
Unraveling Drug Response from Pharmacogenomic Data to Advance Systems Pharmacology Decisions in Tumor Therapeutics

 ,
,  ,
,  and
and
Abstract
:1. Introduction
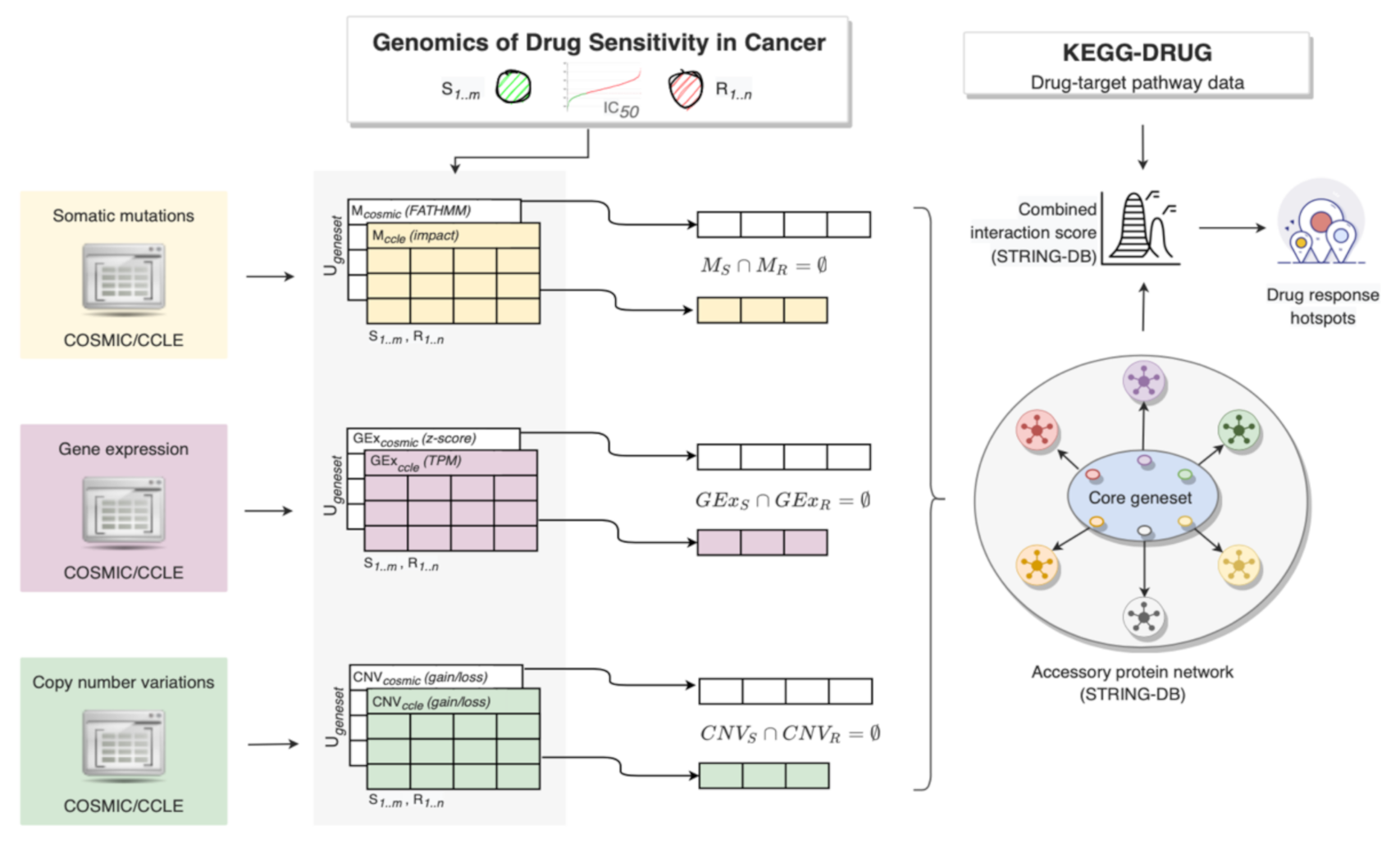
2. Materials and Methods
2.1. Cancer Cell Line Selection
2.2. Dinstict Mutation, Gene Expression and Copy Number Profiles
2.2.1. Mutation Profiles
2.2.2. Gene Expression Profiles
2.2.3. Copy Number Profiles
2.3. Interaction Network with Anticancer Drug Targets
3. Results
3.1. Breast Cancer and Afatinib
3.1.1. Breast Cancer Cell Lines with Extreme Responses
3.1.2. High-Confidence Drug-Response Gene Markers
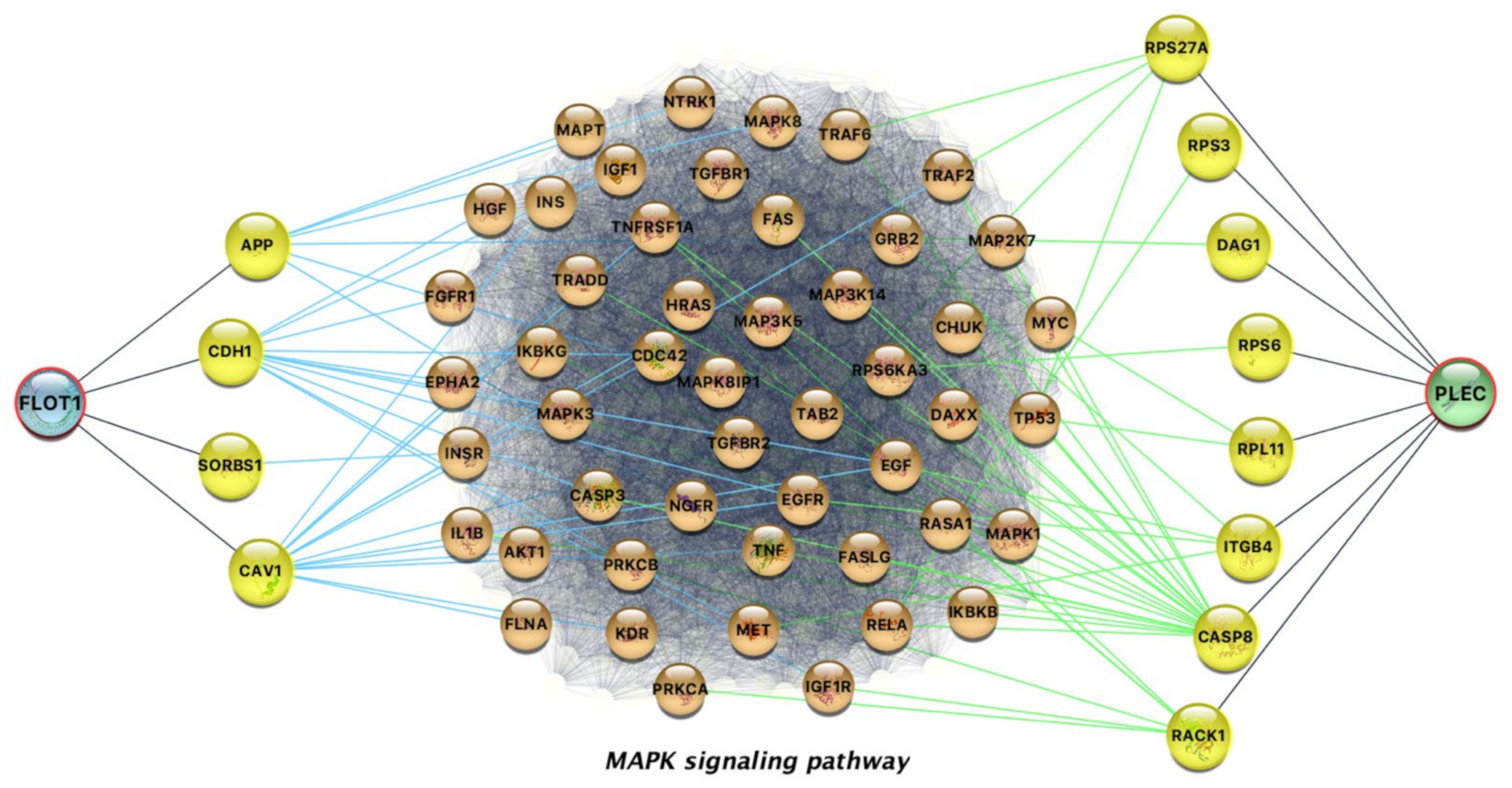
3.1.3. Interpretability of the Afatinib-Gene Interactions
3.2. Glioblastoma Multiforme and Trametinib
3.2.1. Glioblastoma Resistant and Sensitive Cancer Cell Lines
3.2.2. High-Confidence Trametinib-Response Gene Markers
3.3. Skin Cutaneous Melanoma and Dabrafenib
3.3.1. Dabrafenib’s Sensitive and Resistant Skin Cutaneous Melanoma Cell Lines
3.3.2. High-Confidence Dabrafenib-Response Gene Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vizirianakis, I.S.; Miliotou, A.N.; Mystridis, G.A.; Andriotis, E.G.; Andreadis, I.I.; Papadopoulou, L.C.; Fatouros, D.G. Tackling pharmacological response heterogeneity by PBPK modeling to advance precision medicine productivity of nanotechnology and genomics therapeutics. Expert Rev. Precis. Med. Drug Dev. 2019, 4, 139–151. [Google Scholar] [CrossRef]
- Vizirianakis, I.; Fatouros, D.G. Personalized nanomedicine: Paving the way to the practical clinical utility of genomics and nanotechnology advancements. Adv. Drug Deliv. Rev. 2012, 64, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Kinch, M.S. An analysis of FDA-approved drugs for oncology. Drug Discov. Today 2014, 19, 1831–1835. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Russell, L.E.; Zhou, Y.; Almousa, A.A.; Sodhi, J.K.; Nwabufo, C.K.; Lauschke, V.M. Pharmacogenomics in the Era of next Generation Sequencing–from Byte to Bedside. Drug Metab. Rev. 2021, 53, 253–278. [Google Scholar] [CrossRef]
- Pritchard, D.E.; Moeckel, F.; Villa, M.S.; Housman, L.T.; McCarty, C.A.; McLeod, H.L. Strategies for integrating personalized medicine into healthcare practice. Pers. Med. 2017, 14, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- Wang, W.; Malyutina, A.; Pessia, A.; Saarela, J.; Heckman, C.A.; Tang, J. Combined gene essentiality scoring improves the prediction of cancer dependency maps. EBioMedicine 2019, 50, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Li, H.; Li, S.; Leung, K.-S. Improving prediction of phenotypic drug response on cancer cell lines using deep convolutional network. BMC Bioinform. 2019, 20, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Dorman, S.N.; Baranova, K.; Knoll, J.H.; Urquhart, B.L.; Mariani, G.; Carcangiu, M.L.; Rogan, P.K. Genomic signatures for paclitaxel and gemcitabine resistance in breast cancer derived by machine learning. Mol. Oncol. 2015, 10, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Tang, T.; Zhang, B.; Li, G. A Prognostic Signature Based on Immunogenomic Profiling Offers Guidance for Esophageal Squamous Cell Cancer Treatment. Front. Oncol. 2021, 11, 603634. [Google Scholar] [CrossRef]
- Li, Y.; Umbach, D.M.; Krahn, J.M.; Shats, I.; Li, X.; Li, L. Predicting tumor response to drugs based on gene-expression biomarkers of sensitivity learned from cancer cell lines. BMC Genom. 2021, 22, 1–18. [Google Scholar] [CrossRef]
- Sotudian, S.; Paschalidis, I.C. Machine Learning for Pharmacogenomics and Personalized Medicine: A Ranking Model for Drug Sensitivity Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 1. [Google Scholar] [CrossRef]
- Kusch, N.; Schuppert, A. Two-step multi-omics modelling of drug sensitivity in cancer cell lines to identify driving mechanisms. PLoS ONE 2020, 15, e0238961. [Google Scholar] [CrossRef]
- Tognetti, M.; Gabor, A.; Yang, M.; Cappelletti, V.; Windhager, J.; Rueda, O.M.; Charmpi, K.; Esmaeilishirazifard, E.; Bruna, A.; de Souza, N.; et al. Deciphering the signaling network of breast cancer improves drug sensitivity prediction. Cell Syst. 2021, 12, 401–418. [Google Scholar] [CrossRef]
- Kim, P.; Li, H.; Wang, J.; Zhao, Z. Landscape of drug-resistance mutations in kinase regulatory hotspots. Briefings Bioinform. 2021, 22, bbaa108. [Google Scholar] [CrossRef]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. {COSMIC}: Exploring the World’s Knowledge of Somatic Mutations in Human Cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef]
- A Shihab, H.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.; Gaunt, T.R. Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum. Genom. 2014, 8, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., III; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dungo, R.T.; Keating, G.M. Afatinib: First Global Approval. Drugs 2013, 73, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Wind, S.; Schnell, D.; Ebner, T.; Freiwald, M.; Stopfer, P. Clinical Pharmacokinetics and Pharmacodynamics of Afatinib. Clin. Pharmacokinet. 2017, 56, 235–250. [Google Scholar] [CrossRef] [Green Version]
- Wecker, H.; Waller, C.F. Afatinib. Recent Results in Cancer Research; Martens, U., Ed.; Springer: Cham, Switzerland, 2018; Volume 211, pp. 199–215. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Young, N.R.; Soneru, C.; Liu, J.; Grushko, T.A.; Hardeman, A.; Olopade, O.I.; Baum, A.; Solca, F.; Cohen, E. Afatinib efficacy against squamous cell carcinoma of the head and neck cell lines in vitro and in vivo. Target. Oncol. 2015, 10, 501–508. [Google Scholar] [CrossRef] [Green Version]
- Ebert, K.; Zwingenberger, G.; Barbaria, E.; Keller, S.; Heck, C.; Arnold, R.; Hollerieth, V.; Mattes, J.; Geffers, R.; Raimúndez, E.; et al. Determining the effects of trastuzumab, cetuximab and afatinib by phosphoprotein, gene expression and phenotypic analysis in gastric cancer cell lines. BMC Cancer 2020, 20, 1–19. [Google Scholar] [CrossRef]
- Hanker, A.; Brewer, M.R.; Sheehan, J.H.; Koch, J.P.; Sliwoski, G.R.; Nagy, R.; Lanman, R.; Berger, M.F.; Hyman, D.M.; Solit, D.B.; et al. An Acquired HER2T798I Gatekeeper Mutation Induces Resistance to Neratinib in a Patient with HER2 Mutant–Driven Breast Cancer. Cancer Discov. 2017, 7, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Tamura, S.; Wang, Y.; Veeneman, B.; Hovelson, D.; Bankhead, A.; Broses, L.J.; Hiles, G.L.; Liebert, M.; Rubin, J.R.; Day, K.C.; et al. Molecular Correlates of In Vitro Responses to Dacomitinib and Afatinib in Bladder Cancer. Bl. Cancer 2018, 4, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, S.K.; Cabrera, R.; Mao, S.P.; Christin, J.R.; Wu, B.; Guo, W.; Bravo-Cordero, J.J.; Condeelis, J.S.; Segall, J.E.; Hodgson, L. Rac3 regulates breast cancer invasion and metastasis by controlling adhesion and matrix degradation. J. Cell Biol. 2017, 216, 4331–4349. [Google Scholar] [CrossRef] [PubMed]
- Kavarthapu, R.; Anbazhagan, R.; Dufau, M.L. Crosstalk between PRLR and EGFR/HER2 Signaling Pathways in Breast Cancer. Cancers 2021, 13, 4685. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McGee, J.; Chen, X.; Doman, T.N.; Gong, X.; Zhang, Y.; Hamm, N.; Ma, X.; Higgs, R.E.; Bhagwat, S.V.; et al. Identification of Druggable Cancer Driver Genes Amplified across TCGA Datasets. PLoS ONE 2014, 9, e98293. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.-S.; Yuan, L.-T.; Kuei, C.-H.; Lin, H.-Y.; Chen, Y.-L.; Chiu, H.-W.; Lin, Y.-F. RGL2 Drives the Metastatic Progression of Colorectal Cancer via Preventing the Protein Degradation of β-Catenin and KRAS. Cancers 2021, 13, 1763. [Google Scholar] [CrossRef]
- Murakami, Y.; Sonoda, K.; Abe, H.; Watari, K.; Kusakabe, D.; Azuma, K.; Kawahara, A.; Akiba, J.; Oneyama, C.; Pachter, J.A.; et al. The activation of SRC family kinases and focal adhesion kinase with the loss of the amplified, mutated EGFR gene contributes to the resistance to afatinib, erlotinib and osimertinib in human lung cancer cells. Oncotarget 2017, 8, 70736–70751. [Google Scholar] [CrossRef] [Green Version]
- Stanley, A.; Ashrafi, G.H.; Seddon, A.M.; Modjtahedi, H. Synergistic effects of various Her inhibitors in combination with IGF-1R, C-MET and Src targeting agents in breast cancer cell lines. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Canonici, A.; Browne, A.L.; Ibrahim, M.F.K.; Fanning, K.P.; Roche, S.; Conlon, N.T.; O’Neill, F.; Meiller, J.; Cremona, M.; Morgan, C.; et al. Combined targeting EGFR and SRC as a potential novel therapeutic approach for the treatment of triple negative breast cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835919897546. [Google Scholar] [CrossRef]
- Conlon, N.; Canonici, A.; Morgan, C.; Cremona, M.; Hennessey, B.; Eustace, A.; O’Brien, N.; Slamon, D.; Crown, J.; O’Donovan, N. Targeting Src Kinase Blocks Development of Afatinib Resistance in HER2-Positive Breast Cancer. In Proceedings of the 2017 San Antonio Breast Cancer Symposium, San Antonio, TX; Philadelphia, PA, USA, 5–9 December 2017. [Google Scholar] [CrossRef]
- Manoharan, N.; Choi, J.; Chordas, C.; Zimmerman, M.A.; Scully, J.; Clymer, J.; Filbin, M.; Ullrich, N.J.; Bandopadhayay, P.; Chi, S.N.; et al. Trametinib for the treatment of recurrent/progressive pediatric low-grade glioma. J. Neuro-Oncol. 2020, 149, 253–262. [Google Scholar] [CrossRef]
- Grossauer, S.; Koeck, K.; Murphy, N.E.; Meyers, I.D.; Daynac, M.; Truffaux, N.; Truong, A.; Nicolaides, T.P.; McMahon, M.; Berger, M.S.; et al. Concurrent MEK targeted therapy prevents MAPK pathway reactivation during BRAFV600E targeted inhibition in a novel syngeneic murine glioma model. Oncotarget 2016, 7, 75839–75853. [Google Scholar] [CrossRef] [Green Version]
- Pudewell, S.; Wittich, C.; Jasemi, N.S.K.; Bazgir, F.; Ahmadian, M.R. Accessory proteins of the RAS-MAPK pathway: Moving from the side line to the front line. Commun. Biol. 2021, 4, 1–10. [Google Scholar] [CrossRef]
- Stegh, A.H.; Herrmann, H.; Lampel, S.; Weisenberger, D.; Andrä, K.; Seper, M.; Wiche, G.; Krammer, P.H.; Peter, M.E. Identification of the Cytolinker Plectin as a Major Early In Vivo Substrate for Caspase 8 during CD95- and Tumor Necrosis Factor Receptor-Mediated Apoptosis. Mol. Cell. Biol. 2000, 20, 5665–5679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czyz, M. Fibroblast Growth Factor Receptor Signaling in Skin Cancers. Cells 2019, 8, 540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of Profound Feedback Inhibition of Mitogenic Signaling by RAF Inhibitors Attenuates Their Activity in BRAFV600E Melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [Green Version]
- Bowyer, S.; Lee, R.; Fusi, A.; Lorigan, P. Dabrafenib and its use in the treatment of metastatic melanoma. Melanoma Manag. 2015, 2, 199–208. [Google Scholar] [CrossRef]
- Vizirianakis, I.S. Clinical Translation of Genotyping and Haplotyping Data: Implementation of in Vivo Pharmacology Experience Leading Drug Prescription to Pharmacotyping. Clin. Pharmacokinet. 2007, 46, 807–824. [Google Scholar] [CrossRef]
- Pirmohamed, M. Personalized Pharmacogenomics: Predicting Efficacy and Adverse Drug Reactions. Annu. Rev. Genom. Hum. Genet. 2014, 15, 349–370. [Google Scholar] [CrossRef]
- Ahn, K.; Luo, J.; Berg, A.; Keefe, D.; Wu, R. Functional mapping of drug response with pharmacodynamic–pharmacokinetic principles. Trends Pharmacol. Sci. 2010, 31, 306–311. [Google Scholar] [CrossRef]
- Astras, G.; Papagiannopoulos, C.I.; Kyritsis, K.A.; Markitani, C.; Vizirianakis, I. Pharmacogenomic Testing to Guide Personalized Cancer Medicine Decisions in Private Oncology Practice: A Case Study. Front. Oncol. 2020, 10, 521. [Google Scholar] [CrossRef]
- Tafazoli, A.; Guchelaar, H.-J.; Miltyk, W.; Kretowski, A.J.; Swen, J.J. Applying Next-Generation Sequencing Platforms for Pharmacogenomic Testing in Clinical Practice. Front. Pharmacol. 2021, 12, 2025. [Google Scholar] [CrossRef]
- Kyriakidis, K.; Charalampidou, A.; Natsiavas, P.; Vizirianakis, I.S.; Malousi, A. Linking exome sequencing data with drug response aberrations. Stud. Health Technol. Inform. 2019, 264, 1845–1846. [Google Scholar] [CrossRef] [PubMed]
- Kirouac, D.C.; Schaefer, G.; Chan, J.; Merchant, M.; Orr, C.; Huang, S.-M.A.; Moffat, J.; Liu, L.; Gadkar, K.; Ramanujan, S. Clinical responses to ERK inhibition in BRAF V600E-mutant colorectal cancer predicted using a computational model. NPJ Syst. Biol. Appl. 2017, 3, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Albeck, J.G.; Burke, J.M.; Aldridge, B.; Zhang, M.; Lauffenburger, D.A.; Sorger, P.K. Quantitative Analysis of Pathways Controlling Extrinsic Apoptosis in Single Cells. Mol. Cell 2008, 30, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Paananen, J.; Fortino, V. An omics perspective on drug target discovery platforms. Brief. Bioinform. 2020, 21, 1937–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi-Noghabi, H.; Jahangiri-Tazehkand, S.; Smirnov, P.; Hon, C.; Mammoliti, A.; Nair, S.K.; Mer, A.S.; Ester, M.; Haibe-Kains, B. Drug sensitivity prediction from cell line-based pharmacogenomics data: Guidelines for developing machine learning models. Briefings Bioinform. 2021, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Dang, C.C.; Ballester, P.J. Systematic Assessment of Multi-Gene Predictors of Pan-Cancer Cell Line Sensitivity to Drugs Exploiting Gene Expression Data. F1000Research 2017, 5, 2927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, J.P.; Soellner, M.B.; Merajver, S.D.; Li, J.Z. Impact of between-tissue differences on pan-cancer predictions of drug sensitivity. PLoS Comput. Biol. 2021, 17, e1008720. [Google Scholar] [CrossRef] [PubMed]
- Vizirianakis, I.S.; Chatzopoulou, M.; Bonovolias, I.D.; Nicolaou, I.; Demopoulos, V.J.; Tsiftsoglou, A.S. Toward the Development of Innovative Bifunctional Agents To Induce Differentiation and To Promote Apoptosis in Leukemia: Clinical Candidates and Perspectives. J. Med. Chem. 2010, 53, 6779–6810. [Google Scholar] [CrossRef]
- Noorbakhsh, J.; Vazquez, F.; McFarland, J.M. Bridging the gap between cancer cell line models and tumours using gene expression data. Br. J. Cancer 2021, 125, 311–312. [Google Scholar] [CrossRef]
- Lavacchi, D.; Roviello, G.; D’Angelo, A. Tumor-Agnostic Treatment for Cancer: When How is Better than Where. Clin. Drug Investig. 2020, 40, 519–527. [Google Scholar] [CrossRef]
- Adashek, J.J.; Subbiah, V.; Kurzrock, R. From Tissue-Agnostic to N-of-One Therapies: (R)Evolution of the Precision Paradigm. Trends Cancer 2021, 7, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Olsen, N.D.; Wagner, J.; McDaniel, J.; Stephens, S.H.; Westreich, S.T.; Prasanna, A.; Johanson, E.; Boja, E.; Maier, E.J.; Serang, O.; et al. PrecisionFDA Truth Challenge V2: Calling Variants from Short- and Long-Reads in Difficult-to-Map Regions. Biorxiv 2020, 11, 380741. [Google Scholar]
- Hsu, C.-H.; Tomiyasu, H.; Liao, C.-H.; Lin, C.-S. Genome-wide DNA methylation and RNA-seq analyses identify genes and pathways associated with doxorubicin resistance in a canine diffuse large B-cell lymphoma cell line. PLoS ONE 2021, 16, e0250013. [Google Scholar] [CrossRef] [PubMed]
- Krushkal, J.; Silvers, T.; Reinhold, W.C.; Sonkin, D.; Vural, S.; Connelly, J.; Varma, S.; Meltzer, P.S.; Kunkel, M.; Rapisarda, A.; et al. Epigenome-wide DNA methylation analysis of small cell lung cancer cell lines suggests potential chemotherapy targets. Clin. Epigenetics 2020, 12, 1–28. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| (a) | |||||
|---|---|---|---|---|---|
| Gene | Cell Line Status 1 | Source | Analysis | Alteration | Pathway Gene |
| FGFR1 | R | COSMIC CCLE | Mutation | Pathogenic/non-conserving, silent | MAPK signaling pathway, Pathways in cancer |
| MAP4K4, ARRB1, CACNA1H, IRAK4 | R | COSMIC | Mutation | Neutral | MAPK signaling pathway |
| RET, ARHGEF1, GSTO2, WNT7A | R | COSMIC | Mutation | Neutral | Pathways in cancer |
| FASLG | R | COSMIC | Mutation | Neutral | MAPK signaling pathway, Pathways in cancer |
| CDK2 | R | COSMIC | Expression | Over | Pathways in cancer |
| BIRC7 | R | COSMIC | Expression | Over | Pathways in cancer |
| (b) | |||||
| Gene | Description1 | Interactions | |||
| DLG1 | R/COSMIC/M | CACNG2,CACNG3,CACNG4,CACNG8,HRAS,KRAS,NRAS,MAPK12,SRC,PIK3CA,PIK3R1,CDH1,CTNNB1,CTNNA1,APC,PTEN,MDM2 | |||
| COL1A2 | R/COSMIC/M(P) | VEGFD,FLT4,LAMA5,FN1,ITGA2,ITGAV,ITGB1,IL4,IL13,MMP2,CASP8 | |||
| CDK2 | R/COSMIC/E | MAPK1,MAPK3,MYC,CDC25B,TP53,ABL1,CDKN1B,CDKN1A,CCND1,RHOA,FOXO1,MDM2,CDK4,CCNA2,CCNA1,CEBPA,E2F1,E2F2,E2F3,CDK6,CCND2,CCND3,CKS1B,CKS2,SKP1,CUL1,SKP2,CCNE1,CCNE2,RB1,TERT,SMAD3,SMAD4,HDAC1,BRCA2 | |||
| MAP3K9 | R/CCLE/M | RAC1,CDC42,MAP2K4,MAP2K6 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kardamiliotis, K.; Karanatsiou, E.; Aslanidou, I.; Stergiou, E.; Vizirianakis, I.S.; Malousi, A. Unraveling Drug Response from Pharmacogenomic Data to Advance Systems Pharmacology Decisions in Tumor Therapeutics. Future Pharmacol. 2022, 2, 31-44. https://doi.org/10.3390/futurepharmacol2010003
Kardamiliotis K, Karanatsiou E, Aslanidou I, Stergiou E, Vizirianakis IS, Malousi A. Unraveling Drug Response from Pharmacogenomic Data to Advance Systems Pharmacology Decisions in Tumor Therapeutics. Future Pharmacology. 2022; 2(1):31-44. https://doi.org/10.3390/futurepharmacol2010003
Chicago/Turabian StyleKardamiliotis, Konstantinos, Evangelina Karanatsiou, Ioanna Aslanidou, Eirini Stergiou, Ioannis S. Vizirianakis, and Andigoni Malousi. 2022. "Unraveling Drug Response from Pharmacogenomic Data to Advance Systems Pharmacology Decisions in Tumor Therapeutics" Future Pharmacology 2, no. 1: 31-44. https://doi.org/10.3390/futurepharmacol2010003
APA StyleKardamiliotis, K., Karanatsiou, E., Aslanidou, I., Stergiou, E., Vizirianakis, I. S., & Malousi, A. (2022). Unraveling Drug Response from Pharmacogenomic Data to Advance Systems Pharmacology Decisions in Tumor Therapeutics. Future Pharmacology, 2(1), 31-44. https://doi.org/10.3390/futurepharmacol2010003