
Role of Oxygen Radicals in Alzheimer’s Disease: Focus on Tau Protein





{kind=link}
{kind=link}
Abstract
1. Introduction: Why This Study?
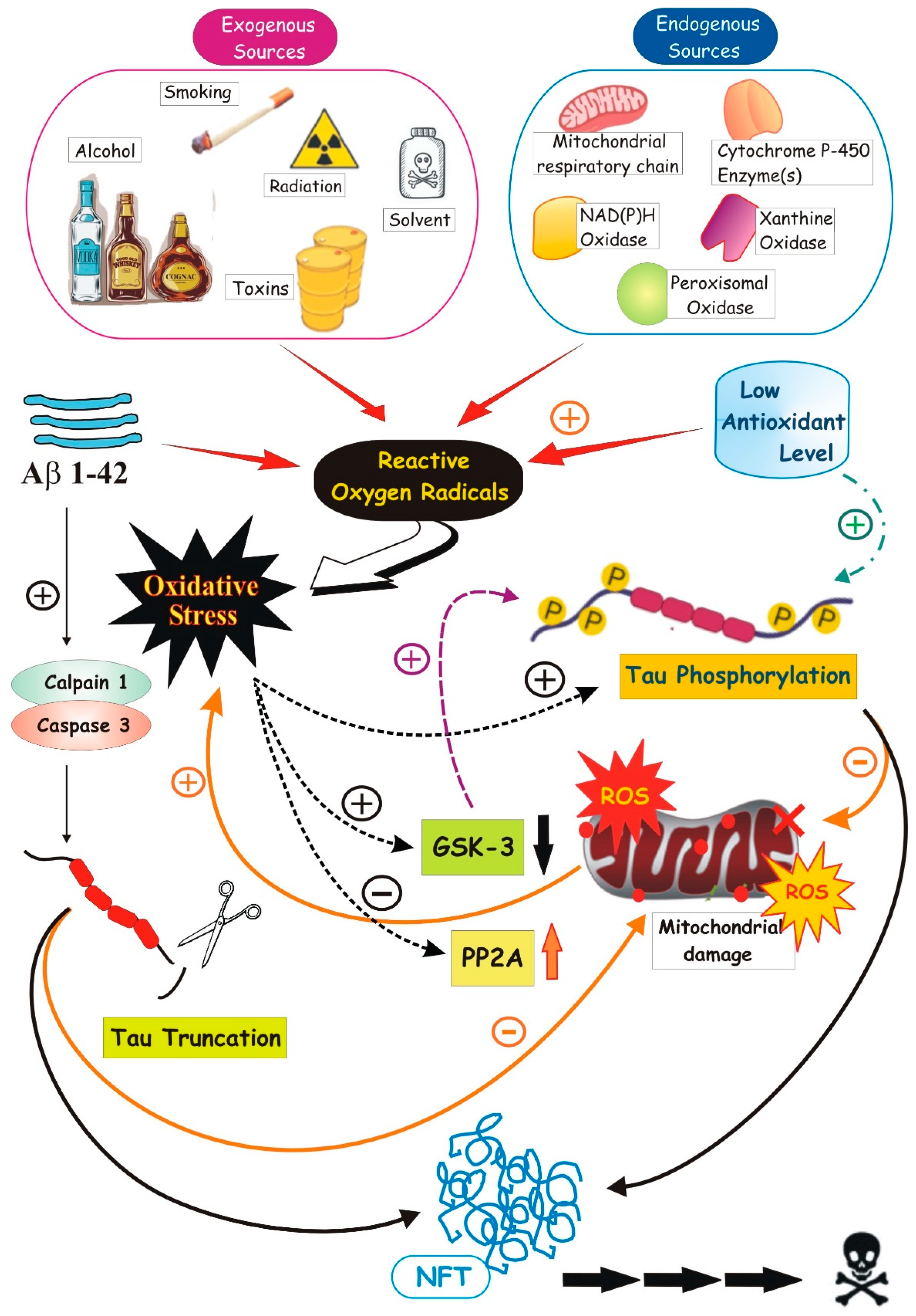
2. Oxidative Stress, a Real Cruelty to the Brain
2.1. Bioenergetic Efficiency of the Brain Is Vital for Normal Functioning of the Central Nervous System (CNS)
3. Characteristics of Alzheimer’s Disease: An Overview
3.1. Misfolded Proteins in the Mechanism of Neurodegeneration
3.2. Mitochondrial Dysfunction and Oxidative Stress Are Early Features of AD Brains
4. Tau–ROS Interplay
4.1. What Is Tau?
4.2. Tau Isoforms
4.3. Molecular Mechanisms Underlying the Pathology of the Tau Protein
4.4. Tau Hyperphosphorylation and Truncation
4.5. Hyperphosphorylation of the Tau Protein Is Essentially Interlinked with the OS. Aβ Production and Mitochondrial Dysfunction Participate in the Process
4.5.1. OS-Induced Tau Phosphorylation
4.5.2. Tau Phosphorylation-Induced OS
4.6. Pathological Tau Affects Mitochondrial Function in AD
4.7. Aβ and Tau Act Synergistically on Cellular Processes or Organelles and Can Amplify Each Other’s Neurotoxic Effects
4.8. 12A12: A Monoclonal Antibody Able of Specifically Binding the Toxic Tau
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 27, 2181–2192. [Google Scholar] [CrossRef]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef]
- Liu, Z.; Li, T.; Li, P.; Wei, N.; Zhao, Z.; Liang, H.; Ji, X.; Chen, W.; Xue, M.; Wei, J. The Ambiguous Relationship of Oxidative Stress, Tau Hyperphosphorylation, and Autophagy Dysfunction in Alzheimer’s Disease. Oxid. Med. Cell Longev. 2015, 2015, 352723. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Ayton, S.; Bush, A.I. The essential elements of Alzheimer’s disease. J. Biol. Chem. 2021, 296, 100105. [Google Scholar] [CrossRef] [PubMed]
- Sukhorukov, V.S.; Mudzhiri, N.M.; Voronkova, A.S.; Baranich, T.I.; Glinkina, V.V.; Illarioshkin, S.N. Mitochondrial Disorders in Alzheimer’s Disease. Biochemistry 2021, 86, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Ciotti, M.T.; Costanzi, M.; Cestari, V.; Calissano, P.; Canu, N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc. Natl. Acad. Sci. USA 2006, 103, 2892–2897. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, V.; Amadoro, G.; Gentile, A.; Capsoni, S.; Ciotti, M.T.; Cencioni, M.T.; Atlante, A.; Canu, N.; Rohn, T.T.; Cattaneo, A.; et al. Identification of a caspase-derived N-terminal tau fragment in cellular and animal Alzheimer’s disease models. Mol. Cell. Neurosci. 2008, 38, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Amadoro, G.; Bobba, A.; de Bari, L.; Corsetti, V.; Pappalardo, G.; Marra, E.; Calissano, P.; Passarella, S. A peptide containing residues 26–44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim. Biophys. Acta 2008, 1777, 1289–12300. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Valenti, D. A Walk in the Memory, from the First Functional Approach up to Its Regulatory Role of Mitochondrial Bioenergetic Flow in Health and Disease: Focus on the Adenine Nucleotide Translocator. Int. J. Mol. Sci. 2021, 22, 4164. [Google Scholar] [CrossRef]
- Kirdajova, D.B.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity from the Perspective of Glial Cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef]
- Amadoro, G.; Serafino, A.L.; Barbato, C.; Ciotti, M.T.; Sacco, A.; Calissano, P.; Canu, N. Role of N-terminal tau domain integrity on the survival of cerebellar granule neurons. Cell Death Differ. 2004, 11, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Connolly, N.M.C.; Prehn, J.H.M. The metabolic response to excitotoxicity-lessons from single-cell imaging. J. Bioenerg. Biomembr. 2015, 47, 75–88. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH(2)-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33, e1–e25. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Giannattasio, S.; Bobba, A.; Gagliardi, S.; Petragallo, V.; Calissano, P.; Marra, E.; Passarella, S. An increase in the ATP levels occurs in cerebellar granule cells en route to apoptosis in which ATP derives from both oxidative phosphorylation and anaerobic glycolysis. Biochim. Biophys. Acta 2005, 1708, 50–62. [Google Scholar] [CrossRef]
- Atlante, A.; Bobba, A.; Calissano, P.; Passarella, S.; Marra, E. The apoptosis/necrosis transition in cerebellar granule cells depends on the mutual relationship of the antioxidant and the proteolytic systems which regulate ROS production and cytochrome c release en route to death. J. Neurochem. 2003, 84, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Vercesi, A.E.; Kowaltowski, A.J.; Grijalba, M.T.; Meinicke, A.R.; Castilho, R.F. The role of reactive oxygen species in mitochondrial permeability transition. Biosci. Rep. 1997, 17, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Passarella, S.; Quagliariello, E.; Moreno, G.; Salet, C. Haematoporphyrin derivative (Photofrin II) photosensitization of isolated mitochondria: Inhibition of ADP/ATP translocator. J. Photochem. Photobiol. 1989, B4, 35–46. [Google Scholar] [CrossRef]
- Fonyò, A. SH-group reagents as tools in the study of mitochondrial anion transport. J. Bioenerg. Biomembr. 1978, 10, 171–194. [Google Scholar] [CrossRef]
- Aprille, J.R.; Asimakis, G.K. Postnatal development of rat liver mitochondria: State 3 respiration, adenine nucleotide translocase activity, and the net accumulation of adenine nucleotides. Arch. Biochem. Biophys. 1980, 201, 564–575. [Google Scholar] [CrossRef]
- Austin, J.; Aprille, J.R. Carboxyatractyloside-insensitive influx and efflux of adenine nucleotides in rat liver mitochondria. J. Biol. Chem. 1984, 259, 154–160. [Google Scholar] [CrossRef]
- Bobba, A.; Amadoro, G.; Petragallo, V.A.; Calissano, P.; Atlante, A. Dissecting the molecular mechanism by which NH2htau and Aβ1-42 peptides impair mitochondrial ANT-1 in Alzheimer disease. Biochim. Biophys. Acta 2013, 1827, 848–860. [Google Scholar] [CrossRef]
- Bobba, A.; Amadoro, G.; Azzariti, A.; Pizzuto, R.; Atlante, A. Extracellular ADP prevents neuronal apoptosis via activation of cell antioxidant enzymes and protection of mitochondrial ANT-1. Biochim. Biophys. Acta 2014, 1837, 1338–1349. [Google Scholar] [CrossRef]
- Atlante, A.; Passarella, S.; Quagliariello, E.; Moreno, G.; Salet, C. Carrier thiols are targets of Photofrin II photosensitization of isolated rat liver mitochondria. J. Photochem. Photobiol. 1990, B7, 21–32. [Google Scholar] [CrossRef]
- Corti, A.; De Tata, V.; Pompella, A. Agenti e meccanismi di stress ossidativo nella patologia umana. Ligand Assay 2009, 14, 9–16. [Google Scholar]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef] [PubMed]
- Morelli, A.M.; Ravera, S.; Panfoli, I. The aerobic mitochondrial ATP synthesis from a comprehensive point of view. Open Biol. 2020, 10, 200224. [Google Scholar] [CrossRef] [PubMed]
- Papuć, E.; Rejdak, K. The role of myelin damage in Alzheimer’s disease pathology. Arch. Med. Sci. 2018, 16, 345–351. [Google Scholar] [CrossRef] [PubMed]
- LaRosa, V.; Remacle, C. Insights into the respiratory chain and oxidative stress. Biosci. Rep. 2018, 38, BSR20171492. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Y.; Chen, S.; Lin, Y.; Lai, H.; Chen, B.; Chen, T. Biomedical Application of Reactive Oxygen Species–Responsive Nanocarriers in Cancer, Inflammation, and Neurodegenerative Diseases. Front. Chem. 2020, 8, 838. [Google Scholar] [CrossRef] [PubMed]
- Surgucheva, I.; Sharov, V.S.; Surguchov, A. γ-Synuclein: Seeding of α-synuclein aggregation and transmission between cells. Biochemistry 2012, 51, 4743–4754. [Google Scholar] [CrossRef] [PubMed]
- Banafsheh, A.A.; Sirous, G. Studies on oxidants and antioxidants with a brief glance at their relevance to the immune system. Life Sci. 2016, 146, 163–173. [Google Scholar] [CrossRef]
- Bhatt, S.; Puli, L.; Patil, C.R. Role of reactive oxygen species in the progression of Alzheimer’s disease. Drug Discov. Today 2021, 26, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Redox Potential of GSH/GSSG Couple: Assay and Biological Significance. Methods Enzymol. 2002, 348, 93–112. [Google Scholar] [PubMed]
- Hernández-Almanza, A.; Montañez, J.; Martínez, G.; Aguilar-Jiménez, A.; Contreras-Esquivel, J.C.; Aguilar, C.N. Lycopene: Progress in microbial production. Trends Food Sci. Technol. 2016, 56, 142–148. [Google Scholar] [CrossRef]
- Salehi, B.; Lopez-Jornet, P.; Pons-Fuster López, E.; Calina, D.; Sharifi-Rad, M.; Ramírez-Alarcón, K.; Forman, K.; Fernández, M.; Martorell, M.; Setzer, W.N.; et al. Plant-derived bioactives in oral mucosal lesions: A key emphasis to curcumin, lycopene, chamomile, aloe vera, green tea and coffee properties. Biomolecules 2019, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Imam, M.U.; Zhang, S.; Ma, J.; Wang, H.; Wang, F. Antioxidants mediate both iron homeostasis and oxidative stress. Nutrients 2017, 9, 671. [Google Scholar] [CrossRef]
- Calabrese, V.; Scapagnini, G.; Stella, A.G.; Bates, T.E.; Clark, J.B. Mitochondrial involvement in brain function and dysfunction: Relevance to aging, neurodegenerative disorders and longevity. Neurochem. Res. 2001, 26, 739–764. [Google Scholar] [CrossRef]
- Chamberlain, K.A.; Sheng, Z.-H. Mechanisms for the maintenance and regulation of axonal energy supply. J. Neurosci. Res. 2019, 97, 897–913. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative stress in neurodegenerative diseases: From molecular mechanisms to clinical applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Castelli, V.; Benedetti, E.; Antonosante, A.; Catanesi, M.; Pitari, G.; Ippoliti, R.; Cimini, A.; d’Angelo, M. Neuronal cells rearrangement during aging and neurodegenerative disease: Metabolism, oxidative stress and organelles dynamic. Front. Mol. Neurosci. 2019, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Behl, C. Vitamin E and other antioxidants in neuroprotection. Int. J. Vitam. Nutr. Res. 1999, 69, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Poitelon, Y.; Kopec, A.M.; Belin, S. Myelin fat facts: An overview of lipids and fatty acid metabolism. Cells 2020, 9, 812. [Google Scholar] [CrossRef]
- Icer, M.A.; Arslan, N.; Gezmen-Karadag, M. Effects of vitamin E on neurodegenerative diseases: An update. Acta Neurobiol. Exp. 2021, 81, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, R.A.; Tapia-Monsalves, C. Current the Role of Mitochondrial Impairment in Alzheimer’s Disease Neurodegeneration: The Tau Connection. Neuropharmacology 2020, 18, 1076–1091. [Google Scholar] [CrossRef]
- Kumar, D.; Ganeshpurkar, A.; Kumar, D.; Modi, G.; Gupta, S.K.; Singh, S.K. Secretase inhibitors for the treatment of Alzheimer’s disease: Long road ahead. Eur. J. Med. Chem. 2018, 148, 436–452. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid β-protein. J. Alzheimer’s Dis. 2001, 3, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.K.; Chao, S.P.; Hu, C.J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef]
- Lozupone, M.; Solfrizzi, V.; D’Urso, F.; Di Gioia, I.; Sardone, R.; Dibello, V.; Stallone, R.; Liguori, A.; Ciritella, C.; Daniele, A.; et al. Anti-amyloid-beta protein agents for the treatment of Alzheimer’s disease: An update on emerging drugs. Expert Opin. Emerg. Drugs 2020, 25, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Jeremic, D.; Jiménez-Díaz, L.; Navarro-López, J.D. Past, present and future of therapeutic strategies against amyloid-beta peptides in Alzheimer’s disease: A systematic review. Ageing Res. Rev. 2021, 72, 101496. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Greengard, P.; Gandy, S. Regulated formation of Golgi secretory vesicles containing Alzheimer β-amyloid precursor protein. J. Biol. Chem. 1995, 270, 23243–23245. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.P.; Corbett, N.J.; Kellett, K.A.B.; Hooper, N.M. Tau Proteolysis in the Pathogenesis of Tauopathies: Neurotoxic Fragments and Novel Biomarkers. J. Alzheimer’s Dis. 2018, 63, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Engmann, O.; Giese, K.P. Crosstalk between Cdk5 and GSK3β: Implications for Alzheimer’s Disease. Mol. Neurosci. 2009, 2, 2. [Google Scholar] [CrossRef]
- Nizynski, B.; Dzwolak, W.; Nieznanski, K. Amyloidogenesis of Tau protein. Protein Sci. 2017, 26, 2126–2150. [Google Scholar] [CrossRef]
- Liu, M.C.; Kobeissy, F.; Zheng, W.; Zhang, Z.; Hayes, R.L.; Wang, K.K.W. Dual vulnerability of tau to calpains and caspase-3 proteolysis under neurotoxic and neurodegenerative conditions. ASN Neuro. 2011, 3, e00051. [Google Scholar] [CrossRef]
- La Joie, R.; Visani, A.V.; Baker, S.L.; Brown, J.A.; Bourakova, V.; Cha, J.; Chaudhary, K.; Edwards, L.; Iaccarino, L.; Janabi, M.; et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci. Transl. Med. 2020, 12, eaau5732. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 109 (Suppl. S1), 153–159. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Sang, S.; Zhong, C. Brain Energy Improvement as an Emerging Approach for Alzheimer’s Disease Treatment. Neurosci. Bull. 2021, 37, 892–893. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid β, P-tau and mitochondria. Ageing Res. Rev. 2021, 65, 101208. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid β peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-g.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Sanabria-Castro, A.; Alvarado-Echeverría, I.; Monge-Bonilla, C. Molecular Pathogenesis of Alzheimer’s Disease: An Update. Ann. Neurosci. 2017, 24, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Mutisya, E.M.; Bowling, A.C.; Beal, M.F. Cortical cytochrome oxidase activity is reduced in Alzheimer’s disease. J. Neurochem. 1994, 63, 2179–2184. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Atamna, H.; Boyle, K. Amyloid-β peptide binds with heme to form a peroxidase: Relationship to the cytopathologies of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 3381–3386. [Google Scholar] [CrossRef]
- Bobba, A.; Amadoro, G.; Valenti, D.; Corsetti, V.; Lassandro, R.; Atlante, A. Mitochondrial respiratory chain Complexes I and IV are impaired by β-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion 2013, 13, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene 2002, 286, 135–141. [Google Scholar] [CrossRef]
- Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi, V.; Di Carlo, M. Mitochondrial dysfunction: Different routes to Alzheimer’s disease therapy. Oxidative Med. Cell. Longev. 2014, 2014, 780179. [Google Scholar] [CrossRef] [PubMed]
- Ankarcrona, M.; Mangialasche, F.; Winblad, B. Rethinking Alzheimer’s disease therapy: Are mitochondria the key? J. Alzheimer’s Dis. 2010, 20 (Suppl. S2), S579–S590. [Google Scholar] [CrossRef] [PubMed]
- Gillardon, F.; Rist, W.; Kussmaul, L.; Vogel, J.; Berg, M.; Danzer, K.; Kraut, N.; Hengerer, B. Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics 2007, 7, 605–616. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Jia, K.; Du, H. Mitochondrial Permeability Transition: A Pore Intertwines Brain Aging and Alzheimer’s Disease. Cells 2021, 10, 649. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Schmitt, K.; Götz, J. Mitochondrial dysfunction—The beginning of the end in Alzheimer’s disease? Separate and synergistic modes of tau and amyloid-β toxicity. Alzheimers Res. Ther. 2011, 3, 15. [Google Scholar] [CrossRef]
- Asik, R.M.; Suganthy, N.; Aarifa, M.A.; Kumar, A.; Szigeti, K.; Mathe, D.; Gulyás, B.; Archunan, G.; Padmanabhan, P. Alzheimer’s Disease: A Molecular View of β-Amyloid Induced Morbific Events. Biomedicines 2021, 9, 1126. [Google Scholar] [CrossRef]
- Lizard, G.; Rouaud, O.; Demarquoy, J.; Cherkaoui-Malki, M.; Iuliano, L. Potential roles of peroxisomes in Alzheimer’s disease and in dementia of the Alzheimer’s type. J. Alzheimer’s Dis. 2012, 29, 241–254. [Google Scholar] [CrossRef]
- Zarrouk, A.; Nury, T.; El Hajj, H.; Gondcaille, C.; Andreoletti, P.; Moreau, T.; Cherkaoui-Malki, M.; Berger, J.; Hammami, M.; Lizard, G.; et al. Potential Involvement of Peroxisome in Multiple Sclerosis and Alzheimer’s Disease: Peroxisome and Neurodegeneration. Adv. Exp. Med. Biol. 2020, 1299, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Kou, J.; Kovacs, G.G.; Höftberger, R.; Kulik, W.; Brodde, A.; Forss-Petter, S.; Hönigschnabl, S.; Gleiss, A.; Brügger, B.; Wanders, R.; et al. Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathol. 2011, 122, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Dorninger, F.; Moser, A.B.; Kou, J.; Wiesinger, C.; Forss-Petter, S.; Gleiss, A.; Hinterberger, M.; Jungwirth, S.; Fischer, P.; Berger, J. Alterations in the Plasma Levels of Specific Choline Phospholipids in Alzheimer’s Disease Mimic Accelerated Aging. J. Alzheimer’s Dis. 2018, 62, 841–854. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.L.; Quaglio, N.B.; de Souza, G.T.; Garcia, R.T.; Dati, L.M.M.; Moreira, W.L.; Loureiro, A.P.D.M.; de Souza-Talarico, J.N.; Smid, J.; Porto, C.S.; et al. Peripheral oxidative stress biomarkers in mild cognitive impairment and Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 26, 59–68. [Google Scholar] [CrossRef]
- Sultana, R.; Butterfield, D.A. Role of oxidative stress in the progression of Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 19, 341–353. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Reed, T.; Perluigi, M.; De Marco, C.; Coccia, R.; Cini, C.; Sultana, R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci. Lett. 2006, 397, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Schmitt, F.A.; Scheff, S.W.; Ding, Q.; Chen, Q.; Butterfield, D.A.; Markesbery, W.R. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 2005, 64, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Kim, S.R. Linking Oxidative Stress and Proteinopathy in Alzheimer’s Disease. Antioxidants 2021, 10, 1231. [Google Scholar] [CrossRef]
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S.P.; Wu, J.F.; Carney, J.M.; et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem. 1995, 65, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Llanos-González, E.; Henares-Chavarino, Á.A.; Pedrero-Prieto, C.M.; García-Carpintero, S.; Frontiñán-Rubio, J.; Sancho-Bielsa, F.J.; Alcain, F.J.; Peinado, J.R.; Rabanal-Ruíz, Y.; Durán-Prado, M. Interplay Between Mitochondrial Oxidative Disorders and Proteostasis in Alzheimer’s Disease. Front. Neurosci. 2020, 13, 1444. [Google Scholar] [CrossRef] [PubMed]
- Coppede, F.; Migliore, L. DNA damage in neurodegenerative diseases. Mutat. Res. 2015, 776, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Sultana, R.; Boyd-Kimball, D.; Poon, H.F.; Cai, J.; Pierce, W.M.; Klein, J.B.; Merchant, M.; Markesbery, W.R.; Butterfield, D.A. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: An approach to understand pathological and biochemical alterations in AD. Neurobiol. Aging 2006, 27, 1564–1576. [Google Scholar] [CrossRef]
- Resende, R.; Moreira, P.I.; Proença, T.; Deshpande, A.; Busciglio, J.; Pereira, C.; Resende Oliveira, C. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic. Biol. Med. 2008, 44, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Akterin, S.; Cowburn, R.F.; Miranda-Vizuete, A.; Jiménez, A.; Bogdanovic, N.; Winblad, B.; Cedazo-Minguez, A. Involvement of glutaredoxin-1 and thioredoxin-1 in β-amyloid toxicity and Alzheimer’s disease. Cell Death Diff. 2006, 13, 1454–1465. [Google Scholar] [CrossRef]
- Cenini, G.; Sultana, R.; Memo, M.; Butterfield, D.A. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer’s disease. J. Cell. Mol. Med. 2008, 12, 987–994. [Google Scholar] [CrossRef]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain glutathione levels–a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol. Psychiatry 2015, 78, 702–710. [Google Scholar] [CrossRef]
- Persson, T.; Popescu, B.O.; Cedazo-Minguez, A. Oxidative stress in Alzheimer’s disease: Why did antioxidant therapy fail? Oxid. Med. Cell. Longev. 2014, 2014, 427318. [Google Scholar] [CrossRef] [PubMed]
- Testa, G.; Staurenghi, E.; Zerbinati, C.; Gargiulo, S.; Iuliano, L.; Giaccone, G.; Fantò, F.; Poli, G.; Leonarduzzi, G.; Gamba, P. Changes in brain oxysterols at different stages of Alzheimer’s disease: Their involvement in neuroinflammation. Redox Biol. 2016, 10, 24–33. [Google Scholar] [CrossRef]
- Burlot, M.A.; Braudeau, J.; Michaelsen-Preusse, K.; Potier, B.; Ayciriex, S.; Varin, J.; Gautier, B.; Djelti, F.; Audrain, M.; Dauphinot, L.; et al. Cholesterol 24-hydroxylase defect is implicated in memory impairments associated with Alzheimer-like Tau pathology. Hum. Mol. Genet. 2015, 24, 5965–5976. [Google Scholar] [CrossRef] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Borger, E.; Aitken, L.; Du, H.; Zhang, W.; Gunn-Moore, J.F.; Shi Du Yan, S. Is amyloid binding alcohol dehydrogenase a drug target for treating Alzheimer’s disease? Curr. Alzheimer Res. 2013, 10, 21–29. [Google Scholar] [PubMed]
- Bhatia, V.; Sharma, S. Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J. Neurol. Sci. 2021, 421, 117253. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Du, H.; Yan, S.; Fang, F.; Wang, C.; Lue, L.-F.; Guo, L.; Chen, D.; Stern, D.M.; Moore, F.J.G. Inhibition of amyloid-β (Aβ) peptide-binding alcohol dehydrogenase-Aβ interaction reduces Aβ accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 2313–2320. [Google Scholar] [CrossRef]
- Misonou, H.; Morishima-Kawashima, M.; Ihara, Y. Oxidative stress induces intracellular accumulation of amyloid β-protein (Aβ) in human neuroblastoma cells. Biochemistry 2000, 39, 6951–6959. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Xu, H.; Shi, Q.; Chen, L.H.; Pedrini, S.; Pechman, D.; Baker, H.; Beal, M.F.; Gandy, S.E.; Gibson, G.E. Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in Alzheimer’s mouse model. Neurobiol. Aging 2009, 30, 1587–1600. [Google Scholar] [CrossRef]
- Massaad, C.A.; Pautler, R.G.; Klann, E. Mitochondrial superoxide: A key player in Alzheimer’s disease. Aging 2009, 1, 758–761. [Google Scholar] [CrossRef][Green Version]
- Cascella, M.; Bimonte, S.; Muzio, M.R.; Schiavone, V.; Cuomo, A. The efficacy of Epigallocatechin-3-gallate (green tea) in the treatment of Alzheimer’s disease: An overview of pre-clinical studies and translational perspectives in clinical practice. Infect. Agents Cancer 2017, 12, 36. [Google Scholar] [CrossRef]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 2001, 21, 8370–8377. [Google Scholar] [CrossRef]
- Monacelli, F.; Acquarone, E.; Giannotti, G.; Borghi, R.; Nencioni, A. Vitamin C, Aging and Alzheimer’s Disease. Nutrients 2017, 9, 670. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Amadoro, G.; Bobba, A.; Latina, V. Functional Foods: An Approach to Modulate Molecular Mechanisms of Alzheimer’s Disease. Cells 2020, 9, 2347. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, V.; Borreca, A.; Latina, V.; Giacovazzo, G.; Pignataro, A.; Krashia, P.; Natale, F.; Cocco, S.; Rinaudo, M.; Malerba, F.; et al. Passive immunotherapy for N-truncated tau ameliorates the cognitive deficits in two mouse Alzheimer’s disease models. Brain Commun. 2020, 2, fcaa039. [Google Scholar] [CrossRef]
- Amadoro, G.; Latina, V.; Balzamino, B.O.; Squitti, R.; Varano, M.; Calissano, P.; Micera, A. Nerve Growth Factor-Based Therapy in Alzheimer’s Disease and Age-Related Macular Degeneration. Front. Neurosci. 2021, 15, 735928. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Jara, C.; Quintanilla, R.A. Contribution of Tau Pathology to Mitochondrial Impairment in Neurodegeneration. Front. Neurosci. 2018, 12, 441. [Google Scholar] [CrossRef]
- Hanger, D.P.; Wray, S. Tau cleavage and tau aggregation in neurodegenerative disease. Biochem. Soc. Trans. 2010, 38, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Kolarova, M.; Garcia-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimer’s Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef]
- Dorostkar, M.M.; Zou, C.; Blazquez-Llorca, L.; Herms, J. Analyzing dendritic spine pathology in Alzheimer’s disease: Problems and opportunities. Acta Neuropathol. 2015, 130, 1–19. [Google Scholar] [CrossRef]
- Matsuo, E.S.; Shin, R.-W.; Billingsley, M.L.; Van deVoorde, A.; O’Connor, M.; Trojanowski, J.Q.; Lee, V.M. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron 1994, 13, 989–1002. [Google Scholar] [CrossRef]
- Cheng, Y.; Bai, F. The Association of Tau with Mitochondrial Dysfunction in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 163. [Google Scholar] [CrossRef] [PubMed]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef]
- Amos, L.A. Microtubule structure and its stabilisation. Org. Biomol. Chem. 2004, 2, 2153–2160. [Google Scholar] [CrossRef]
- Lee, G.; Neve, R.L.; Kosik, K.S. The microtubule binding domain of tau protein. Neuron 1989, 2, 1615–1624. [Google Scholar] [CrossRef]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Iqbal, K. Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, J.H.; Jeon, J.H.; Lee, M.J. Degradation or aggregation: The ramifications of post-translational modifications on tau. BMB Rep. 2018, 51, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, T.; Uryu, K.; Giasson, B.I.; Ischiropoulos, H.; LightFoot, R.; Bellmann, C.; Richter-Landsberg, C.; Lee, V.M.-Y.; Trojanowski, J.Q. Nitration of tau protein is linked to neurodegeneration in tauopathies. Am. J. Pathol. 2003, 163, 1021–1031. [Google Scholar] [CrossRef]
- Ledesma, M.D.; Bonay, P.; Avila, J. τ Protein from Alzheimer’s disease patients is glycated at its tubulin-binding domain. J. Neurochem. 1995, 65, 1658–1664. [Google Scholar] [CrossRef]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M.Y. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef]
- Naini, S.M.A.; Soussi-Yanicostas, N. Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies? Oxid. Med. Cell. Longev. 2015, 2015, 151979. [Google Scholar] [CrossRef]
- De Calignon, A.; Fox, L.M.; Pitstick, R.; Carlson, G.A.; Bacskai, B.J.; Spires-Jones, T.L.; Hyman, B.T. Caspase activation precedes and leads to tangles. Nature 2010, 464, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Rojas, C.; Cabezas-Opazo, F.; Deaton, C.A.; Vergara, E.H.; Johnson, G.V.W.; Quintanilla, R.A. It’s all about tau. Prog. Neurobiol. 2019, 175, 54–76. [Google Scholar] [CrossRef] [PubMed]
- Sinsky, J.; Pichlerova, K.; Hanes, J. Tau Protein Interaction Partners and Their Roles in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2021, 22, 9207. [Google Scholar] [CrossRef] [PubMed]
- Melov, S.; Adlard, P.A.; Morten, K.; Johnson, F.; Golden, T.R.; Hinerfeld, D.; Schilling, B.; Mavros, C.; Masters, C.L.; Volitakis, I.; et al. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS ONE 2007, 2, e536. [Google Scholar] [CrossRef] [PubMed]
- Schulz, K.L.; Eckert, A.; Rhein, V.; Mai, S.; Haase, W.; Reichert, A.S.; Jendrach, M.; Müller, W.E.; Leuner, K. A new link to mitochondrial impairment in tauopathies. Mol. Neurobiol. 2012, 46, 205–216. [Google Scholar] [CrossRef]
- Yao, Y.; Chen, X.; Bao, Y.; Wu, Y. Puerarin inhibits beta amyloid peptide 1–42 induced tau hyperphosphorylation via the wnt/betacatenin signaling pathway. Mol. Med. Rep. 2017, 16, 9081–9085. [Google Scholar] [CrossRef]
- Sun, Y.; Xiao, Q.; Luo, C.; Zhao, Y.; Pu, D.; Zhao, K.; Chen, J.; Wang, M.; Liao, Z. High-glucose induces tau hyperphosphorylation through activation of tlr9-p38mapk pathway. Exp. Cell Res. 2017, 359, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, B.; Tung, E.J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur. J. Neurosci. 2007, 26, 3429–3436. [Google Scholar] [CrossRef]
- Ercan-Herbst, E.; Ehrig, J.; Schöndorf, D.C.; Behrendt, A.; Klaus, B.; Ramos, B.G.; Oriol, N.P.; Weber, C.; Ehrnhoefer, D.E. A post-translational modification signature defines changes in soluble tau correlating with oligomerization in early stage Alzheimer’s disease brain. Acta Neuropathol. Commun. 2019, 7, 192. [Google Scholar] [CrossRef]
- Luna-Viramontes, N.I.; Campa-Córdoba, B.B.; Ontiveros-Torres, M.; Harrington, C.R.; Villanueva-Fierro, I.; Guadarrama-Ortíz, P.; Garcés-Ramírez, L.; De La Cruz, F.; Hernandes-Alejandro, M.; Martínez-Robles, S.; et al. PHF-Core Tau as the Potential Initiating Event for Tau Pathology in Alzheimer’s Disease. Front. Cell. Neurosci. 2020, 14, 247. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Tammineni, P. Mitochondrial aspects of synaptic dysfunction in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1087–1103. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. Oxidative stress in Alzheimer disease as a target for therapy. Bratisl. Med. J. 2018, 119, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.; Hanger, D.P.; Miller, C.C.; Lovestone, S. The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 2013, 4, 83. [Google Scholar] [CrossRef]
- Amadoro, G.; Latina, V.; Corsetti, V.; Calissano, P. N-terminal tau truncation in the pathogenesis of Alzheimer’s disease (AD): Developing a novel diagnostic and therapeutic approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165584. [Google Scholar] [CrossRef]
- Gamblin, T.C.; Chen, F.; Zambrano, A.; Abraha, A.; Lagalwar, S.; Guillozet, A.L.; Lu, M.; Fu, Y.; Garcia-Sierra, F.; Lapointe, N.; et al. Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 10032–10037. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Corsetti, V.; Stringaro, A.; Colone, M.; D’Aguanno, S.; Meli, G.; Ciotti, M.; Sancesario, G.; Cattaneo, A.; Bussani, R.; et al. A NH2 tau fragment targets neuronal mitochondria at AD synapses: Possible implications for neurodegeneration. J. Alzheimer’s Dis. 2010, 21, 445–470. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.M.; Murale, D.P.; Kim, Y.K.; Lee, J.S. Crosstalk between Oxidative Stress and Tauopathy. Int. J. Mol. Sci. 2019, 20, 1959. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Xia, Y.; Yu, G.; Shu, X.; Ge, H.; Zeng, K.; Wang, J.; Wang, X. Cleavage of GSK-3β by calpain counteracts the inhibitory effect of Ser9 phosphorylation on GSK-3β activity induced by H2O2. J. Neurochem. 2013, 126, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The Key Roles of GSK-3beta in Regulating Mitochondrial Activity. Cell. Physiol. Biochem. 2017, 44, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Kim, J.; Park, K.; Lee, J.; Park, S.; Lee, S.; Kim, J.; Hong, I.; Song, B.; Choi, S. GSK-3β activation is required for ZIP-induced disruption of learned fear. Sci. Rep. 2020, 10, 18227. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Koh, S.-H.; Noh, M.Y.; Park, K.-W.; Lee, Y.J.; Kim, S.H. Glycogen synthase kinase-3β activity plays very important roles in determining the fate of oxidative stress inflicted neuronal cells. Brain Res. 2007, 1129, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Morel, M.; Authelet, M.; Dedecker, R.; Brion, J.P. Glycogen synthase kinase-3beta and the p25 activator of cyclin dependent kinase 5 increase pausing of mitochondria in neurons. Neuroscience 2010, 167, 1044–1056. [Google Scholar] [CrossRef]
- Mudher, A.; Shepherd, D.; Newman, T.A.; Mildren, P.; Jukes, J.P.; Squire, A.; Mears, A.; Berg, S.; MacKay, D.; Asuni, A.A.; et al. GSK-3b inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol. Psychiatry 2004, 9, 522–530. [Google Scholar] [CrossRef]
- Ibáñez-Salazar, A.; Bañuelos-Hernandez, B.; Rodriguez-Leyva, I.; Chi-Ahumada, E.; Monreal-Escalante, E.; Jiménez-Capdeville, M.E.; Rosales-Mendoza, S. Oxidative stress modifies the levels and phosphorylation state of tau protein in human fibroblasts. Front. Neurosci. 2017, 11, 495. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Xiong, S.; Xie, C.; Davies, P.; Markesbery, W.R. Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. J. Alzheimer’s Dis. 2004, 6, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, X.; Lee, H.-G.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. J. 2010, 468, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.K.; Jara, C.; Olesen, M.A.; Tapia-Rojas, C. Pathologically phosphorylated tau at S396/404 (PHF-1) is accumulated inside of hippocampal synaptic mitochondria of aged Wild-type mice. Sci. Rep. 2021, 11, 4448. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.D.; Kaech, S.; Banker, G. Selective microtubule-based transport of dendritic membrane proteins arises in concert with axon specification. J. Neurosci. 2014, 34, 4135–4147. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Kundel, F.; Amodeo, G.F.; Pavlov, E.V.; Klenerman, D.; Abramov, A.Y. Insoluble tau aggregates induce neuronal death through modification of membrane ion conductance, activation of voltage-gated calcium channels and NADPH oxidase. FEBS J. 2021, 288, 127–141. [Google Scholar] [CrossRef]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in disease: Timing is everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef] [PubMed]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef]
- Lavich, I.; de Freitas, B.; Kist, L.; Falavigna, L.; Dargél, V.; Köbe, L.; Aguzzoli, C.; Piffero, B.; Florian, P.; Bogo, M.; et al. Sulforaphane rescues memory dysfunction and synaptic and mitochondrial alterations induced by brain iron accumulation. Neuroscience 2015, 301, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Villaflores, O.B.; Chen, Y.J.; Chen, C.P.; Yeh, J.M.; Wu, T.Y. Effects of curcumin and demethoxy curcumin on amyloid-β precursor and tau proteins through the internal ribosome entry sites: A potential therapeutic for Alzheimer’s disease. Taiwan J. Obstet. Gynecol. 2012, 51, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.-L.; Zuo, X.; Yang, F.; Ubeda, O.J.; Gant, D.J.; Alaverdyan, M.; Teng, E.; Hu, S.; Chen, P.-P.; Maiti, P.; et al. Curcumin suppresses soluble tau dimers and corrects molecular chaperone, synaptic, and behavioral deficits in aged human tau transgenic mice. J. Biol. Chem. 2013, 288, 4056–4065. [Google Scholar] [CrossRef] [PubMed]
- Stuerenburg, H.J.; Ganzer, S.; Müller-Thomsen, T. Plasma beta carotene in Alzheimer’s disease. Association with cerebrospinal fluid beta-amyloid 1–40, (A eta40), beta-amyloid 1–42 (A eta42) and total Tau. Neuro Endocrinol. Lett. 2005, 26, 696–698. [Google Scholar] [PubMed]
- Chen, Y.; Wang, C.; Hu, M.; Pan, J.; Chen, J.; Duan, P.; Zhai, T.; Ding, J.; Xu, C. Effects of ginkgolide A on okadaic acid-induced tau hyperphosphorylation and the PI3K-Akt signaling pathway in N2a cells. Planta Med. 2012, 78, 1337–1341. [Google Scholar] [CrossRef]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Cente, M.; Filipcik, P.; Mandakova, S.; Zilka, N.; Krajciova, G.; Novak, M. Expression of a truncated human tau protein induces aqueous-phase free radicals in a rat model of tauopathy: Implications for targeted antioxidative therapy. J. Alzheimer’s Dis. 2009, 17, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-C.; Xia-Chun, L.; Wang, Z.-H.; Luo, Y.; Zhang, Y.; Liu, X.-P.; Feng, Q.; Wang, Q.; Ye, K.; Liu, G.-P.; et al. Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.-G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Bonda, D.J.; Wang, X.; Perry, G.; Smith, M.A.; Zhu, X. Mitochondrial dynamics in Alzheimer’s disease: Opportunities for future treatment strategies. Drugs Aging 2010, 27, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Nat. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, R.A.; Matthews-Roberson, T.A.; Dolan, P.J.; Johnson, G.V. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: Implications for the pathogenesis of Alzheimer disease. J. Biol. Chem. 2009, 284, 18754–18766. [Google Scholar] [CrossRef] [PubMed]
- Briston, T.; Selwood, D.L.; Szabadkai, G.; Duchen, M.R. Mitochondrial Permeability Transition: A Molecular Lesion with Multiple Drug Targets. Trends Pharmacol. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, R.A.; Von Bernhardi, R.; Godoy, J.A.; Inestrosa, N.C.; Johnson, G.V. Phosphorylated tau potentiates Aβ-induced mitochondrial damage in mature neurons. Neurobiol. Dis. 2014, 71, 260–269. [Google Scholar] [CrossRef]
- Corsetti, V.; Florenzano, F.; Atlante, A.; Bobba, A.; Ciotti, M.; Natale, F.; Della Valle, F.; Borreca, A.; Manca, A.; Meli, G.; et al. NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: Implications in Alzheimer’s disease. Hum. Mol. Genet. 2015, 24, 3058–3081. [Google Scholar] [CrossRef] [PubMed]
- Di Monte, D.A.; Tokar, I.; Langston, J.W. Impaired glutamate clearance as a consequence of energy failure caused by MPP(+) in astrocytic cultures. Toxicol. Appl. Pharmacol. 1999, 158, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef]
- Schmitt, K.; Grimm, A.; Kazmierczak, A.; Strosznajder, J.B.; Götz, J.; Eckert, A. Insights into mitochondrial dysfunction: Aging, amyloid-β, and tau-A deleterious trio. Antioxid Redox Signal 2012, 16, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Bolmont, T.; Clavaguera, F.; Meyer-Luehmann, M.; Herzig, M.; Radde, R.; Staufenbiel, M.; Lewis, J.; Hutton, M.; Tolnay, M.; Jucker, M. Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice. Am. J. Pathol. 2007, 171, 2012–2020. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Gotz, J. Amyloid-beta and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef]
- Campion, D.; Pottier, C.; Nicolas, G.; Le Guennec, K.; Rovelet-Lecrux, A. Alzheimer disease: Modeling an Aβ-centered biological network. Mol. Psychiatry 2016, 21, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Gotz, J.; Ittner, L.M.; Kins, S. Do axonal defects in tau and amyloid precursor protein transgenic animals model axonopathy in Alzheimer’s disease? J. Neurochem. 2006, 98, 993–1006. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Pritchard, S.M.; Dolan, P.J.; Vitkus, A.; Johnson, G.V.W. The toxicity of tau in Alzheimer disease: Turnover, targets and potential therapeutics. J. Cell. Mol. Med. 2011, 15, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Kontsekova, E.; Zilka, N.; Novak, M. Ten years of tau-targeted immunotherapy: The path walked and the roads ahead. Front. Neurosci. 2018, 12, 798. [Google Scholar] [CrossRef] [PubMed]
- Latina, V.; Giacovazzo, G.; Cordella, F.; Balzamino, B.O.; Micera, A.; Varano, M.; Marchetti, C.; Malerba, F.; Florio, R.; Ercole, B.B.; et al. Systemic delivery of a specific antibody targeting the pathological N-terminal truncated tau peptide reduces retinal degeneration in a mouse model of Alzheimer’s Disease. Acta Neuropathol. Commun. 2021, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Mendola, J.D.; Cronin-Golomb, A.; Corkin, S.; Growdon, J.H. Prevalence of visual deficits in Alzheimer’s disease. Optom. Vis. Sci. 1995, 72, 155–167. [Google Scholar] [CrossRef]
- Chiasseu, M.; Alarcon-Martinez, L.; Belforte, N.; Quintero, H.; Dotigny, F.; Destroismaisons, L.; Velde, C.V.; Panayi, F.; Louis, C.; Di Polo, A. Tau accumulation in the retina promotes early neuronal dysfunction and precedes brain pathology in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 58. [Google Scholar] [CrossRef]
- Stover, K.R.; Campbell, M.A.; Van Winssen, C.M.; Brown, R.E. Early detection of cognitive deficits in the 3xTg-AD mouse model of Alzheimer’s disease. Behav. Brain Res. 2015, 289, 29–38. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Florenzano, F.; Atlante, A.; Ciotti, M.; Mongiardi, M.; Bussani, R.; Nicolin, V.; Nori, S.; Campanella, M.; et al. AD-linked, toxic NH2 human tau affects the quality control of mitochondria in neurons. Neurobiol. Dis. 2014, 62, 489–507. [Google Scholar] [CrossRef] [PubMed]
- Florenzano, F.; Veronica, C.; Ciasca, G.; Ciotti, M.T.; Pittaluga, A.; Olivero, G.; Feligioni, M.; Iannuzzi, F.; Latina, V.; Sciacca, M.F.M.; et al. Extracellular truncated tau causes early presynaptic dysfunction associated with Alzheimer’s disease and other tauopathies. Oncotarget 2017, 8, 64745–64778. [Google Scholar] [CrossRef]
- Latina, V.; Giacovazzo, G.; Calissano, P.; Atlante, A.; La Regina, F.; Malerba, F.; Dell’Aquila, M.; Stigliano, E.; Balzamino, B.O.; Micera, A.; et al. Tau Cleavage Contributes to Cognitive Dysfunction in Strepto-Zotocin-Induced Sporadic Alzheimer’s Disease (sAD) Mouse Model. Int. J. Mol. Sci. 2021, 22, 12158. [Google Scholar] [CrossRef]
- Kamat, P.-K.; Kalani, A.; Rai, S.; Tota, S.-K.; Kumar, A.; Ahmad, A.-S. Streptozotocin Intracerebroventricular-Induced Neurotoxicity and Brain Insulin Resistance: A Therapeutic Intervention for Treatment of Sporadic Alzheimer’s Disease (sAD)-Like Pathology. Mol. Neurobiol. 2016, 53, 4548–4562. [Google Scholar] [CrossRef]
- Reeta, K.-H.; Singh, D.; Gupta, Y.-K. Edaravone attenuates intracerebroventricular streptozotocin-induced cognitive impairment in rats. Eur. J. Neurosci. 2017, 45, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Reeta, K.-H.; Singh, D.; Gupta, Y.-K. Chronic treatment with taurine after intracerebroventricular streptozotocin injection improves cognitive dysfunction in rats by modulating oxidative stress, cholinergic functions and neuroinflammation. Neurochem. Int. 2017, 108, 146–156. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atlante, A.; Valenti, D.; Latina, V.; Amadoro, G. Role of Oxygen Radicals in Alzheimer’s Disease: Focus on Tau Protein. Oxygen 2021, 1, 96-120. https://doi.org/10.3390/oxygen1020010
Atlante A, Valenti D, Latina V, Amadoro G. Role of Oxygen Radicals in Alzheimer’s Disease: Focus on Tau Protein. Oxygen. 2021; 1(2):96-120. https://doi.org/10.3390/oxygen1020010
Chicago/Turabian StyleAtlante, Anna, Daniela Valenti, Valentina Latina, and Giuseppina Amadoro. 2021. "Role of Oxygen Radicals in Alzheimer’s Disease: Focus on Tau Protein" Oxygen 1, no. 2: 96-120. https://doi.org/10.3390/oxygen1020010
APA StyleAtlante, A., Valenti, D., Latina, V., & Amadoro, G. (2021). Role of Oxygen Radicals in Alzheimer’s Disease: Focus on Tau Protein. Oxygen, 1(2), 96-120. https://doi.org/10.3390/oxygen1020010