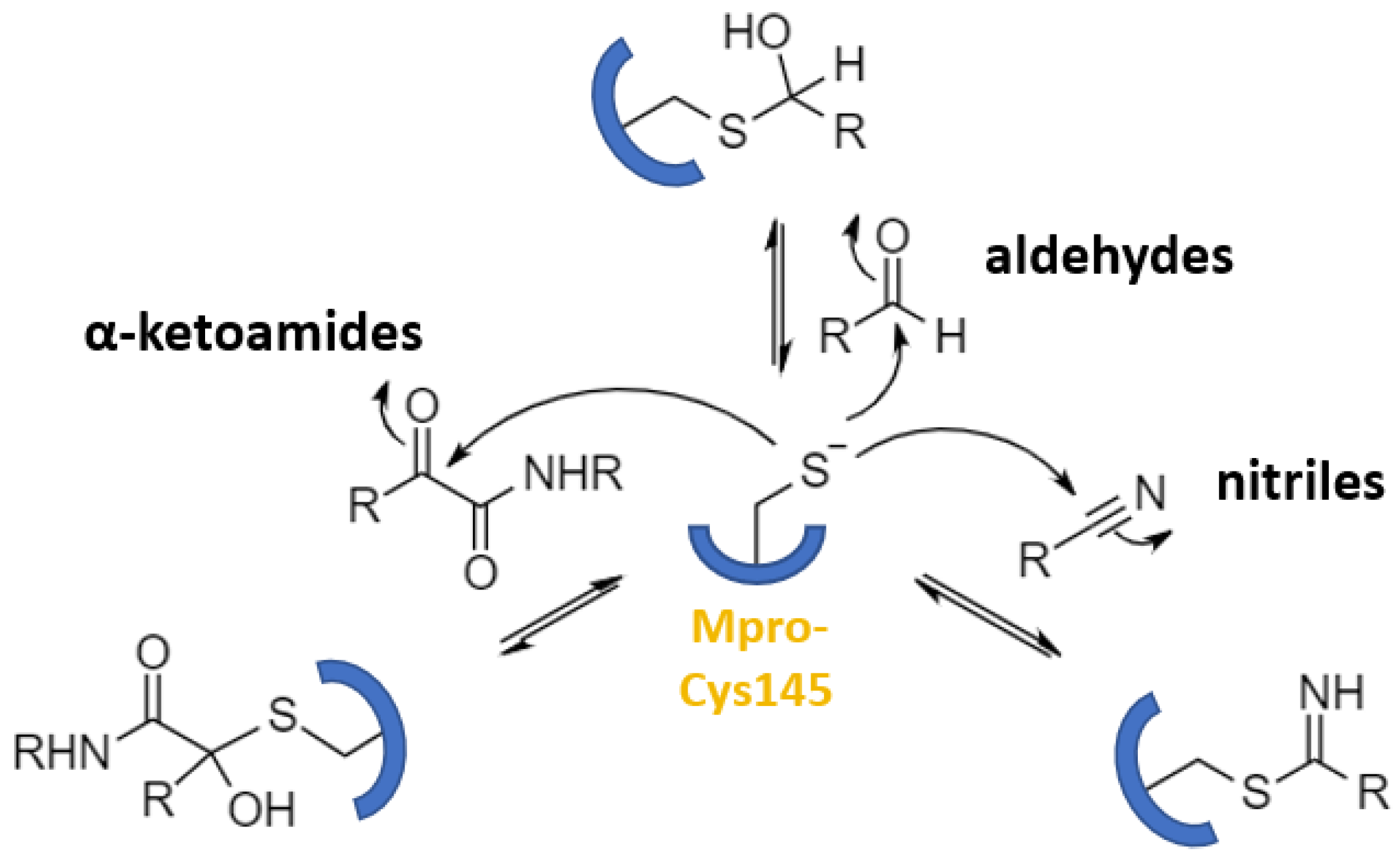
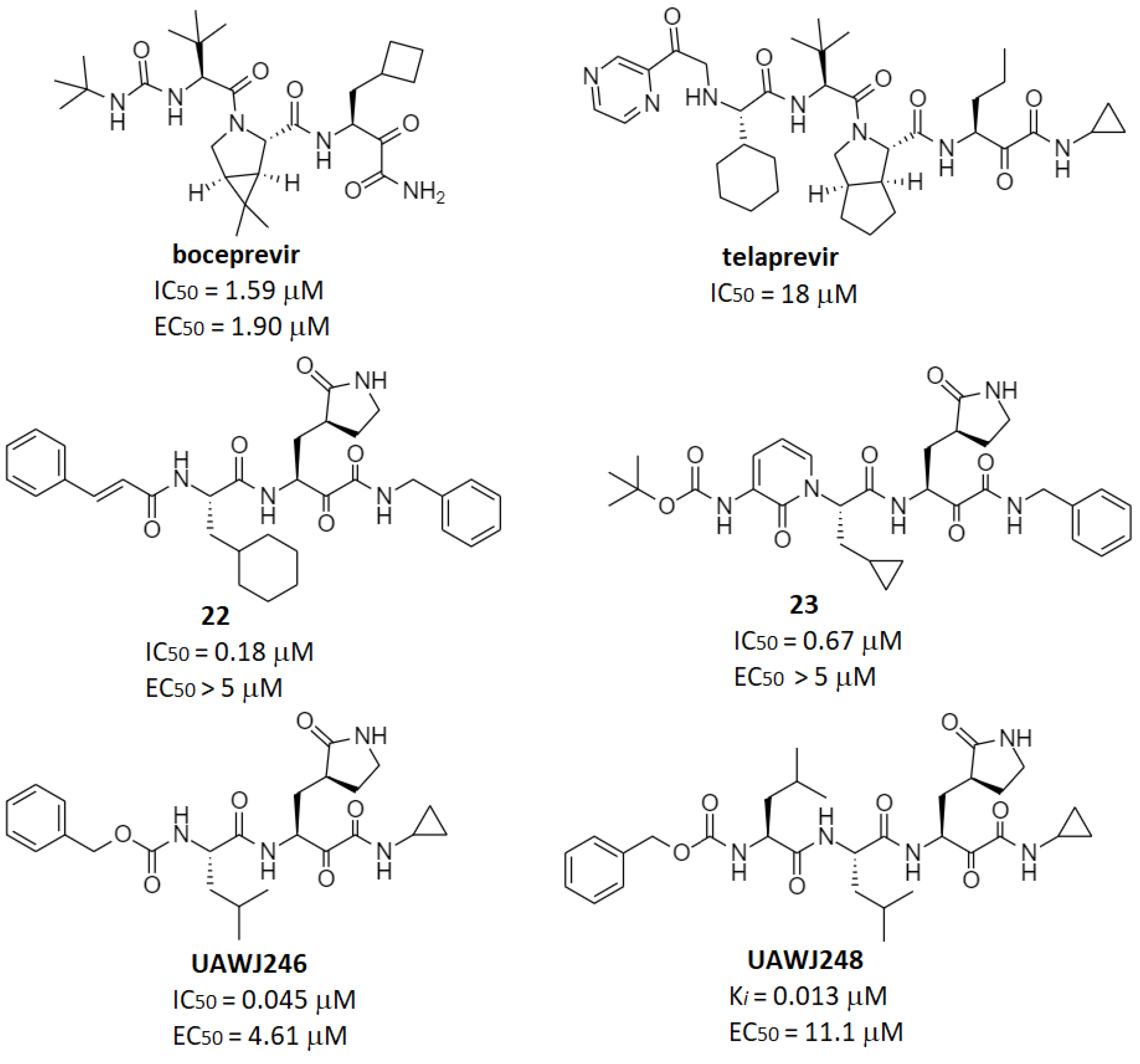
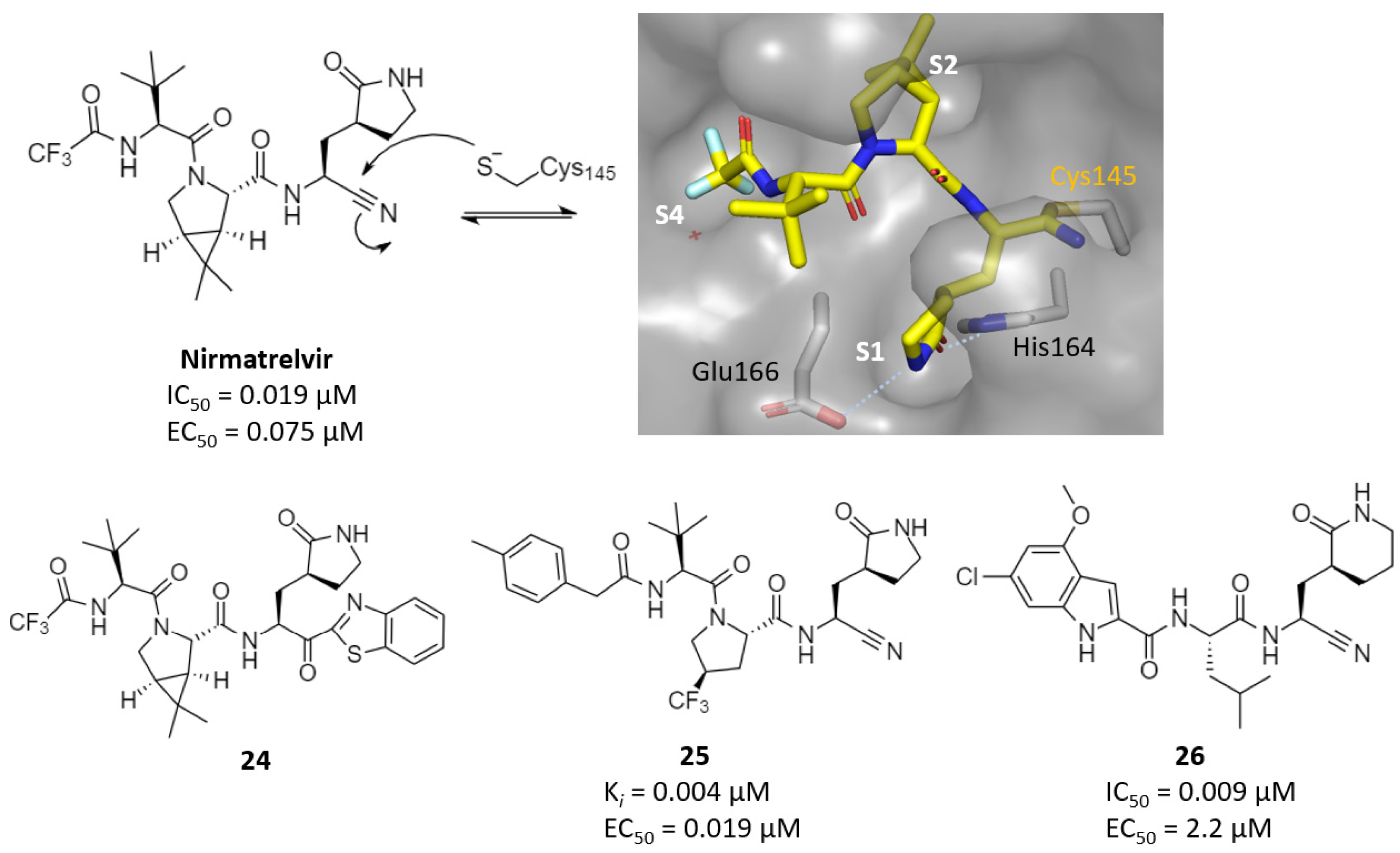
2.2. Reversible Warheads

| Warhead | Drug Example | Efficacy | Potency | Toxicology | Pharmacology | Lit. | ||
|---|---|---|---|---|---|---|---|---|
| IC50 (µM) | EC50 (µM) | CC50 (µM) | t0.5 (h) | Cmax (ng/mL) | Clear. (mL/min/kg) | |||
| Aldehydes | 13–21, GC376, MPI8, MI-23 | GC376: 0.19 | GC376: 0.92 | GC376: >100 (Vero6) | GC376: 1.1 (mouse), 3.9 (rat) | GC376: 46700 (mouse), 12560 (rat) | GC376: 33.1 (mouse), 20.1 (rat) | [43] [46] |
| Ketoamides | 23, UAWJ246 | 23: 0.67 UAWJ246: 0.045 | 23: 4–5 UAWJ246: 4.61 | UAWJ246: >250 (Vero6) | 23: 1.0 (mouse) | 23: 334 (mouse) | 23: 566 (mouse) | [7] [49] |
| Nitriles | nirmatrelvir | 0.019 | 0.075 | >100 (Vero6) | 1.5 (rat, P.O.) | 1290 (mouse, P.O.) | - | [68] |
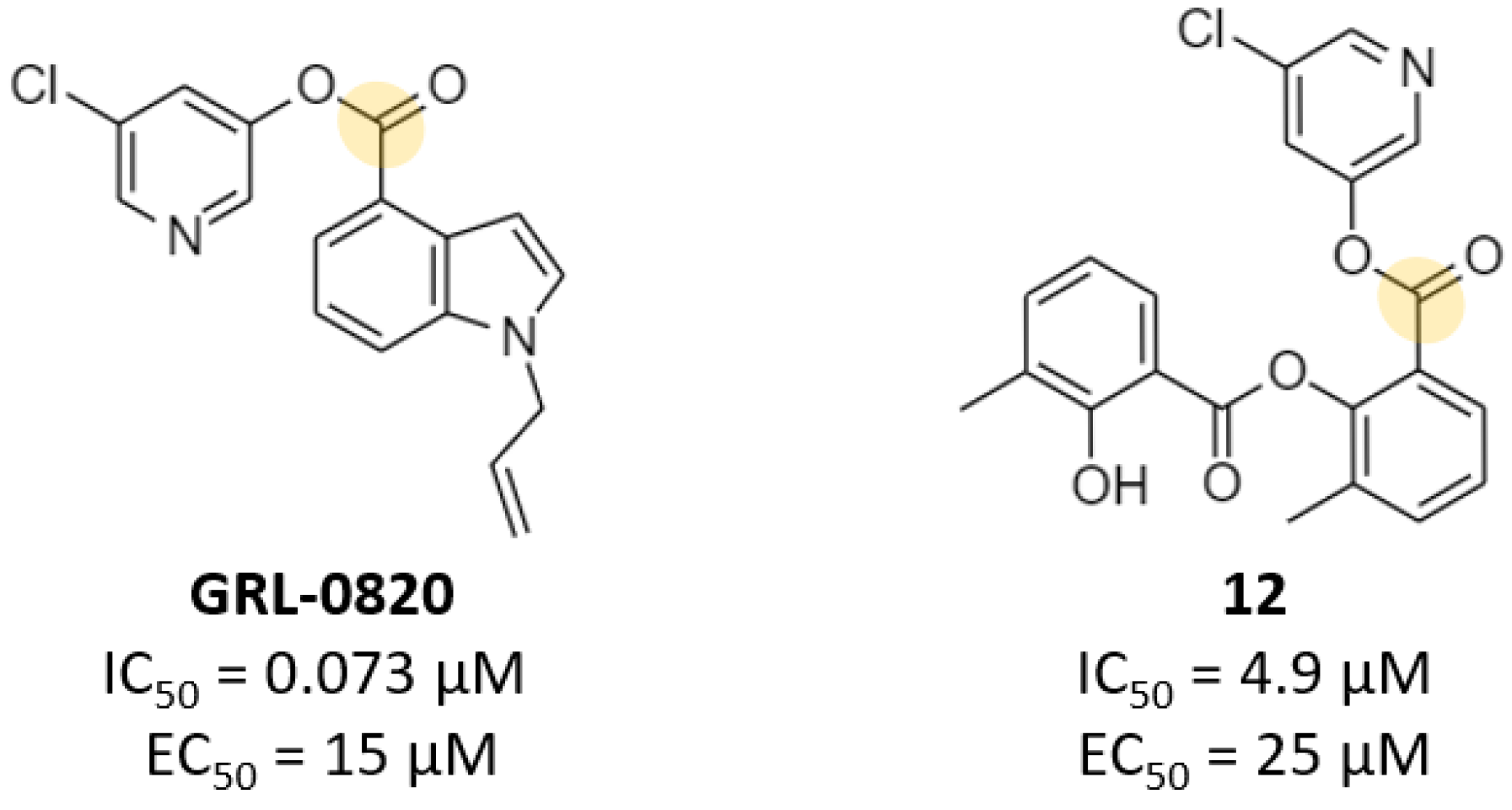
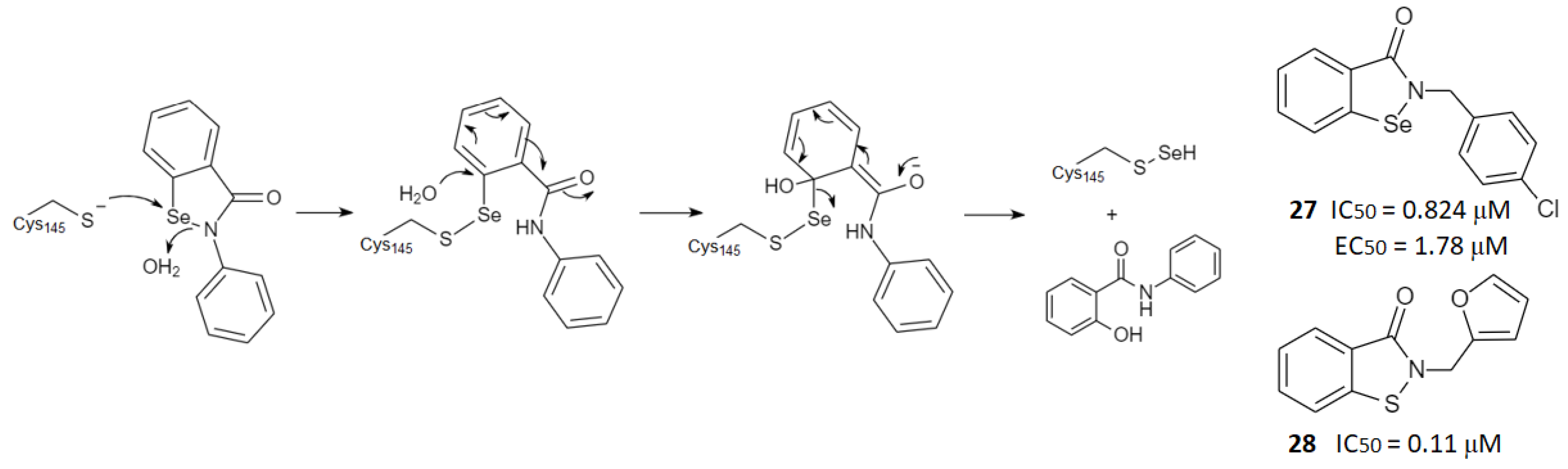
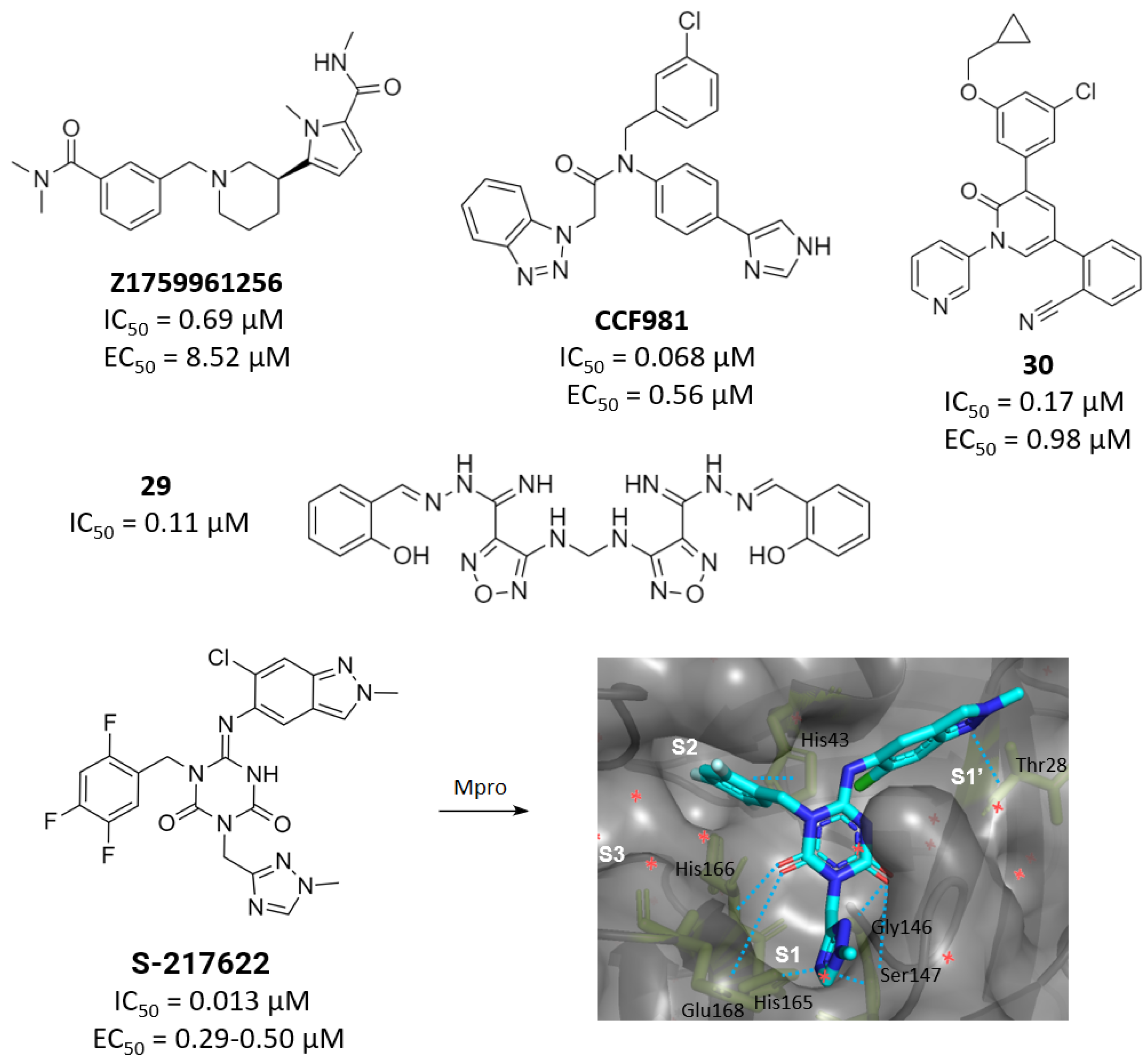
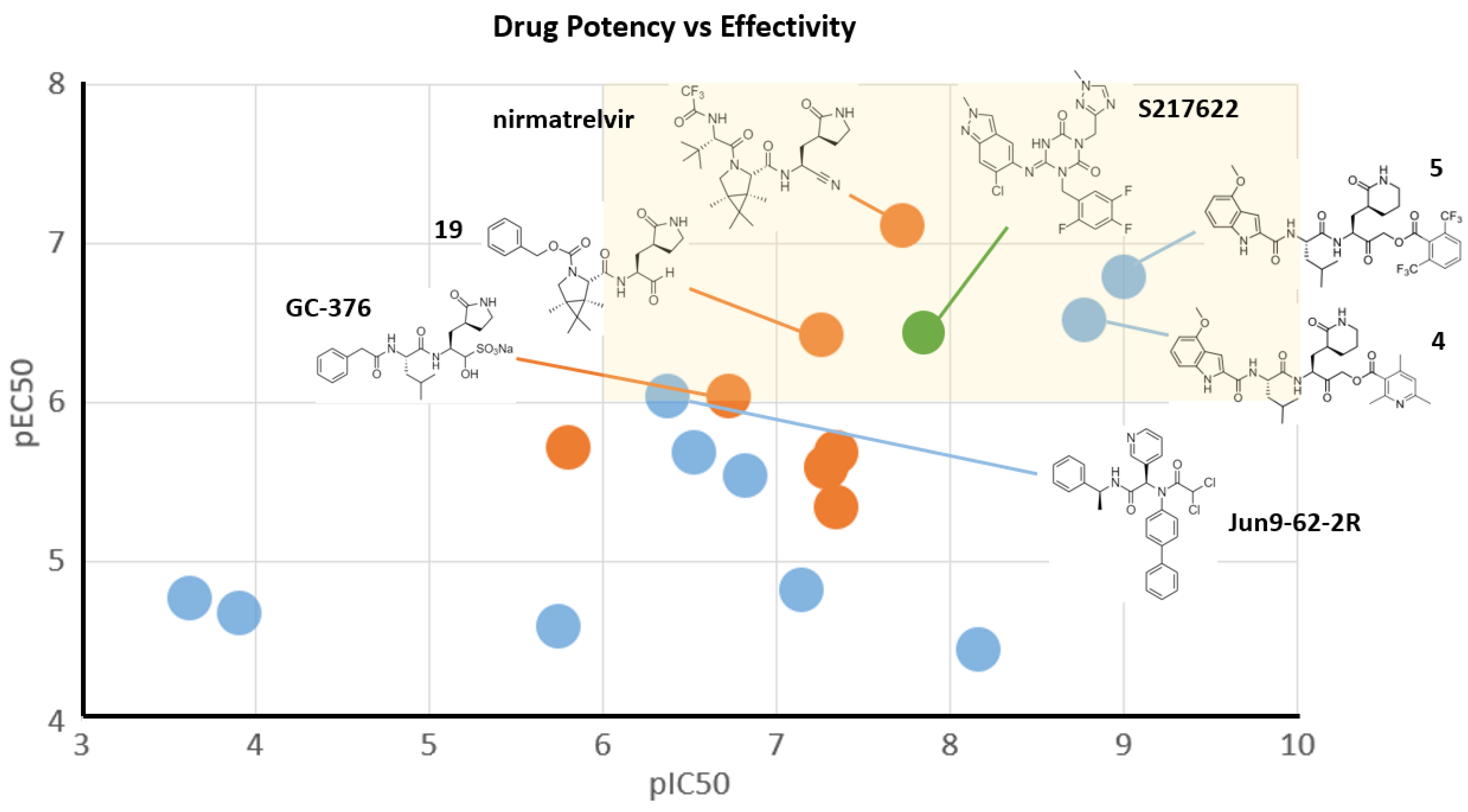
2.3. Noncovalent Active Site Inhibitors
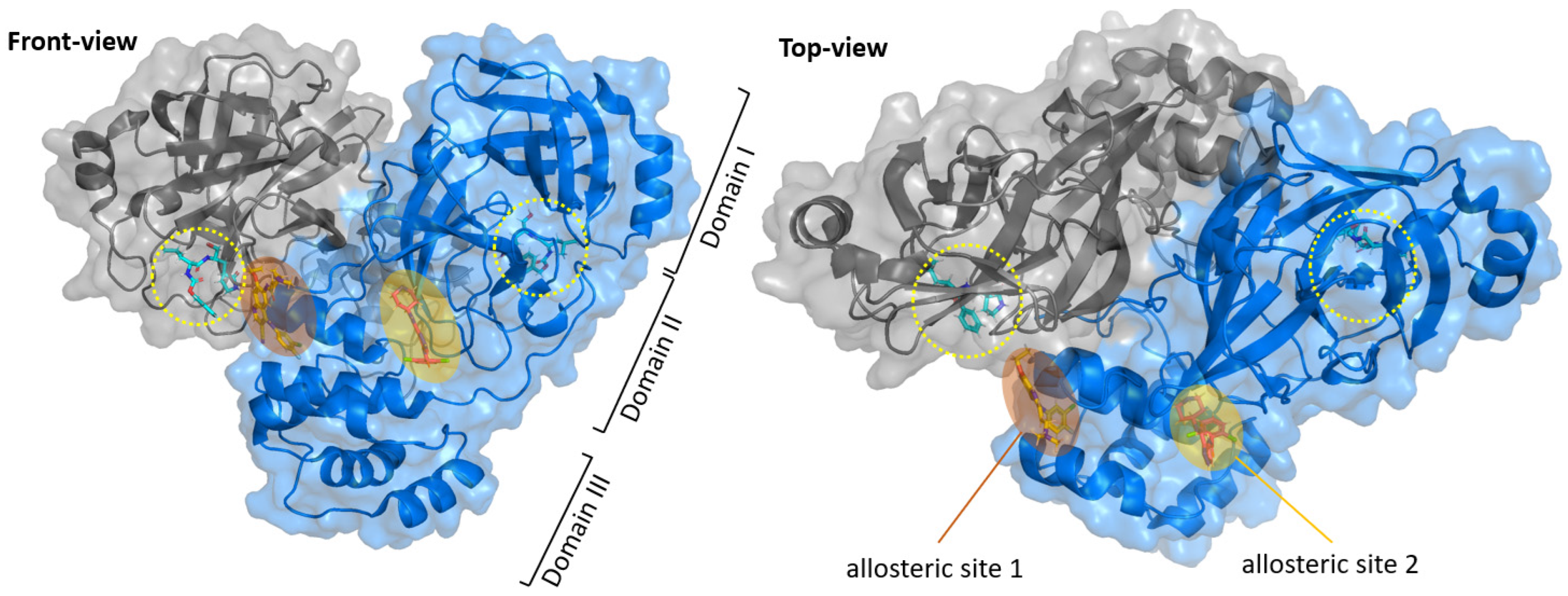
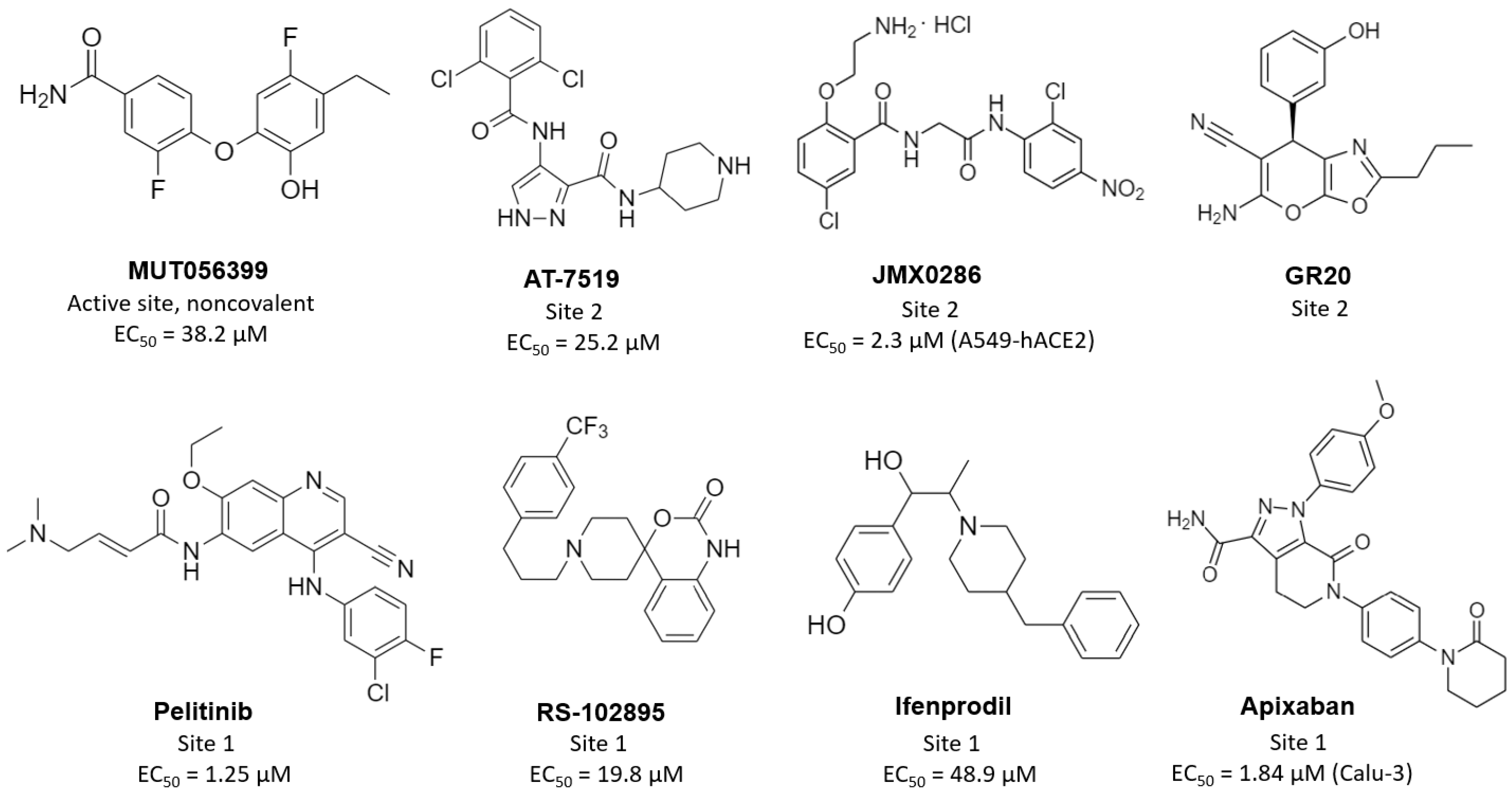
3. Allosteric Inhibitors
| Inhibitor | Non-Competitive Binding in: a | IC50 (µM) | EC50 (µM) b | CC50 (µM) b | PDB Code | Ref. |
|---|---|---|---|---|---|---|
| MUT056399 | active site | - | 38.2 | >100 | 7ap6 | [89] |
| pelitinib | allosteric site 1 | - | 1.25 | 14 | 7axm | [89] |
| ifenprodil | allosteric site 1 | - | 46.9 | >100 | 7aqi | [89] |
| RS-102895 | allosteric site 1 | - | 19.8 | 55 | 7abu | [89] |
| AT7519 | allosteric site 2 | - | 25.2 | >100 | 7aga | [89] |
| JMX0286 | allosteric site 2 * | 4.8 | 2.3 (A549-hACE2) | 53(A549-hACE2) | - | [92] |
| apixaban | allosteric site 1 * | 0.01 | 1.84 (Calu-3) | 491(Calu-3) | - | [94] |
| GR20 | allosteric site 2 * | 91.8 | - | - | - | [95] |
| chebulagic acid | alternative site | 9.76 | 9.09 | ~100 | - | [96] |
| punicalagin | alternative site | 7.2 | 4.62 | ~100 | [96] | |
| colloidal bismuth-subcitrate (CBS) | alternative site | 0.93 | 177.3 | - | - | [97] |
| NB1A2 | alternative site | 0.19 | - | - | 7vfa | [99] |
| NB2B4 | alternative site | 0.12 | - | - | 7vfb | [99] |
4. Inhibitors in Action—Drug Performance in the Clinic
5. Future Strategies/Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- WHO Coronavirus (COVID-19) Dashboard | WHO Coronavirus (COVID-19) Dashboard with Vaccination Data. Available online: https://covid19.who.int/ (accessed on 12 December 2022).
- Paul, D.; Pyne, N.; Paul, S. Mutation profile of SARS-CoV-2 spike protein and identification of potential multiple epitopes within spike protein for vaccine development against SARS-CoV-2. Virusdisease 2021, 32, 703–726. [Google Scholar] [CrossRef] [PubMed]
- Tuekprakhon, A.; Nutalai, A.; Dijokaite-Guraliuc, A.; Zhou, D.; Ginn, H.M.; Selvaraj, M.; Liu, C.; Mentzer, A.J.; Supasa, P.; Duyvesteyn, H.M.E.; et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 2022, 185, 2422–2433. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.; Irfan, M.; Ahsan, H.; Ahmed, A.; Kaushik, A.C.; Khan, A.S.; Chinnasamy, S.; Ali, A.; Wei, D.-Q. Structures of SARS-CoV-2 RNA-Binding Proteins and Therapeutic Targets. Intervirology 2021, 64, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Amicone, M.; Borges, V.; Alves, M.J.; Isidro, J.; Zé-Zé, L.; Duarte, S.; Vieira, L.; Guiomar, R.; Gomes, J.P.; Gordo, I. Mutation rate of SARS-CoV-2 and emergence of mutators during experimental evolution. Evol. Med. Public Health 2022, 10, 142–155. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B 2021, 11, 237–245. [Google Scholar] [CrossRef]
- Goyal, B.; Goyal, D. Targeting the Dimerization of the main protease of Coronaviruses: A potential broad-spectrum therapeutic strategy. ACS Comb. Sci. 2020, 22, 297–305. [Google Scholar] [CrossRef]
- Tan, H.; Hu, Y.; Jadhav, P.; Tan, B.; Wang, J. Progress and challenges in targeting the SARS-CoV-2 papain-like protease. J. Med. Chem. 2022, 65, 7561–7580. [Google Scholar] [CrossRef]
- Ferreira, J.C.; Fadl, S.; Villanueva, A.J.; Rabeh, W.M. Catalytic Dyad Residues His41 and Cys145 Impact the Catalytic Activity and Overall Conformational Fold of the Main SARS-CoV-2 Protease 3-Chymotrypsin-Like Protease. Front. Chem. 2021, 9, 692168. [Google Scholar] [CrossRef]
- Gisdon, F.J.; Bombarda, E.; Ullmann, G.M. Serine and Cysteine Peptidases: So Similar, Yet Different. How the Active-Site Electrostatics Facilitates Different Reaction Mechanisms. J. Phys. Chem. B 2022, 22, 4035–4048. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Atkins, W.M. A Quantitative index of substrate promiscuity. Biochemistry 2008, 47, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gui, C.; Luo, X.; Yang, Q.; Günther, S.; Scandella, E.; Drosten, C.; Bai, D.; He, X.; Ludewig, B.; et al. Cinanserin is an inhibitor of the 3C-like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. J. Virol. 2005, 79, 7095–7103. [Google Scholar] [CrossRef]
- Iketani, S.; Forouhar, F.; Liu, H.; Hong, S.J.; Lin, F.Y.; Nair, M.S.; Zask, A.; Huang, Y.; Xing, L.; Stockwell, B.R.; et al. Lead compounds for the development of SARS-CoV-2 3CL protease inhibitors. Nat Commun. 2021, 12, 2016. [Google Scholar] [CrossRef]
- Amendola, G.; Ettari, R.; Previti, S.; Di Chio, C.; Messere, A.; Di Maro, S.; Hammerschmidt, S.J.; Zimmer, C.; Zimmermann, R.A.; Schirmeister, T.; et al. Lead discovery of SARSCoV-2 main protease inhibitors through covalent docking-based virtual screening. J. Chem. Inf. Model. 2021, 61, 2062–2073. [Google Scholar] [CrossRef]
- Manandhar, A.; Srinivasulu, V.; Mohamad, H.; Tarazi, H.; Omar, H.; Colussi, D.J.; Gordon, J.; Childers, W.; Klein, M.L.; Al-Tel, T.H.; et al. Discovery of Novel Small-Molecule Inhibitors of SARS-CoV-2 Main Protease as Potential Leads for COVID-19 Treatment. J. Chem. Inf. Model. 2021, 61, 4745–4757. [Google Scholar] [CrossRef]
- Zaidman, D.; Gehrtz, P.; Filep, M.; Fearon, D.; Gabizon, R.; Douangamath, A.; Prilusky, J.; Duberstein, S.; Cohen, G.; Owen, C.D.; et al. An automatic pipeline for the design of irreversible derivatives identifies a potent SARS-CoV-2 Mpro inhibitor. Cell Chem. Biol. 2021, 28, 1795–1806. [Google Scholar] [CrossRef]
- Gossen, J.; Albani, S.; Hanke, A.; Joseph, B.P.; Bergh, C.; Kuzikov, M.; Costanzi, E.; Manelfi, C.; Storici, P.; Gribbon, P.; et al. A blueprint for high affinity SARS-CoV-2 Mpro inhibitors from activity-based compound library screening guided by analysis of protein dynamics. ACS Pharmacol. Transl. Sci. 2021, 4, 1079–1095. [Google Scholar] [CrossRef]
- Chaves, O.A.; Fintelman-Rodrigues, N.; Wang, X.; Sacramento, C.Q.; Temerozo, J.R.; Ferreira, A.C.; Mattos, M.; Pereira-Dutra, F.; Bozza, P.T.; Castro-Faria-Neto, H.C.; et al. Commercially Available Flavonols Are Better SARS-CoV-2 Inhibitors than Isoflavone and Flavones. Viruses 2022, 14, 1458. [Google Scholar] [CrossRef]
- Su, H.; Yao, S.; Zhao, W.; Zhang, Y.; Liu, J.; Shao, Q.; Wang, Q.; Li, M.; Xie, H.; Shang, W.; et al. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat. Commun. 2021, 12, 3623. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Cui, M.; Zheng, C.; Wang, M.; Sun, R.; Gao, D.; Bao, J.; Ren, S.; Yang, B.; Lin, J.; et al. Myricetin inhibits SARS-CoV-2 viral replication by targeting Mpro and ameliorates pulmonary inflammation. Front. Pharmacol. 2021, 12, 669642. [Google Scholar] [CrossRef]
- Jin, Z.; Zhao, Y.; Sun, Y.; Zhang, B.; Wang, H.; Wu, Y.; Zhu, Y.; Zhu, C.; Hu, T.; Du, X.; et al. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020, 27, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Hu, Y.; Townsend, J.A.; Lagarias, P.I.; Marty, M.T.; Kolocouris, A.; Wang, J. Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin Are Nonspecific Promiscuous SARS-CoV-2 Main Protease Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutler, N.; Binder, J.; Chen, E.; Eng, H.; et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID-19. Nat. Commun. 2021, 12, 6055. [Google Scholar] [CrossRef]
- Bai, B.; Belovodskiy, A.; Hena, M.; Kandadai, A.S.; Joyce, M.A.; Saffran, H.A.; Shields, J.A.; Khan, M.B.; Arutyunova, E.; Lu, J.; et al. Peptidomimetic α-Acyloxymethylketone Warheads with Six-Membered Lactam P1 Glutamine Mimic: SARS-CoV-2 3CL Protease Inhibition, Coronavirus Antiviral Activity, and in vitro Biological Stability. J. Med. Chem. 2022, 65, 2905–2925. [Google Scholar] [CrossRef]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef]
- Rasnick, D. Synthesis of peptide fluoromethyl ketones and the inhibition of human cathepsin B. Anal. Biochem. 1985, 149, 461–465. [Google Scholar] [CrossRef]
- Eichhold, T.H.; Hookfin, E.B.; Taiwo, Y.O.; De, B.; Wehmeyer, K.R. Isolation and quantification of fluoroacetate in rat tissues, following dosing of Z-Phe-Ala-CH2-F, a peptidyl fluoromethyl ketone protease inhibitor. J. Pharm. Biomed. Anal. 1997, 16, 459–467. [Google Scholar] [CrossRef]
- Stille, J.K.; Tjutrins, J.; Wang, G.; Venegas, F.A.; Hennecker, C.; Rueda, A.M.; Sharon, I.; Blaine, N.; Miron, C.E.; Pinus, S.; et al. Design, synthesis and in vitro evaluation of novel SARS-CoV-2 3CLpro covalent inhibitors. Eur. J. Med. Chem. 2022, 5, 114046. [Google Scholar] [CrossRef]
- Ma, C.; Xia, Z.; Sacco, M.D.; Hu, Y.; Townsend, J.A.; Meng, X.; Choza, J.; Tan, H.; Jang, J.; Gongora, M.V.; et al. Discovery of Di- and Trihaloacetamides as Covalent SARS-CoV-2 Main Protease Inhibitors with High Target Specificity. J. Am. Chem. Soc. 2021, 143, 20697–20709. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://cdn.dwi.gov.uk/wp-content/uploads/2020/10/27111054/DWI70_2_243.pdf (accessed on 12 December 2022).
- Yamane, D.; Onitsuka, S.; Re, S.; Isogai, H.; Hamada, R.; Hiramoto, T.; Kawanishi, E.; Mizuguchi, K.; Shindo, N.; Ojida, A. Selective covalent targeting of SARS-CoV-2 main protease by enantiopure chlorofluoroacetamide. Chem. Sci. 2022, 13, 3027–3034. [Google Scholar] [CrossRef] [PubMed]
- Moon, P.; Boike, L.; Dovala, D.; Henning, N.J.; Knapp, M.; Spradlin, J.N.; Ward, C.C.; Wolleb, H.; Zammit, C.M.; Fuller, D.; et al. Discovery of Potent Pyrazoline-Based Covalent SARS-CoV-2 Main Protease Inhibitors. bioRxiv 2022. [Google Scholar] [CrossRef]
- Xiong, M.; Nie, T.; Shao, Q.; Li, M.; Su, H.; Xu, Y. In silico screening-based discovery of novel covalent inhibitors of the SARS-CoV-2 3CL protease. Eur. J. Med. Chem. 2022, 231, 114130. [Google Scholar] [CrossRef]
- Ettari, R.; Bonaccorso, C.; Micale, N.; Heindl, C.; Schirmeister, T.; Calabrò, M.L.; Grasso, S.; Zappalà, M. Development of Novel Peptidomimetics Containing a Vinyl Sulfone Moiety as Proteasome Inhibitors. ChemMedChem 2011, 6, 1228–1237. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Raghavaiah, J.; Shahabi, D.; Yadav, M.; Anson, B.J.; Lendy, E.K.; Hattori, S.; Higashi-Kuwata, N.; Mitsuya, H.; Mesecar, A.D. Indole Chloropyridinyl Ester-Derived SARS-CoV-2 3CLpro Inhibitors: Enzyme Inhibition, Antiviral Efficacy, Structure–Activity Relationship, and X-ray Structural Studies. J. Med. Chem. 2021, 64, 14702–14714. [Google Scholar] [CrossRef]
- Breidenbach, J.; Lemke, C.; Pillaiyar, T.; Schäkel, L.; Al Hamwi, G.; Diett, M.; Gedschold, R.; Geiger, N.; Lopez, V.; Mirza, S.; et al. Targeting the Main Protease of SARS-CoV-2: From the Establishment of High Throughput Screening to the Design of Tailored Inhibitors. Angew. Chem. Int. Ed. Engl. 2021, 60, 10423–10429. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Shahabi, D.; Yadav, M.; Kovela, S.; Anson, B.J.; Lendy, E.K.; Bonham, C.; Sirohi, D.; Brito-Sierra, C.A.; Hattori, S.I.; et al. Chloropyridinyl Esters of Nonsteroidal Anti-Inflammatory Agents and Related Derivatives as Potent SARS-CoV-2 3CL Protease Inhibitors. Molecules 2021, 26, 5782. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef]
- Dai, W.; Jochmans, D.; Xie, H.; Yang, H.; Li, J.; Su, H.; Chang, D.; Wang, J.; Peng, J.; Zhu, L.; et al. Design, Synthesis, and Biological Evaluation of Peptidomimetic Aldehydes as Broad-Spectrum Inhibitors against Enterovirus and SARS-CoV-2. J. Med. Chem. 2022, 65, 2794–2808. [Google Scholar] [CrossRef]
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; Van Belkum, M.J.; Joyce, M.A.; et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef] [PubMed]
- Cáceres, C.J.; Cardenas-Garcia, S.; Carnaccini, S.; Seibert, B.; Rajao, D.S.; Wang, J.; Perez, D.R. Efficacy of GC-376 against SARS-CoV-2 virus infection in the K18 hACE2 transgenic mouse model. Sci. Rep. 2021, 11, 9609. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Shuai, L.; Wen, Z.; Wang, C.; Yan, Y.; Jiao, Z.; Guo, F.; Fu, Z.F.; Chen, H.; Bu, Z.; et al. The preclinical inhibitor GS441524 in combination with GC376 efficaciously inhibited the proliferation of SARS-CoV-2 in the mouse respiratory tract. Emerg. Microbes Infect. 2021, 10, 481–492. [Google Scholar] [CrossRef]
- Arutyunova, E.; Khan, M.B.; Fischer, C.; Lu, J.; Lamer, T.; Vuong, W.; van Belkum, M.J.; McKay, R.T.; Tyrrell, D.L.; Vederas, J.C.; et al. N-Terminal Finger Stabilizes the S1 Pocket for the Reversible Feline Drug GC376 in the SARS-CoV-2 Mpro Dimer. J. Mol. Biol. 2021, 433, 167003. [Google Scholar] [CrossRef]
- Vuong, W.; Fischer, C.; Khan, M.B.; Van Belkum, M.J.; Lamer, T.; Willoughby, K.D.; Lu, J.; Arutyunova, E.; Joyce, M.A.; Saffran, H.A.; et al. Improved SARS-CoV-2 Mpro inhibitors based on feline antiviral drug GC376: Structural enhancements, increased solubility, and micellar studies. Eur. J. Med. Chem. 2021, 222, 113584. [Google Scholar] [CrossRef]
- Sacco, M.D.; Ma, C.; Lagarias, P.; Gao, A.; Townsend, J.A.; Meng, X.; Dube, P.; Zhang, X.; Hu, Y.; Kitamura, N.; et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci. Adv. 2020, 6, eabe0751. [Google Scholar] [CrossRef]
- Liu, H.; Iketani, S.; Zask, A.; Khanizeman, N.; Bednarova, E.; Forouhar, F.; Fowler, B.; Hong, S.J.; Mohri, H.; Nair, M.S.; et al. Development of optimized drug-like small molecule inhibitors of the SARS-CoV-2 3CL protease for treatment of COVID-19. Nat. Commun. 2022, 13, 1891. [Google Scholar] [CrossRef]
- Dampalla, C.S.; Kim, Y.; Bickmeier, N.; Rathnayake, A.D.; Nguyen, H.N.; Zheng, J.; Kashipathy, M.M.; Baird, M.A.; Battaile, K.P.; Lovell, S.; et al. Structure-Guided Design of Conformationally Constrained Cyclohexane Inhibitors of Severe Acute Respiratory Syndrome Coronavirus-2 3CL Protease. J. Med. Chem. 2021, 64, 10047–10058. [Google Scholar] [CrossRef]
- Xia, Z.; Sacco, M.; Hu, Y.; Ma, C.; Meng, X.; Zhang, F.; Szeto, T.; Xiang, Y.; Chen, Y.; Wang, J. Rational Design of Hybrid SARS-CoV-2 Main Protease Inhibitors Guided by the Superimposed Cocrystal Structures with the Peptidomimetic Inhibitors GC-376, Telaprevir, and Boceprevir. ACS Pharmacol. Transl. Sci. 2021, 4, 1408–1421. [Google Scholar] [CrossRef]
- Qiao, J.; Li, Y.S.; Zeng, R.; Liu, F.L.; Luo, R.H.; Huang, C.; Wang, Y.F.; Zhang, J.; Quan, B.; Shen, C.; et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371, 1374–1378. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.S.; Ma, X.R.; Ma, Y.; Alugubelli, Y.R.; Scott, D.A.; Vatansever, E.C.; Drelich, A.K.; Sankaran, B.; Geng, Z.Z.; Blankenship, L.R.; et al. A Quick Route to Multiple Highly Potent SARS-CoV-2 Main Protease Inhibitors. ChemMedChem 2021, 16, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.R.; Alugubelli, Y.R.; Ma, Y.; Vatansever, E.C.; Scott, D.A.; Qiao, Y.; Yu, G.; Xu, S.; Liu, W.R. MPI8 is Potent against SARS-CoV-2 by Inhibiting Dually and Selectively the SARS-CoV-2 Main Protease and the Host Cathepsin L. ChemMedChem 2022, 17, e202100456. [Google Scholar] [CrossRef] [PubMed]
- Costanzi, E.; Kuzikov, M.; Esposito, F.; Albani, S.; Demitri, N.; Giabbai, B.; Camasta, M.; Tramontano, E.; Rossetti, G.; Zaliani, A.; et al. Structural and Biochemical Analysis of the Dual Inhibition of MG-132 against SARS-CoV-2 Main Protease (Mpro/3CLpro) and Human Cathepsin-L. Int. J. Mol. Sci. 2021, 22, 11779. [Google Scholar] [CrossRef]
- Kuzikov, M.; Costanzi, E.; Reinshagen, J.; Esposito, F.; Vangeel, L.; Wolf, M.; Ellinger, B.; Claussen, C.; Geisslinger, G.; Corona, A.; et al. Identification of Inhibitors of SARS-CoV-2 3CL-Pro Enzymatic Activity Using a Small Molecule in vitro Repurposing Screen. ACS Pharmacol. Transl. Sci. 2021, 4, 1096–1110. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Hu, Y.; Ma, C.; Szeto, T.; Hurst, B.; Tarbet, B.; Wang, J. Boceprevir, Calpain Inhibitors II and XII, and GC-376 Have Broad-Spectrum Antiviral Activity against Coronaviruses. ACS Infect. Dis. 2021, 7, 586–597. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; Von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Oerlemans, R.; Ruiz-Moreno, A.J.; Cong, Y.; Dinesh Kumar, N.; Velasco-Velazquez, M.A.; Neochoritis, C.G.; Smith, J.; Reggiori, F.; Groves, M.R.; Dömling, A. Repurposing the HCV NS3-4A protease drug boceprevir as COVID-19 therapeutics. RSC Med. Chem. 2020, 12, 370–379. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; Weiss, K.L.; Zhang, Q.; Coates, L.; Kovalevsky, A. Direct Observation of Protonation State Modulation in SARS-CoV-2 Main Protease upon Inhibitor Binding with Neutron Crystallography. J. Med. Chem. 2021, 64, 4991–5000. [Google Scholar] [CrossRef]
- Kneller, D.W.; Galanie, S.; Phillips, G.; O’Neill, H.M.; Coates, L.; Kovalevsky, A. Malleability of the SARS-CoV-2 3CL Mpro active-site cavity facilitates binding of clinical antivirals. Structure 2020, 28, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.; Mostafa, A.; Al-Karmalawy, A.A.; Zidan, A.; Abulkhair, H.S.; Mahmoud, S.H.; Shehata, M.; Elhefnawi, M.M.; Ali, M.A. Telaprevir is a potential drug for repurposing against SARS-CoV-2: Computational and in vitro studies. Heliyon 2021, 7, e07962. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liang, B.; Chen, Y.; Fuk-Woo Chan, J.; Yuan, S.; Ye, H.; Nie, L.; Zhou, J.; Wu, Y.; Wu, M.; et al. A new class of α-ketoamide derivatives with potent anticancer and anti-SARS-CoV-2 activities. Eur. J. Med. Chem. 2021, 215, 113267. [Google Scholar] [CrossRef] [PubMed]
- Chuck, C.; Chen, C.; Ke, Z.; Wan, D.C.; Chow, H.; Wong, K. Design, synthesis and crystallographic analysis of nitrile-based broad-spectrum peptidomimetic inhibitors for coronavirus 3C-like proteases. Eur. J. Med. Chem. 2013, 59, 1–6. [Google Scholar] [CrossRef]
- Halford, B. The Path to Paxlovid. ACS Cent. Sci. 2022, 8, 405–407. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Zhao, Y.; Fang, C.; Zhang, Q.; Zhang, R.; Zhao, X.; Duan, Y.; Wang, H.; Zhu, Y.; Feng, L.; Zhao, J.; et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell 2022, 13, 689–693. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Foo, C.S.; Jochmans, D.; Vangeel, L.; De Jonghe, S.; Augustijns, P.; Mols, R.; Weynand, B.; Wattanakul, T.; Hoglund, R.M.; et al. The oral protease inhibitor (PF-07321332) protects syrian hamsters against infection with SARS-CoV-2 variants of concern. Nat. Commun. 2022, 13, 719. [Google Scholar] [CrossRef]
- Li, P.; Wang, Y.; Lavrijsen, M.; Lamers, M.M.; De Vries, A.C.; Rottier, R.J.; Bruno, M.J.; Peppelenbosch, M.P.; Haagmans, B.L.; Pan, Q. SARS-CoV-2 Omicron variant is highly sensitive to molnupiravir, nirmatrelvir, and the combination. Cell Res. 2022, 32, 322–324. [Google Scholar] [CrossRef]
- Rai, D.K.; Yurgelonis, I.; McMonagle, P.; Rothan, H.A.; Hao, L.; Gribenko, A.; Titova, E.; Kreiswirth, B.; White, K.M.; Zhu, Y.; et al. Nirmatrelvir, an orally active Mpro inhibitor, is a potent inhibitor of SARS-CoV-2 variants of concern. bioRxiv 2022. [Google Scholar] [CrossRef]
- Greasley, S.E.; Noell, S.; Plotnikova, O.; Ferre, R.; Liu, W.; Bolanos, B.; Fennell, K.; Nicki, J.; Craig, T.; Zhu, Y.; et al. Structural basis for the in vitro efficacy of nirmatrelvir against SARS-CoV-2 variants. J. Biol. Chem. 2022, 298, 101972. [Google Scholar] [CrossRef] [PubMed]
- Rosales, R.; McGovern, B.L.; Rodriguez, M.L.; Rai, D.K.; Cardin, R.D.; Anderson, A.S.; PSP Study Group; Sordillo, E.M.; van Bakel, H.; Simon, V.; et al. Nirmatrelvir, molnupiravir, and remdesivir maintain potent in vitro activity against the SARS-CoV-2 Omicron variant. bioRxiv 2022. [Google Scholar] [CrossRef]
- Singh, R.S.P.; Toussi, S.S.; Hackman, F.; Chan, P.L.; Rao, R.; Allen, R.; Van Eyck, L.; Pawlak, S.; Kadar, E.P.; Clark, F.; et al. Innovative Randomized Phase I Study and Dosing Regimen Selection to Accelerate and Inform Pivotal COVID-19 Trial of Nirmatrelvir. Clin. Pharmacol. Ther. 2022, 112, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Toussi, S.S.; Neutel, J.M.; Navarro, J.; Preston, R.A.; Shi, H.; Kavetska, O.; LaBadie, R.R.; Binks, M.; Chan, P.L.S.; Demers, N.; et al. Pharmacokinetics of Oral Nirmatrelvir/Ritonavir, a Protease Inhibitor for Treatment of COVID-19, in Subjects with Renal Impairment. Clin. Pharmacol. Ther. 2022, 112, 892–900. [Google Scholar] [CrossRef]
- Owen, D.R.; Pettersson, M.Y.; Reese, M.R.; Sammons, M.F.; Tuttle, J.B.; Verhoest, P.R.; Wei, L.; Yang, Q.; Yang, X. Nitrile-Containing Antiviral Compounds, World Intellectual Property Organization International. Bureau Patent WO 2021/250648A1, 16 December 2021. [Google Scholar]
- Bai, B.; Arutyunova, E.; Khan, M.B.; Lu, J.; Joyce, M.A.; Saffran, H.A.; Shields, J.A.; Kandadai, A.S.; Belovodskiy, A.; Hena, M.; et al. Peptidomimetic nitrile warheads as SARS-CoV-2 3CL protease inhibitors. RSC Med. Chem. 2021, 12, 1722–1730. [Google Scholar] [CrossRef] [PubMed]
- Amporndanai, K.; Meng, X.; Shang, W.; Jin, Z.; Rogers, M.; Zhao, Y.; Rao, Z.; Liu, Z.J.; Yang, H.; Zhang, L.; et al. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 2021, 12, 3061. [Google Scholar] [CrossRef]
- Madabeni, A.; Nogara, P.A.; Omage, F.B.; Rocha, J.B.T.; Orian, L. Mechanistic insight into SARS-CoV-2 Mpro inhibition by organoselenides: The ebselen case study. Appl. Sci. 2021, 11, 6291. [Google Scholar] [CrossRef]
- Parise, A.; Romeo, I.; Russo, N.; Marino, T. The Se-S bond formation in the covalent inhibition mechanism of SARS-CoV-2 main protease by ebselen-like inhibitors: A computational study. Int. J. Mol. Sci. 2021, 22, 9792. [Google Scholar] [CrossRef]
- Yang, J.; Lin, X.; Xing, N.; Zhang, Z.; Zhang, H.; Wu, H.; Xue, W. Structure-Based Discovery of Novel Nonpeptide Inhibitors Targeting SARS-CoV-2 Mpro. J. Chem. Inf. Model. 2021, 61, 3917–3926. [Google Scholar] [CrossRef]
- Han, S.H.; Goins, C.M.; Arya, T.; Shin, W.J.; Maw, J.; Hooper, A.; Sonawane, D.P.; Porter, M.R.; Bannister, B.E.; Crouch, R.D.; et al. Structure-Based Optimization of ML300-Derived, Noncovalent Inhibitors Targeting the Severe Acute Respiratory Syndrome Coronavirus 3CL Protease (SARS-CoV-2 3CLpro). J. Med. Chem. 2022, 65, 2880–2904. [Google Scholar] [CrossRef]
- Elseginy, S.A.; Fayed, B.; Hamdy, R.; Mahrous, N.; Mostafa, A.; Almehdi, A.M.; Soliman, S.S.M. Promising anti-SARS-CoV-2 drugs by effective dual targeting against the viral and host proteases. Bioorg. Med. Chem. Lett. 2021, 43, 128099. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.H.; Stone, E.A.; Deshmukh, M.; Ippolito, J.A.; Ghahremanpour, M.M.; Tirado-Rives, J.; Spasov, K.A.; Zhang, S.; Takeo, Y.; Kudalkar, S.N.; et al. Potent Noncovalent Inhibitors of the Main Protease of SARS-CoV-2 from Molecular Sculpting of the Drug Perampanel Guided by Free Energy Perturbation Calculations. ACS Cent. Sci. 2021, 7, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.; Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.; et al. Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19. J. Med. Chem. 2022, 65, 6499–6512. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Tabata, K.; Kishimoto, M.; Itakura, Y.; Kobayashi, H.; Ariizumi, T.; Uemura, K.; Toba, S.; Kusakabe, S.; Maruyama, Y.; et al. S-217622, a SARS-CoV-2 main protease inhibitor, decreases viral load and ameliorates COVID-19 severity in hamsters. Sci. Transl. Med. 2022, 15, eabq4064. [Google Scholar] [CrossRef]
- Alzyoud, L.; Ghattas, M.A.; Atatreh, N. Allosteric Binding Sites of the SARS-CoV-2 Main Protease: Potential Targets for Broad-Spectrum Anti-Coronavirus Agents. Drug Des. Devel. Ther. 2022, 16, 2463–2478. [Google Scholar] [CrossRef]
- Günther, S.; Reinke, P.Y.A.; Fernández-García, Y.; Lieske, J.; Lane, T.J.; Ginn, H.M.; Koua, F.H.M.; Ehrt, C.; Ewert, W.; Oberthuer, D.; et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 2021, 372, 642–646. [Google Scholar] [CrossRef]
- Safety and Efficacy of NP-120 (Ifenprodil) for the Treatment of Hospitalized Patient with Confirmed COVID-19 Disease. Available online: https://beta.clinicaltrials.gov/study/NCT04382924?tab=results (accessed on 12 December 2022).
- Xu, J.; Shi, P.Y.; Li, H.; Zhou, J. Broad Spectrum Antiviral Agent Niclosamide and Its Therapeutic Potential. ACS Infect. Dis. 2020, 6, 909–915. [Google Scholar] [CrossRef]
- Samrat, S.K.; Xu, J.; Xie, X.; Gianti, E.; Chen, H.; Zou, J.; Pattis, J.G.; Elokely, K.; Lee, H.; Li, Z.; et al. Allosteric inhibitors of the main protease of SARS-CoV-2. Antiviral Res. 2022, 205, 105381. [Google Scholar] [CrossRef]
- Cairns, D.M.; Dulko, D.; Griffiths, J.K.; Golan, Y.; Cohen, T.; Trinquart, L.; Price, L.L.; Beaulac, K.R.; Selker, H.P. Efficacy of Niclosamide vs Placebo in SARS-CoV-2 Respiratory Viral Clearance, Viral Shedding, and Duration of Symptoms Among Patients with Mild to Moderate COVID-19: A Phase 2 Randomized Clinical Trial. JAMA Netw. Open. 2022, 5, e2144942. [Google Scholar] [CrossRef]
- Chaves, O.A.; Sacramento, C.Q.; Fintelman-Rodrigues, N.; Temerozo, J.R.; Pereira-Dutra, F.; Mizurini, D.M.; Monteiro, R.Q.; Vazquez, L.; Bozza, P.T.; Castro-Faria-Neto, H.C.; et al. Apixaban, an orally available anticoagulant, inhibits SARS-CoV-2 replication and its major protease in a non-competitive way. J. Mol. Cell Biol. 2022, 14, mjac039. [Google Scholar] [CrossRef]
- Mahgoub, R.E.; Mohamed, F.E.; Alzyoud, L.; Ali, B.R.; Ferreira, J.; Rabeh, W.M.; AlNeyadi, S.S.; Atatreh, N.; Ghattas, M.A. The Discovery of Small Allosteric and Active Site Inhibitors of the SARS-CoV-2 Main Protease via Structure-Based Virtual Screening and Biological Evaluation. Molecules 2022, 27, 6710. [Google Scholar] [CrossRef]
- Du, R.; Cooper, L.; Chen, Z.; Lee, H.; Rong, L.; Cui, Q. Discovery of chebulagic acid and punicalagin as novel allosteric inhibitors of SARS-CoV-2 3CLpro. Antiviral Res. 2021, 190, 105075. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Zhang, L.; Du, L.; Liao, R.; Cai, H.; Lu, K.; Zhao, Z.; Xie, Y.; Wang, P.H.; Pan, J.A.; et al. Allosteric inhibition of SARS-CoV-2 3CL protease by colloidal bismuth subcitrate. Chem. Sci. 2021, 12, 14098–14102. [Google Scholar] [CrossRef] [PubMed]
- El-Baba, T.J.; Lutomski, C.A.; Kantsadi, A.L.; Malla, T.R.; John, T.; Mikhailov, V.; Bolla, J.R.; Schofield, C.J.; Zitzmann, N.; Vakonakis, I.; et al. Allosteric Inhibition of the SARS-CoV-2 Main Protease: Insights from Mass Spectrometry Based Assays. Angew. Chem. Int. Ed. 2020, 59, 23544–23548. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, L.; Li, X.; Fan, C.; Xu, J.; Shi, Z.; Qiao, H.; Lan, Z.; Zhang, X.; Li, L.; et al. An extended conformation of SARS-CoV-2 main protease reveals allosteric targets. Proc. Natl. Acad. Sci. USA 2022, 119, e2120913119. [Google Scholar] [CrossRef]
- Brüssow, H. Clinical trials with antiviral drugs against COVID-19: Some progress and many shattered hopes. Environ. Microbiol. 2021, 23, 6364–6376. [Google Scholar] [CrossRef]
- Cully, M. A tale of two antiviral targets—And the COVID-19 drugs that bind them. Nat. Rev. Drug Discov. 2022, 21, 3–5. [Google Scholar] [CrossRef]
- COVID-19 Therapeutics Thresholds, Orders, and Replenishment by Jurisdiction. Available online: https://aspr.hhs.gov/COVID-19/Therapeutics/Orders/Pages/default.aspx (accessed on 12 December 2022).
- Wen, W.; Chen, C.; Tang, J.; Wang, C.; Zhou, M.; Cheng, Y.; Zhou, X.; Wu, Q.; Zhang, X.; Feng, Z.; et al. Efficacy and safety of three new oral antiviral treatment (molnupiravir, fluvoxamine and Paxlovid) for COVID-19: A meta-analysis. Ann. Med. 2022, 54, 516–523. [Google Scholar] [CrossRef]
- Najjar-Debbiny, R.; Gronich, N.; Weber, G.; Khoury, J.; Amar, M.; Stein, N.; Goldstein, L.H.; Saliba, W. Effectiveness of Paxlovid in Reducing Severe COVID-19 and Mortality in High-Risk Patients. Clin. Infect. Dis. 2022, ciac443. [Google Scholar] [CrossRef]
- Heilmann, E.; Costacurta, F.; Moghadasi, S.A.; Ye, C.; Pavan, M.; Bassani, D.; Volland, A.; Ascher, C.; Weiss, A.K.H.; Bante, D.; et al. SARS-CoV-2 3CLpro mutations selected in a VSV-based system confer resistance to nirmatrelvir, ensitrelvir, and GC376. Sci. Transl. Med. 2022, eabq7360. [Google Scholar] [CrossRef]
- Lowe, D. Paxlovid Resistance: Is It Just a Matter of Time Now? | Science | AAAS. Available online: https://www.science.org/content/blog-post/paxlovid-resistance-it-just-matter-time-now (accessed on 12 December 2022).
- Pandit, J.A.; Radin, J.M.; Chiang, D.; Spencer, E.; Pawelek, J.; Diwan, M.; Roumani, L.; Mina, M. The paxlovid rebound study: A prospective cohort study to evaluate viral and symptom rebound differences between Paxlovid and untreated COVID-19 participants. medRxiv 2022. [Google Scholar] [CrossRef]
- Carlin, A.F.; Clark, A.E.; Chaillon, A.; Garretson, A.F.; Bray, W.; Porrachia, M.; Santos, A.L.T.; Rana, T.M.; Smith, D.M. Virologic and immunologic characterization of coronavirus diseases 2019 recrudescence after nirmatrelvir/ritonavir treatment. Clin. Infect. Dis. 2022, ciac496. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Z.; Yan, Y.; Chen, Y.; Wang, G.; Wang, X.; Li, L.; Yang, M.; Hu, X.; Guo, Y.; Shi, Y.; et al. Adaptive Mutation in the Main Protease Cleavage Site of Feline Coronavirus Renders the Virus More Resistant to Main Protease Inhibitors. J. Virol. 2022, 96, e0090722. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, P.; Lavrijsen, M.; Li, Y.; Ma, Z.; Peppelenbosch, M.P.; Baig, M.S.; Pan, Q. Differing pan-coronavirus antiviral potency of boceprevir and GC376 in vitro despite discordant molecular docking predictions. Arch. Virol. 2022, 167, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- A Study to Compare S-217622 with Placebo in Non-Hospitalized Participants with COVID-19 (SCORPIO-HR). Available online: https://clinicaltrials.gov/ct2/show/NCT05305547 (accessed on 12 December 2022).
- The COVID Moonshot Consortium; Achdout, H.; Aimon, A.; Bar-David, E.; Barr, H.; Ben-Shmuel, A.; Bennett, J.; Bobby, M.L.; Borden, B.; Bowman, G.R.; et al. Open science discovery of oral non-covalent SARS-CoV-2 main protease inhibitor therapeutics. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kulkarni, S.A.; Ingale, K. In silico approaches for drug repurposing for SARS-CoV-2 infection. In The Coronavirus Pandemic and the Future: Virology, Epidemiology, Translational Toxicology and Therapeutics, 1st ed.; Waters, M.D., Dhawan, A., Marrs, T., Anderson, D., Warren, S., Hughes, C.L., Eds.; RSC Publishing: Croydon, UK, 2022; Volume 2, pp. 1–80. [Google Scholar]
- Gidari, A.; Sabbatini, S.; Schiaroli, E.; Bastianelli, S.; Pierucci, S.; Busti, C.; Comez, L.; Libera, V.; Macchiarulo, A.; Paciaroni, A.; et al. The Combination of Molnupiravir with Nirmatrelvir or GC376 Has a Synergic Role in the Inhibition of SARS-CoV-2 Replication In Vitro. Microorganisms 2022, 10, 1475. [Google Scholar] [CrossRef] [PubMed]
- Wagoner, J.; Herring, S.; Hsiang, T.Y.; Ianevski, A.; Biering, S.B.; Xu, S.; Hoffmann, M.; Pöhlmann, S.; Gale, M., Jr.; Aittokallio, T.; et al. Combinations of Host- and Virus-Targeting Antiviral Drugs Confer Synergistic Suppression of SARS-CoV-2. Microbiol. Spectr. 2022, 10, e0333122. [Google Scholar] [CrossRef]
- Vankadara, S.; Dawson, M.D.; Fong, J.Y.; Oh, Q.Y.; Ang, Q.A.; Liu, B.; Chang, H.Y.; Koh, J.; Koh, X.; Tan, Q.W.; et al. A Warhead Substitution Study on the Coronavirus Main Protease Inhibitor Nirmatrelvir. ACS Med. Chem. Lett. 2022, 13, 1345–1350. [Google Scholar]
- Vázquez-Mendoza, L.H.; Mendoza-Figueroa, H.L.; García-Vázquez, J.B.; Correa-Basurto, J.; García-Machorro, J. In Silico Drug Repositioning to Target the SARS-CoV-2 Main Protease as Covalent Inhibitors Employing a Combined Structure-Based Virtual Screening Strategy of Pharmacophore Models and Covalent Docking. Int. J. Mol. Sci. 2022, 23, 3987. [Google Scholar] [CrossRef]
- Williams, R. Improved Formulations to Enable Stable Delivery of Biologics. BioPharm. Int. 2022, 35, 46–49. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, C.; Feys, J.R. SARS-CoV-2 Mpro Inhibitors: Achieved Diversity, Developing Resistance and Future Strategies. Future Pharmacol. 2023, 3, 80-107. https://doi.org/10.3390/futurepharmacol3010006
Fischer C, Feys JR. SARS-CoV-2 Mpro Inhibitors: Achieved Diversity, Developing Resistance and Future Strategies. Future Pharmacology. 2023; 3(1):80-107. https://doi.org/10.3390/futurepharmacol3010006
Chicago/Turabian StyleFischer, Conrad, and Jenson R. Feys. 2023. "SARS-CoV-2 Mpro Inhibitors: Achieved Diversity, Developing Resistance and Future Strategies" Future Pharmacology 3, no. 1: 80-107. https://doi.org/10.3390/futurepharmacol3010006
APA StyleFischer, C., & Feys, J. R. (2023). SARS-CoV-2 Mpro Inhibitors: Achieved Diversity, Developing Resistance and Future Strategies. Future Pharmacology, 3(1), 80-107. https://doi.org/10.3390/futurepharmacol3010006