Abstract
In Nuclear Magnetic Resonance (NMR) spectroscopy, the isotropic chemical shift is a measure of the electron density around the observed nuclide. For characterization of solid materials and compounds, it is desirable to find correlations between and structural parameters such as coordination numbers and distances to neighboring atoms. Correlations of good quality are easier to find when the coordination sphere is formed by only one element, as the electron density is obviously strongly dependent on the atomic number. The current study is therefore restricted to nuclides in pure oxygen coordination. It is shown that the isotropic shift correlates well with the average oxygen distances (as defined by the coordination sphere) for the nuclides Na (with spin ), Al (), and Ca (), using literature data for a range of periodic solids. It has been previously suggested for Pb () that may alternatively be related to the shortest oxygen distance in the structure, and our study corroborates this also for the nuclides considered here. While the correlation with the minimal distance is not always better, it has the advantage of being uniquely defined. In contrast, the average distance is strongly dependent on the designation of the oxygen coordination sphere, which may be contentious in some crystal structures.
1. Introduction
Nuclear Magnetic Resonance (NMR) spectroscopy probes the response of the magnetic moments of nuclei in an external magnetic field. Since its inception in 1946 [1], it has developed into a powerful analytical tool, with a wide range of applications in chemistry, physics, and materials science [2,3,4]. In particular, NMR spectroscopy of inorganic solids has become an established method for the elucidation of structure and dynamics, as a technique complementary to X-ray diffraction (XRD) [4,5,6,7]. The successful application of solid-state NMR to crystallographic problems has led to the establishment of a ‘Commission on NMR Crystallography and Related Methods’ in the International Union of Crystallography [8]. Many crystallographic questions, such as the determination of asymmetric units, or the assignment of space groups may be answered by considering the isotropic chemical shift of the NMR-observed nuclide in the corresponding structures [5,6]. The chemical shift reflects the modification of the resonance frequency by the electronic charges surrounding the nucleus, which obviously encapsulates information about the coordination sphere, i.e., number and type of neighboring atoms. This three-dimensional electronic shielding is commonly described by a symmetric second-rank tensor. The isotropic chemical shift is the weighted trace of this tensor:
From solution-state NMR spectra, only is available. NMR spectra of solids, whether under static or magic-angle spinning conditions [9], may additionally contain information about the individual tensor elements [10,11,12].
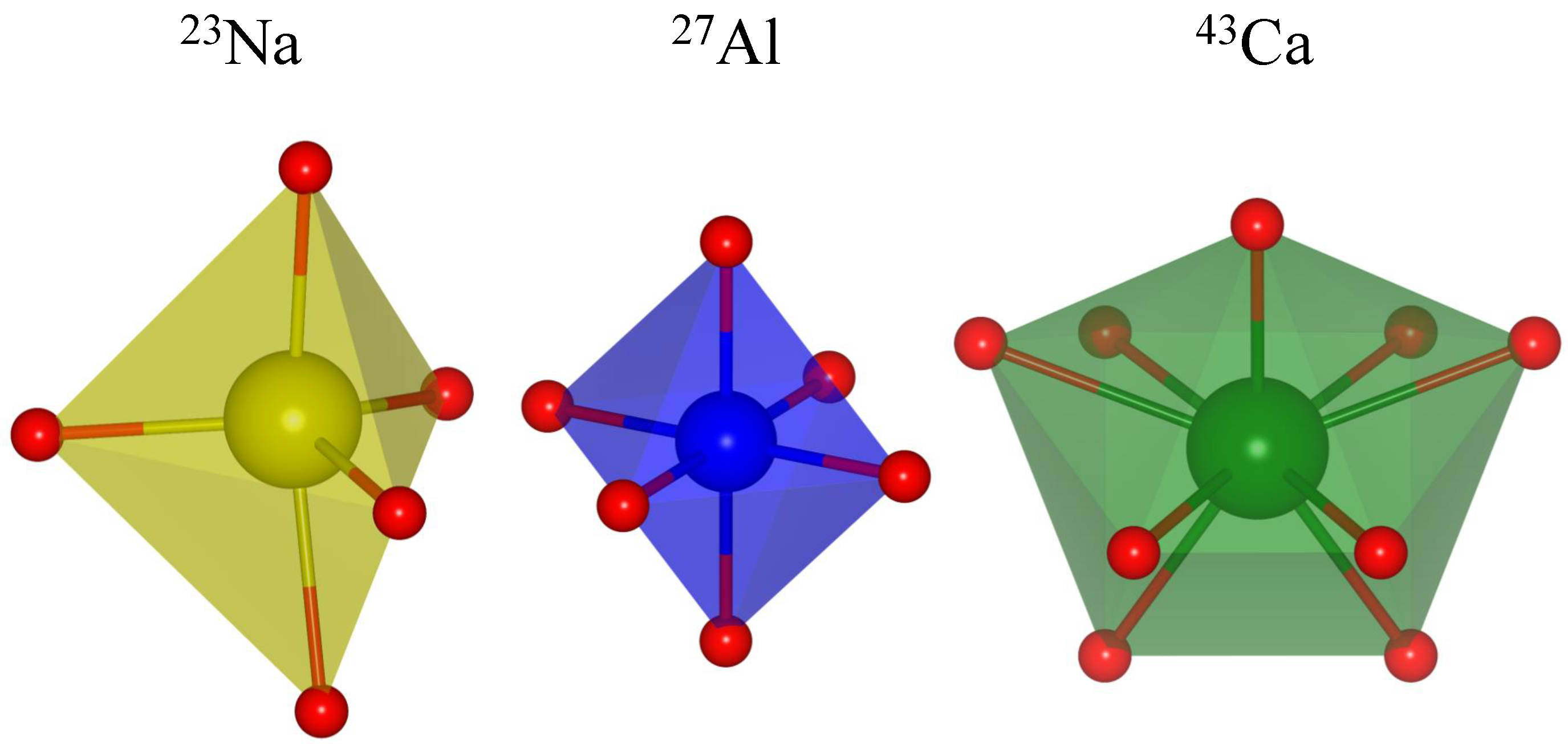
Among other structural information that may be derived from the isotropic chemical shift, one is of particular interest in the context of the current work, namely the attempt to correlate with interatomic distances between the observed nuclide and the coordinating oxygen atoms. Many inorganic compounds contain oxygen, not just in the form of the O ion (as present in oxides) but also in more complex oxyanions such as phosphates PO, sulphates SO, carbonates CO, etc. In many cases, the oxygen atoms form (more or less) regular coordination polyhedra around the elements of the cationic sublattice. Examples for five-, six-, and ninefold coordination by oxygen are depicted in Figure 1.
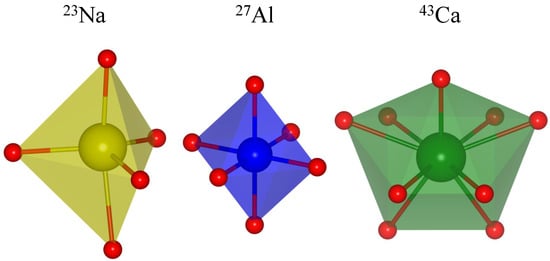
Figure 1.
Oxygen coordination polyhedra for: fivefold-coordinated sodium in NaGeO (ICSD no. 1622), sixfold-coordinated aluminum in YAlO (ICSD no. 88261), and ninefold-coordinated calcium in CaCO (aragonite, ICSD no. 32100).
Obviously, it would be advantageous if one could obtain information about the distances of oxygen coordination via the parameter of the observed nuclide, which is comparatively easy to extract from NMR spectra. Such correlations have indeed been suggested for the oxygen coordination sphere of Na [13,14], Al [15], Ca [16], and Pb [17]. In these papers, it was assumed that the isotropic chemical shift of an observed nuclide X, which is surrounded only by oxygen, depends on the average X–O distance, , calculated over the oxygen atoms coordinating X. However, is distinctly dependent on the choice of this coordination sphere, and the coordination number () defined by it. The question of how to define a coordination sphere in a periodic structure is indeed complex and still a matter of debate among chemists and crystallographers [18]. Recently, it has been suggested to circumvent this problem by using the shortest X–O distance found in the structure, , instead of the average over the neighboring atoms. For Pb (with spin ) in oxygen surroundings, use of leads to improvements in the correlation [19].
In the present work, we extend the concept of correlating of the observed nuclide with its distance to the nearest oxygen to NMR spectroscopy of Na (), Al (), and Ca (). For all three nuclei, it is shown that the quality of the correlations is similar to or better than those obtained by using , while removing the ambiguity inherent in the definition of the coordination sphere.
2. Materials and Methods
Isotropic chemical shift values were collected from the literature, with the specific references listed in Table A1, Table A2 and Table A3. It should be noted that all nuclides considered here have a spin and thus possess a quadrupole moment [20]. For non-cubic electronic surroundings, this affects the NMR energy levels by the so-called quadrupole coupling, which is the interaction between the quadrupole moment of the nucleus and the electric field gradient (EFG) of the surroundings. For large coupling constants, the resonance position of the central transition is affected by it, in addition to the change of position caused by the chemical shift. This “quadrupolar shift” needs to be corrected for when determining . For the literature NMR results used in the current work, we only selected studies where (i) the quadrupolar shift was properly considered (whenever necessary), and (ii) the applied referencing for the chemical shift was clearly defined. The most common reference systems are: 1M NaCl solution for Na, Al in aqueous solution for Al, and 1M CaCl solution for Ca. In some cases, when the relation of different reference material was well known, the originally reported values were corrected to the references listed above. This meant adding 7.2 ppm when solid NaCl was used as reference for Na [21], and adding 8 ppm when a saturated CaCl solution was used [22]. (Theoretically, the change in Larmor frequency needs to be considered when moving a reference; however, for corrections <10 ppm, this makes practically no difference).
For those compounds selected on the grounds of rigorous and sound NMR characterization, the needed structural parameters were extracted from the Inorganic Crystal Structure Database (ICSD, provided by FIZ Karlsruhe GmbH). In case of multiple entries for one compound, the selection criteria were data quality, goodness of R-factor, the age of the publication, and the consistency with other structural models. To determine distances, the implemented ICSD function “Distances and Angles” was employed. The standard settings of this tool define the first coordination sphere by taking bond lengths in the range between 0.7 and 3.0 Å into account, which were accordingly used for our evaluations.
3. Results
3.1. NMR of Na with in Oxygen Coordination
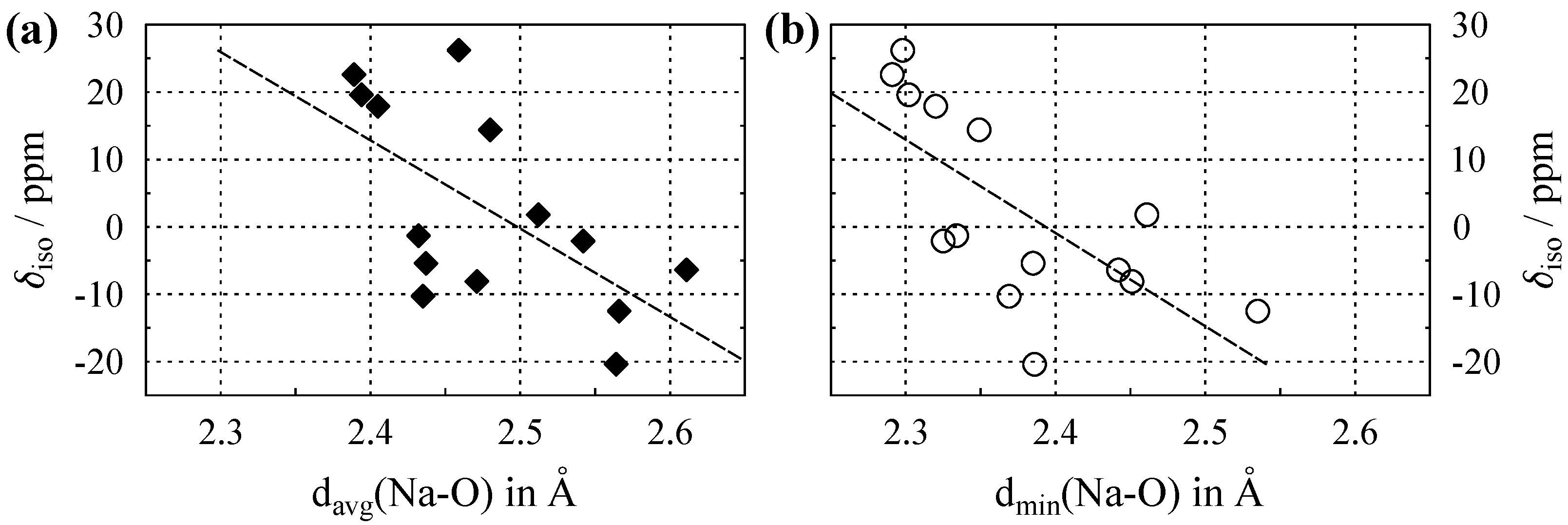
The isotropic chemical shifts () of Na in a number of inorganic compounds are tabulated in Table A1 in Appendix A, together with the parameters of their crystal structures: coordination numbers (), and both the average () and minimal sodium–oxygen () distance within the coordination sphere. Figure 2a shows the plot of versus , and Figure 2b the plot of versus . The data points in both plots display appreciable scatter, leading to Pearson correlation coefficients [23] that are comparatively low. By considering classes of sodium-containing compounds such as silicates, borates, carbonates, etc. in separate plots, these correlations can be substantially improved [14]. However, the purpose of the current work is to compare average to the shortest distance; therefore, members of various compound classes were included in our plots. It can be seen from the caption of Figure 2 that plotting the chemical shifts versus leads to a marginally better correlation coefficient. While this improvement might not be significant, the important advantage of using the minimal distance to oxygen is that, in contrast to the average distance , it is unequivocally defined in the crystal structure.
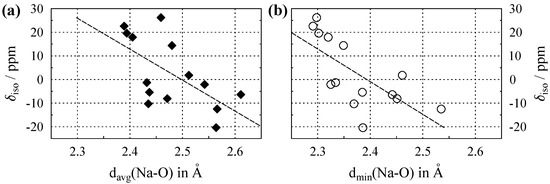
Figure 2.
(a) Evolution of the Na isotropic chemical shift () versus average Na–O distance, , for compounds where sodium is solely coordinated by oxygen. (b) versus the shortest Na–O distance, . Dashed lines show the least-square linear fits, which follow the relations , with a Pearson correlation coefficient of , and , with , respectively. All data are listed in Table A1.
3.2. NMR of Al with in Oxygen Coordination
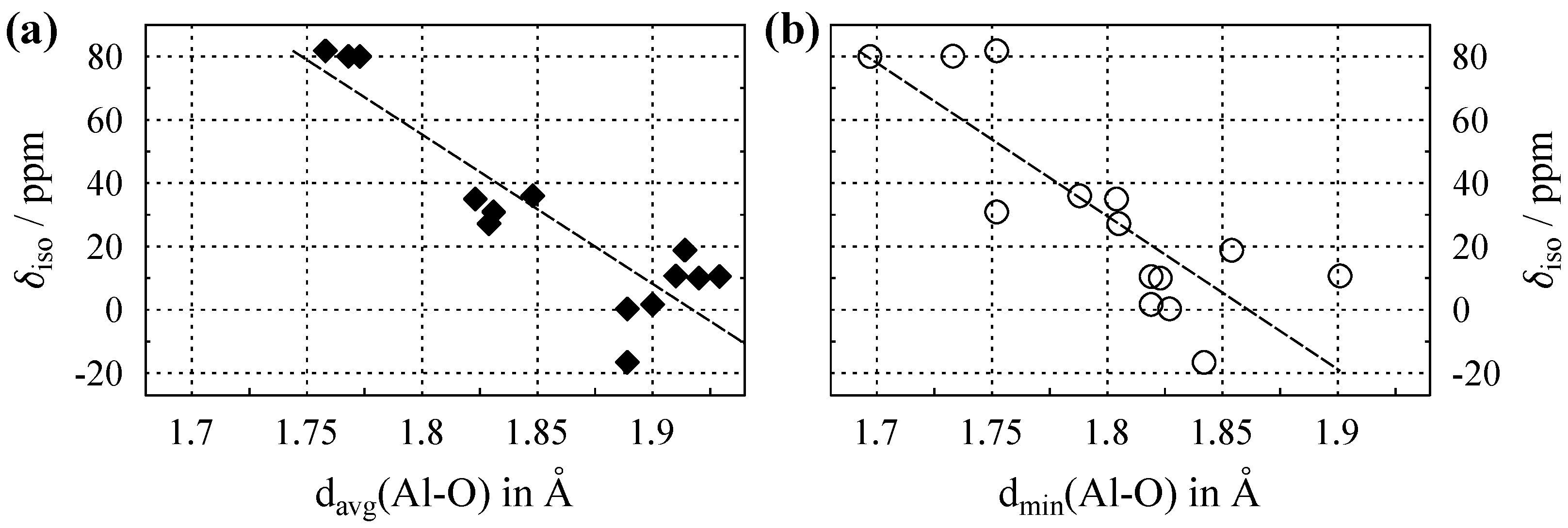
The plots in Figure 3 show the Al isotropic chemical shifts () versus average and shortest aluminum–oxygen distance in the crystal structures, with the relevant data listed in Table A2 in Appendix B. For the listed values of , the absence of digits after the decimal points indicates that the estimated precision was ppm. This may even apply for different crystallographic sites in a single compound, as may be seen from the entry for -AlO in Table A2. Here, the site with the lower coordination number () experiences stronger quadrupole coupling (see also Section 2), which leads to broader lines and hence decreased the accuracy for determination of . Regarding the scaling of the x-axis in Figure 3, it should be noted that for Al, the interatomic distances to oxygen are significantly smaller than those reported for either Na (see above) or Ca (see below). This is due to the fact that Al with its high net electrical charge has a larger atomic number than Na despite both cations possessing an identical electron configuration. Compared to Na, and also to Ca with its additional fully-occupied and orbitals, this leads to a smaller effective ionic radius for Al. This effect is also visible in the graphics of Figure 1. An alternative view of the situation is to consider the role of covalency. Since the difference in electronegativity between oxygen and aluminum (≈1.8) is much lower than those of sodium (≈2.5) and calcium (≈2.4), a higher degree of covalent character is expected for aluminum–oxygen compounds. The more directed nature of covalent bonding leads to shorter distances.
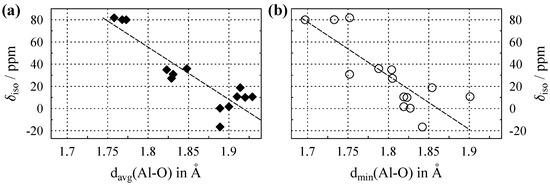
Figure 3.
(a) Evolution of the Al isotropic chemical shift () versus average Al–O distance, , for compounds where aluminum is solely coordinated by oxygen. (b) versus the shortest Al–O distance, . Dashed lines show the least-square linear fits, which follow the relations , with a Pearson correlation coefficient of , and , with , respectively. All data are listed in Table A2.
Figure 3a shows a remarkably strong clustering of the data points according to their coordination number (). The highest shift values are observed for the , the lowest for , with those for clearly separated in between. Apparently, this clustering also leads to correlation coefficients that are much better than those observed for the Na data discussed above. One possible explanation for the clustering is to again consider the non-negligible covalent character in the bond interactions of aluminum discussed above. In a coordination sphere realized only by covalent bonds (i.e., a molecule), the number of coordination partners and the bond lengths are clearly defined. Thus, the presence of some covalent character in the aluminum compounds would narrow the spread of available distances and lead to the cluster effect observed in Figure 3a. For the plot of the minimal distances in Figure 3b, the clustering was less pronounced, with the data points for five- and sixfold-coordination overlapping. From the caption of Figure 3, it may be seen that for Al, use of the shortest distances leads to a correlation coefficient that is slightly worse than that for use of . However, the resulting linear equations are very similar; and again, finding in a given structure is much easier than defining average distances.
3.3. NMR of Ca with in Oxygen Coordination
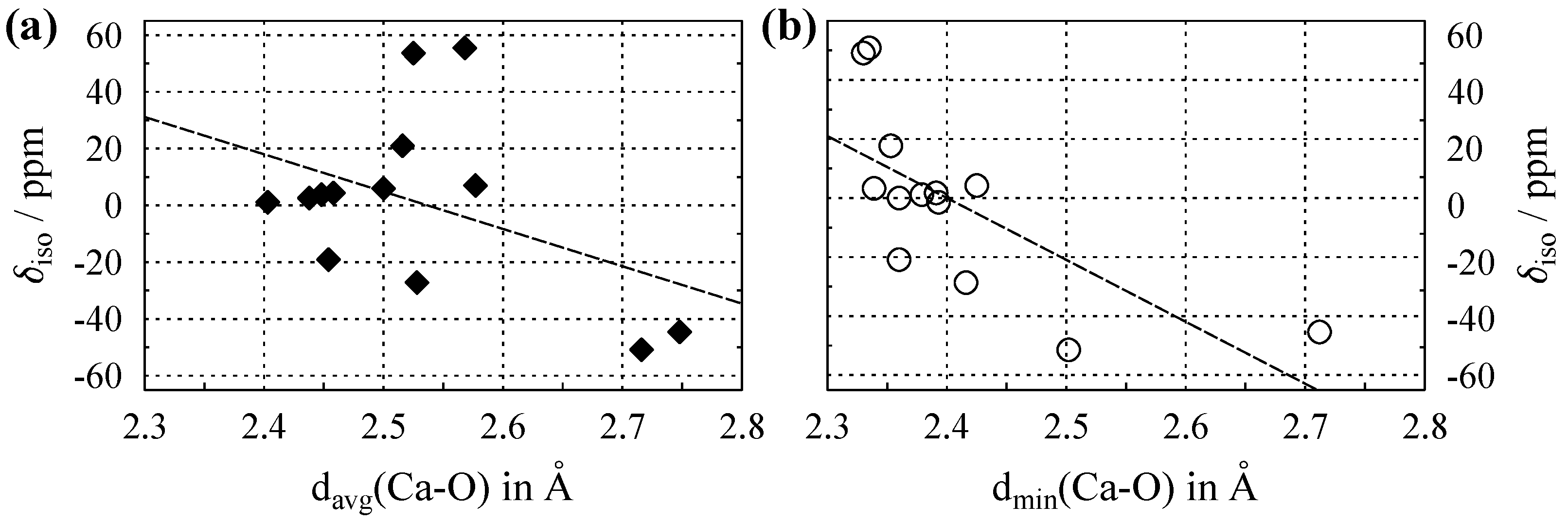
Figure 4 shows values of Ca plotted versus average and shortest calcium–oxygen distance in the crystal structures, with the relevant data listed in Table A3 in Appendix C. In comparison to both Na and Al, the coordination number () of Ca is generally higher, with . This is due to the larger size of the calcium ion (see also Figure 1), which allows for more neighbors in the coordination sphere. Some clustering according to the coordination number can be seen in Figure 4a. For Ca, this only separates off the compounds with , whereas the values for all overlap; hence, the cluster effect is much weaker than that observed for Al, cf. Figure 3a. From the data considered in this paper, the improvement in the correlation when using minimal instead of average distances to oxygen is the largest for Ca (see also Figure 4b), with the Pearson correlation coefficient increasing from to . Similar to Al, the clustering effect in the plots disappears when using instead of .
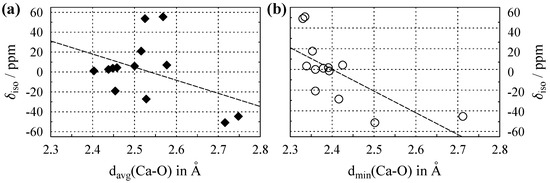
Figure 4.
(a) Evolution of the Ca isotropic chemical shift () versus average Ca–O distance, , for compounds where calcium is solely coordinated by oxygen. (b) versus the shortest Ca–O distance, . Dashed lines show the least-square linear fits, which follow the relations , with a Pearson correlation coefficient of , and , with , respectively. All data are listed in Table A3.
4. Discussion
In the context of relating NMR parameters to structural features in inorganic compounds, we investigated the relation of the isotropic chemical shift () of the nuclides Na (with spin ), Al (), and Ca () in pure oxygen coordination to the nuclide–oxygen distances in their coordination sphere. It could be shown that the correlation of with the shortest oxygen distance [19] works about as well as using the average oxygen distance [13,14,15,16,17] for Na and Al and even better for Ca. Use of the minimal distance has the additional advantage that this parameter is uniquely defined. In contrast, the calculation of an average distance is strongly dependent on the definition of the oxygen coordination sphere, which may be contentious in some crystal structures.
Author Contributions
Conceptualization, O.E.O.Z. and T.B.; methodology, J.S., O.E.O.Z. and T.B.; formal analysis, J.S. and T.B.; writing—original draft preparation, J.S. and T.B.; writing—review and editing, J.S., O.E.O.Z. and T.B.; supervision, T.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are contained within the article.
Acknowledgments
J.S. and T.B. would like to thank Wolfgang Schnick (University of Munich, LMU) for continuing financial support.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CN | Coordination Number |
| ICSD | Inorganic Crystal Structure Database |
| MDPI | Multidisciplinary Digital Publishing Institute |
| NMR | Nuclear Magnetic Resonance |
| XRD | X-ray Diffraction |
Appendix A. Chemical Shifts and Structural Parameters for 23Na

Table A1.
Na isotropic chemical shifts of compounds containing sodium in pure oxygen coordination. The shifts are reported relative (and where necessary, have been converted) to a 1M NaCl solution. Also listed are the coordination numbers (CN) of sodium and the distances to the surrounding oxygens in the crystal structure. The literature references refer to the NMR work and the crystallographic references may be traced to via their entry code in the ICSD.
Table A1.
Na isotropic chemical shifts of compounds containing sodium in pure oxygen coordination. The shifts are reported relative (and where necessary, have been converted) to a 1M NaCl solution. Also listed are the coordination numbers (CN) of sodium and the distances to the surrounding oxygens in the crystal structure. The literature references refer to the NMR work and the crystallographic references may be traced to via their entry code in the ICSD.
| Compound | ICSD Code (year) | Space Group (no.) | CN | /Å (Na–O) | /Å (Na–O) | /ppm | Ref. |
|---|---|---|---|---|---|---|---|
| NaAlO | 14780 (2020) | (33) | 5 | 2.459 | 2.298 | 26.2 | [24] |
| NaGeO | 1622 (1978) | (36) | 5 | 2.398 | 2.291 | 22.6 | [14] |
| NaBO | 10061 (1979) | (15) | 5 | 2.394 | 2.302 | 19.6 | [14] |
| 6 | 2.480 | 2.349 | 14.4 | ||||
| NaCO | 56906 (1981) | (14) | 6 | 2.405 | 2.320 | 17.9 | [25] |
| NaBO | 34645 (1963) | (167) | 7 | 2.512 | 2.461 | 1.8 | [14] |
| NaSO | 28056 (1975) | (70) | 6 | 2.432 | 2.334 | [24] | |
| NaGeO | 68507 (1990) | (88) | 6 | 2.542 | 2.325 | [14] | |
| NaHCO | 18183 (1965) | (14) | 6 | 2.437 | 2.385 | [26] | |
| NaGeO | 189269 (2013) | (165) | 7 | 2.611 | 2.442 | [14] | |
| NaNO | 68707 (1990) | (44) | 6 | 2.471 | 2.451 | [25] | |
| -NaVO | 29450 (1984) | (62) | 6 | 2.435 | 2.369 | [25] | |
| NaIO | 14287 (1970) | (88) | 8 | 2.566 | 2.535 | [26] | |
| NaClO | 200405 (1978) | (63) | 8 | 2.564 | 2.386 | −20.4 | [26] |
Appendix B. Chemical Shifts and Structural Parameters for 27Al
Table A2.
Al isotropic chemical shifts of compounds containing aluminum in pure oxygen coordination. The shifts are reported relative to Al in aqueous solution. Also listed are the coordination numbers (CN) of aluminum and the distances to the surrounding oxygens in the crystal structure. The literature references refer to the NMR work and the crystallographic references may be traced to via their entry code in the ICSD.
Table A2.
Al isotropic chemical shifts of compounds containing aluminum in pure oxygen coordination. The shifts are reported relative to Al in aqueous solution. Also listed are the coordination numbers (CN) of aluminum and the distances to the surrounding oxygens in the crystal structure. The literature references refer to the NMR work and the crystallographic references may be traced to via their entry code in the ICSD.
| Compound | ICSD Code (year) | Space Group (no.) | CN | /Å (Al–O) | /Å (Al–O) | /ppm | Ref. |
|---|---|---|---|---|---|---|---|
| -LiAlO | 430184 (2016) | (92) | 4 | 1.758 | 1.752 | 81.8 | [27] |
| -NaAlO | 14780 (2020) | (33) | 4 | 1.768 | 1.733 | 80.1 | [28] |
| -AlO | 82504 (1996) | (12) | 4 | 1.773 | 1.697 | 80 | [29] |
| 6 | 1.929 | 1.819 | 10.5 | ||||
| AlPO(OH) ·HO (senegalite) | 100542 (1979) | (33) | 5 | 1.848 | 1.788 | 36 | [30] |
| 6 | 1.900 | 1.819 | 1.7 | ||||
| AlSiO (andalusite) | 172728 (2006) | (58) | 5 | 1.823 | 1.804 | 35 | [31] |
| 6 | 1.920 | 1.823 | 10 | ||||
| AlPO(OH) (augelite) | 426641 (2014) | (12) | 5 | 1.831 | 1.752 | 30.9 | [30] |
| 6 | 1.889 | 1.827 | 0.3 | ||||
| AlVO | 55870 (2004) | (2) | 5 | 1.829 | 1.805 | 27.2 | [32] |
| -AlO | 51687 (2001) | (167) | 6 | 1.914 | 1.854 | 18.8 | [33] |
| YAlO | 88261 (1999) | (62) | 6 | 1.910 | 1.901 | 10.7 | [34] |
| KAlPO | 2888 (1973) | (14) | 6 | 1.889 | 1.842 | −16.6 | [35] |
Appendix C. Chemical Shifts and Structural Parameters for 43Ca
Table A3.
Ca isotropic chemical shifts of compounds containing calcium in pure oxygen coordination. The shifts are reported relative (and where necessary, have been converted) to a 1M CaCl solution. Also listed are the coordination numbers (CN) of calcium and the distances to the surrounding oxygens in the crystal structure. The literature references refer to the NMR work and the crystallographic references may be traced to via their entry code in the ICSD.
Table A3.
Ca isotropic chemical shifts of compounds containing calcium in pure oxygen coordination. The shifts are reported relative (and where necessary, have been converted) to a 1M CaCl solution. Also listed are the coordination numbers (CN) of calcium and the distances to the surrounding oxygens in the crystal structure. The literature references refer to the NMR work and the crystallographic references may be traced to via their entry code in the ICSD.
| Compound | ICSD Code (year) | Space Group (no.) | CN | /Å (Ca–O) | /Å (Ca–O) | /ppm | Ref. |
|---|---|---|---|---|---|---|---|
| CaZrO | 97463 (2003) | (62) | 8 | 2.568 | 2.335 | 55.5 | [36] |
| CaAlO | 14270 (1970) | (15) | 7 | 2.525 | 2.330 | 53.7 | [37] |
| CaTiO | 153172 (2005) | (62) | 8 | 2.516 | 2.353 | 21.0 | [38] |
| CaMgSiO | 100736 (1981) | (113) | 8 | 2.577 | 2.425 | 7 | [39] |
| CaBO | 62430 (1987) | (60) | 8 | 2.500 | 2.339 | 6 | [22] |
| CaCO ·2HO | 30783 (1980) | (87) | 8 | 2.458 | 2.391 | 4.4 | [40] |
| CaCO ·3HO | 159351 (1981) | (2) | 8 | 2.448 | 2.379 | 3.8 | [41] |
| CaWO | 5510 (2019) | (88) | 8 | 2.438 | 2.360 | 2.6 | [36] |
| CaMoO | 60556 (1985) | (88) | 8 | 2.403 | 2.393 | 1.1 | [36] |
| CaSO·2HO | 161622 (2008) | (15) | 8 | 2.454 | 2.360 | −19.1 | [38] |
| CaCO (aragonite) | 32100 (1986) | (62) | 9 | 2.528 | 2.416 | −27.2 | [42] |
| CaAlO | 202616 (1988) | (194) | 12 | 2.748 | 2.712 | −44.6 | [37] |
| Ca(NO) | 52351 (1931) | (205) | 12 | 2.716 | 2.502 | −50.9 | [38] |
References
- Bloch, F. Nuclear Induction. Phys. Rev. 1946, 70, 460–474. [Google Scholar] [CrossRef]
- Abragam, A. The Principles of Nuclear Magnetism; Oxford University Press: Oxford, UK, 1961; ISBN 978-0-19-852014-6. [Google Scholar]
- Ernst, R.R.; Bodenhausen, G.; Wokaun, A. Principles of Nuclear Magnetic Resonance in One and Two Dimensions; Clarendon Press: Oxford, UK, 1987; ISBN 0-19-855647-0. [Google Scholar]
- MacKenzie, K.J.D.; Smith, M.E. Multinuclear Solid-State NMR of Inorganic Materials; Pergamon Materials Series v. 6; Pergamon: Oxford, UK, 2002; ISBN 0-08-043787-7. [Google Scholar]
- Harris, R.K. NMR crystallography: The use of chemical shifts. Solid State Sci. 2004, 6, 1025–1037. [Google Scholar] [CrossRef]
- Taulelle, F. NMR crystallography: Crystallochemical formula and space group selection. Solid State Sci. 2004, 6, 1053–1057. [Google Scholar] [CrossRef]
- Bryce, D.L. NMR crystallography: Structure and properties of materials from solid-state nuclear magnetic resonance observables. IUCrJ 2017, 4, 350–359. [Google Scholar] [CrossRef] [Green Version]
- Commission on NMR Crystallography and Related Methods. Available online: https://www.iucr.org/iucr/commissions/nmr-crystallography (accessed on 7 July 2022).
- Andrew, E.R. Magic angle spinning in solid state n.m.r. spectroscopy. Phil. Trans. R. Soc. A 1981, 299, 505–520. [Google Scholar]
- Herzfeld, J.; Berger, A.E. Sideband intensities in NMR spectra of samples spinning at the magic angle. J. Chem. Phys. 1980, 73, 6021–6030. [Google Scholar] [CrossRef]
- Hodgkinson, P.; Emsley, L. The reliability of the determination of tensor parameters by solid-state nuclear magnetic resonance. J. Chem. Phys. 1997, 107, 4808–4816. [Google Scholar] [CrossRef]
- Vosegaard, T. Single-crystal NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectr. 2021, 123, 51–72. [Google Scholar] [CrossRef]
- Xue, X.; Stebbins, J.F. 23Na NMR chemical shifts and local Na coordination environments in silicate crystals, melts and glasses. Phys. Chem. Miner. 1993, 20, 297–307. [Google Scholar] [CrossRef]
- George, A.M.; Sen, S.; Stebbins, J.F. 23Na chemical shifts and local structure in crystalline, glassy, and molten sodium borates and germanates. Solid State Nucl. Magn. Reson. 1997, 10, 9–17. [Google Scholar] [CrossRef]
- Haouas, M.; Taulelle, F.; Martineau, C. Recent advances in application of 27Al NMR spectroscopy to materials science. Prog. Nucl. Magn. Reson. Spectr. 2016, 94–95, 11–36. [Google Scholar] [CrossRef] [PubMed]
- Gambuzzi, E.; Pedone, A.; Menziani, M.C.; Angeli, F.; Florian, P.; Charpentier, T. Calcium environment in silicate and aluminosilicate glasses probed by 43Ca MQMAS NMR experiments and MD-GIPAW calculations. Solid State Nucl. Magn. Reson. 2015, 68–69, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Fayon, F.; Farnan, I.; Bessada, C.; Coutures, J.; Massiot, D.; Coutures, J.P. Empirical Correlations between 207Pb NMR Chemical Shifts and Structure in Solids. J. Am. Chem. Soc. 1997, 119, 6837–6843. [Google Scholar] [CrossRef]
- Hoppe, R. Effective coordination numbers (ECoN) and mean Active ionic radii (MEFIR). Z. Kristallog.-Cryst. Mater. 1979, 150, 23–52. [Google Scholar] [CrossRef]
- Zeman, O.E.O.; Steinadler, J.; Hochleitner, R.; Bräuniger, T. Determination of the Full 207Pb Chemical Shift Tensor of Anglesite, PbSO4, and Correlation of the Isotropic Shift to Lead-Oxygen Distance in Natural Minerals. Crystals 2019, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.H.; Reif, F. Quadrupole effects in nuclear magnetic resonance studies of solids. Solid State Phys. 1957, 5, 321–438. [Google Scholar] [CrossRef]
- Engelhardt, G.; Koller, H. A simple procedure for the determination of the quadrupole interaction parameters and isotropic chemical shifts from magic angle spinning NMR spectra of half-integer spin nuclei in solids. Magn. Reson. Chem. 1991, 29, 941–945. [Google Scholar] [CrossRef]
- Gervais, C.; Laurencin, D.; Wong, A.; Pourpoint, F.; Labram, J.; Woodward, B.; Howes, A.P.; Pike, K.J.; Dupree, R.; Mauri, F.; et al. New perspectives on calcium environments in inorganic materials containing calcium-oxygen bonds: A combined computational-experimental 43Ca NMR approach. Chem. Phys. Lett. 2008, 464, 42–48. [Google Scholar] [CrossRef]
- Pearson, K. Note on regression and inheritance in the case of two parents. Proc. R. Soc. Lond. 1895, 58, 240–242. [Google Scholar]
- Koller, H.; Engelhardt, G.; Kentgens, A.P.M.; Sauer, J. 23Na NMR spectroscopy of solids: Interpretation of quadrupole interaction parameters and chemical shifts. J. Phys. Chem. 1994, 98, 1544–1551. [Google Scholar] [CrossRef]
- Skibsted, J.; Nielsen, N.C.; Bildsøe, H.; Jakobsen, H.J. Satellite transitions in MAS NMR spectra of quadrupolar nuclei. J. Magn. Reson. 1991, 95, 88–117. [Google Scholar] [CrossRef]
- Tabeta, R.; Saito, H. 23Na chemical shifts of some inorganic and organic compounds in the solid state as determined by the magic angle spinning and high power NMR methods. Chem. Lett. 1984, 13, 293–296. [Google Scholar] [CrossRef] [Green Version]
- Bräuniger, T.; Groh, B.; Moudrakovski, I.L.; Indris, S. Local electronic structure in γ-LiAlO2 studied by single-crystal 27Al NMR and DFT calculations. J. Phys. Chem. A 2016, 120, 7839–7846. [Google Scholar] [CrossRef]
- Müller, D.; Gessner, W.; Samoson, A.; Lippmaa, E.; Scheler, G. Solid-state aluminium-27 nuclear magnetic resonance chemical shift and quadrupole coupling data for condensed AlO4 tetrahedra. J. Chem. Soc. Dalton Trans. 1986, 6, 1277–1281. [Google Scholar] [CrossRef]
- O’Dell, L.A.; Savin, S.L.P.; Chadwick, A.V.; Smith, M.E. A 27Al MAS NMR study of a sol-gel produced alumina: Identification of the NMR parameters of the θ-Al2O3 transition alumina phase. Solid State Nucl. Magn. Reson. 2007, 31, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Bleam, W.F.; Dec, S.F.; Frye, J.S. 27Al Solid-state Nuclear Magnetic Resonance Study of Five-Coordinate Aluminum in Augelite and Senegalite. Phys. Chem. Miner. 1989, 16, 817–820. [Google Scholar] [CrossRef]
- Alemany, L.B.; Massiot, D.; Sherriff, B.L.; Smith, M.E.; Taulelle, F. Observation and accurate quantification of 27Al MAS NMR spectra of some Al2SiO5 polymorphs containing sites with large quadrupole interactions. Chem. Phys. Lett. 1991, 177, 301–306. [Google Scholar] [CrossRef]
- Nielsen, U.G.; Boisen, A.; Brorson, M.; Jacobsen, C.J.H.; Jakobsen, H.J.; Skibsted, J. Aluminum orthovanadate (AlVO4): Synthesis and characterization by 27Al and 51V MAS and MQMAS NMR spectroscopy. Inorg. Chem. 2002, 41, 6432–6439. [Google Scholar] [CrossRef]
- Vosegaard, T.; Jakobsen, H.J. 27Al Chemical Shielding Anisotropy. J. Magn. Reson. 1997, 128, 135–137. [Google Scholar] [CrossRef]
- Florian, P.; Gervais, M.; Douy, A.; Massiot, D.; Coutures, J.-P. A Multi-nuclear Multiple-Field Nuclear Magnetic Resonance Study of the Y2O3—Al2O3 Phase Diagram. J. Phys. Chem. B 2001, 105, 379–391. [Google Scholar] [CrossRef]
- Müller, D.; Grunze, I.; Hallas, E.; Ladwig, G. Hochfeld-27Al-NMR-Untersuchungen zur Aluminiumkoordination in kristallinen Aluminiumphosphaten. Z. Anorg. Allg. Chem. 1983, 500, 80–88. [Google Scholar] [CrossRef]
- Lin, Z.; Smith, M.E.; Sowrey, F.E.; Newport, R.J. Probing the local structural environment of calcium by natural-abundance solid-state 43Ca NMR. Phys. Rev. B 2004, 69, 224107. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, K.J.D.; Schmücker, M.; Smith, M.E.; Poplett, I.J.F.; Kemmitt, T. Evolution of crystalline aluminates from hybrid gel-derived precursors studied by XRD and multinuclear solid state MAS NMR IV: Calcium dialuminate, CaAl4O7 and calcium hexaaluminate, CaAl12O19. Thermochim. Acta 2000, 363, 181–188. [Google Scholar] [CrossRef]
- Widdifield, C.M.; Moudrakovski, I.; Bryce, D.L. Calcium-43 chemical shift and electric field gradient tensor interplay: A sensitive probe of structure, polymorphism, and hydration. Phys. Chem. Chem. Phys. 2014, 16, 13340–13359. [Google Scholar] [CrossRef] [Green Version]
- Dupree, R.; Howes, A.P.; Kohn, S.C. Natural abundance solid state 43Ca NMR. Chem. Phys. Lett. 1997, 276, 399–404. [Google Scholar] [CrossRef]
- Bowers, G.M.; Kirkpatrick, R.J. Natural Abundance 43Ca NMR as a Tool for Exploring Calcium Biomineralization: Renal Stone Formation and Growth. Cryst. Growth Des. 2011, 11, 5188–5191. [Google Scholar] [CrossRef]
- Wong, A.; Howes, A.P.; Dupree, R.; Smith, M.E. Natural abundance 43Ca NMR study of calcium-containing organic solids: A model study for Ca-binding biomaterials. Chem. Phys. Lett. 2006, 427, 201–205. [Google Scholar] [CrossRef]
- Huang, Y.-C.; Mou, Y.; Tsai, T.W.-T.; Wu, Y.-J.; Lee, H.-K.; Huang, S.-J.; Chan, J.C.C. Calcium-43 NMR Studies of Polymorphic Transition of Calcite to Aragonite. J. Phys. Chem. B 2012, 116, 14295–14301. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).