
Application of Environmental DNA Metabarcoding to Differentiate Algal Communities by Littoral Zonation and Detect Unreported Algal Species





Abstract
1. Introduction
2. Materials and Methods
2.1. Samples Collecting and Sequencing
2.2. Reads Processing and ASVs Clustering
2.3. Reference Database and Taxonomic Assignment of ASVs
2.4. Alpha and Beta Diversity
2.5. Analyses of Relative Abundances
3. Results
3.1. Alpha Diversity
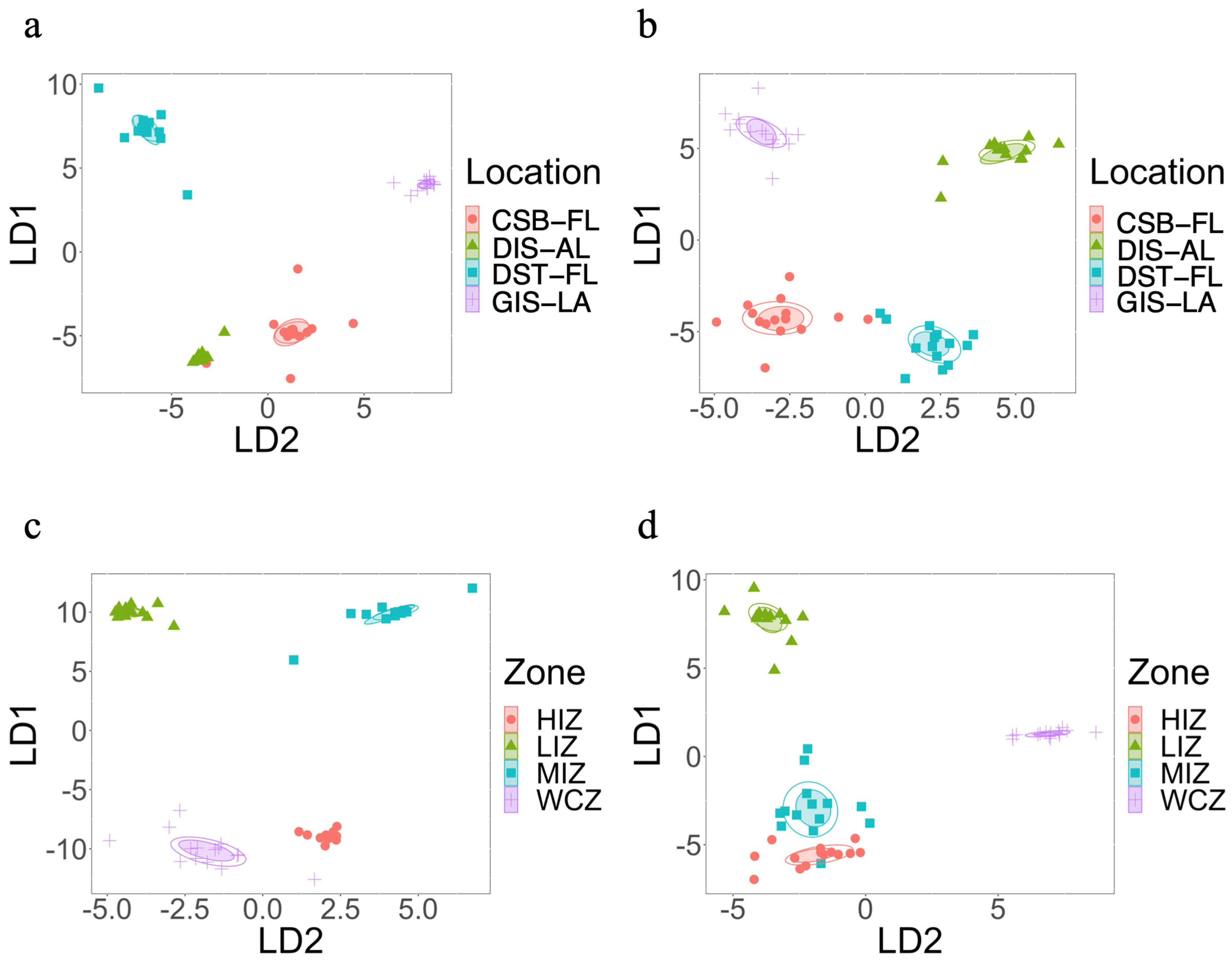
3.2. Beta Diversity Based on Geographical Locations
3.3. Beta Diversity Based on Zonation
3.4. Beta Diversity Based on the Combination of Zone and Location
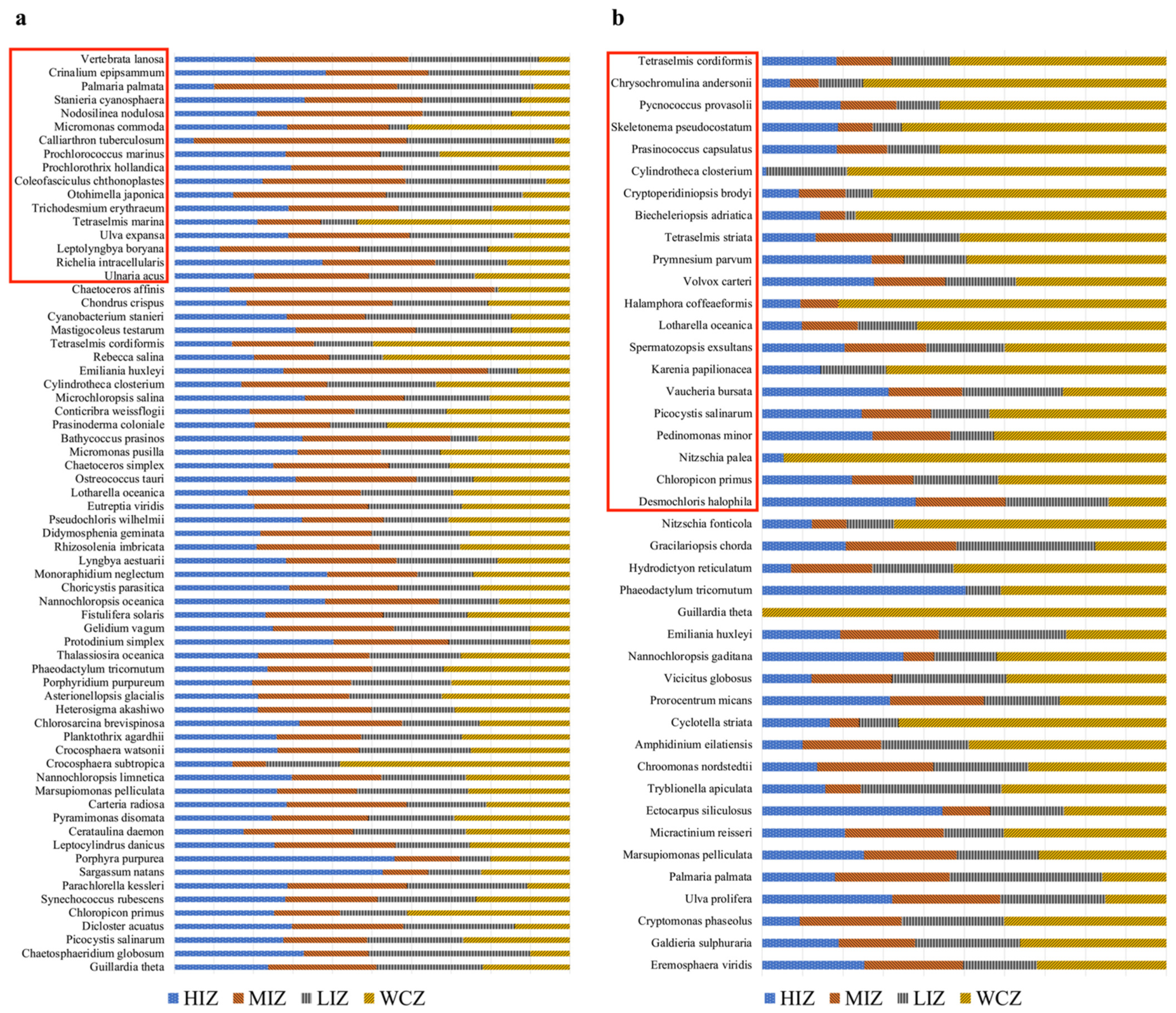
3.5. Algae Richness and Detected Non-Indigenous Species
4. Discussion
4.1. Algal Community Differentiation
4.2. Detected Algal Species and Their Distribution
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ASV | Amplicon sequence variant |
| CSB | Cape San Blas |
| DIS | Dauphin Island |
| DST | Destin |
| eDNA | environmental DNA |
| GIS | Grand Isle |
| HIZ | High-intertidal zone |
| LIZ | Low-intertidal zone |
| MIZ | Middle-intertidal zone |
| NGoM | Northern Gulf of Mexico |
| WCZ | water column zone |
References
- Fautin, D.; Dalton, P.; Incze, L.S.; Leong, J.A.; Pautzke, C.; Rosenberg, A.; Sandifer, P.; Sedberry, G.; Tunnell, J.W., Jr.; Abbott, I.; et al. An overview of marine biodiversity in United States waters. PLoS ONE 2010, 5, e11914. [Google Scholar] [CrossRef]
- Kumpf, H.; Steidinger, K.; Sherman, K. The Gulf of Mexico Large Marine Ecosystem: Assessment, Sustainability, and Management; Blackwell Science: Malden, MA, USA, 1999. [Google Scholar]
- National Oceanic and Atmospheric Administration. The Gulf of Mexico at a Glance: A Second Glance; U.S. Department of Commerce: Washington, DC, USA, 2011. [Google Scholar]
- Tunnell, J.W.; Cato, J.C.; Felder, D.L.; Earle, S.A.; Camp, D.K.; Buster, N.A.; Holmes, C.W. Gulf of Mexico Origin, Waters, and Biota: Volume 2, Ocean and Coastal Economy; Texas A&M University Press: College Station, TX, USA, 2009; Volume 2. [Google Scholar]
- Ward, C.H. Habitats and Biota of the Gulf of Mexico: Before the Deepwater Horizon Oil Spill: Volume 2: Fish Resources, Fisheries, Sea Turtles, Avian Resources, Marine Mammals, Diseases and Mortalities; Springer: Berlin/Heidelberg, Germany, 2017; Volume 2. [Google Scholar]
- Fredericq, S.; Cho, T.O.; Earle, S.A.; Gurgel, C.F.; Krayesky, D.M.; Mateo-Cid, L.E.; Mendoza-González, A.C.; Norris, J.N.; Suárez, A.M. Seaweeds of the Gulf of Mexico. In Gulf of Mexico Origin, Waters and Biota; Texas A&M University Press: College Station, TX, USA, 2009; Volume 1, pp. 187–259. [Google Scholar]
- Davidson, A.D.; Campbell, M.L.; Hewitt, C.L.; Schaffelke, B. Assessing the impacts of non-indigenous marine macroalgae: An update of current knowledge. Bot. Mar. 2015, 58, 55–79. [Google Scholar] [CrossRef]
- Bartolo, A.G.; Zammit, G.; Peters, A.F.; Küpper, F.C. The current state of DNA barcoding of macroalgae in the Mediterranean Sea: Presently lacking but urgently required. Bot. Mar. 2020, 63, 253–272. [Google Scholar] [CrossRef]
- Baker, P.; Baker, S.; Fajans, J. Nonindigenous Marine Species in the Greater Tampa Bay Ecosystem. Tampa Bay Estuary Program Technical Publication, 2004, 02–04. Available online: https://pinellas.wateratlas.usf.edu/upload/documents/NonindigenousMarineSpeciesGreatTampaBayEcosystem.pdf (accessed on 15 February 2021).
- Ji, Y.; Ashton, L.; Pedley, S.M.; Edwards, D.P.; Tang, Y.; Nakamura, A.; Kitching, R.; Dolman, P.M.; Woodcock, P.; Edwards, F.A.; et al. Reliable, verifiable and efficient monitoring of biodiversity via metabarcoding. Ecol. Lett. 2013, 16, 1245–1257. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, J.; Esling, P.; Lejzerowicz, F.; Cedhagen, T.; Wilding, T.A. Environmental monitoring through protist next-generation sequencing metabarcoding: Assessing the impact of fish farming on benthic foraminifera communities. Mol. Ecol. Resour. 2014, 14, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, J.; Lejzerowicz, F.; Apotheloz-Perret-Gentil, L.; Visco, J.; Esling, P. Protist metabarcoding and environmental biomonitoring: Time for change. Eur. J. Protistol. 2016, 55, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Pochon, X.; Wood, S.A.; Keeley, N.B.; Lejzerowicz, F.; Esling, P.; Drew, J.; Pawlowski, J. Accurate assessment of the impact of salmon farming on benthic sediment enrichment using foraminiferal metabarcoding. Mar. Pollut. Bull. 2015, 100, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Ardura, A.; Borrell, Y.J.; Fernández, S.; González Arenales, M.; Martínez, J.L.; Garcia-Vazquez, E. Nuisance Algae in Ballast Water Facing International Conventions. Insights from DNA Metabarcoding in Ships Arriving in Bay of Biscay. Water 2020, 12, 2168. [Google Scholar] [CrossRef]
- Visco, J.A.; Apotheloz-Perret-Gentil, L.; Cordonier, A.; Esling, P.; Pillet, L.; Pawlowski, J. Environmental Monitoring: Inferring the Diatom Index from Next-Generation Sequencing Data. Environ. Sci. Technol. 2015, 49, 7597–7605. [Google Scholar] [CrossRef] [PubMed]
- Apotheloz-Perret-Gentil, L.; Cordonier, A.; Straub, F.; Iseli, J.; Esling, P.; Pawlowski, J. Taxonomy-free molecular diatom index for high-throughput eDNA biomonitoring. Mol. Ecol. Resour. 2017, 17, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Lejzerowicz, F.; Esling, P.; Pillet, L.; Wilding, T.A.; Black, K.D.; Pawlowski, J. High-throughput sequencing and morphology perform equally well for benthic monitoring of marine ecosystems. Sci. Rep. 2015, 5, 13932. [Google Scholar] [CrossRef] [PubMed]
- Aylagas, E.; Rodriguez-Ezpeleta, N. Analysis of Illumina MiSeq Metabarcoding Data: Application to Benthic Indices for Environmental Monitoring. Methods Mol. Biol. 2016, 1452, 237–249. [Google Scholar] [CrossRef]
- Bell, K.L.; Burgess, K.S.; Okamoto, K.C.; Aranda, R.; Brosi, B.J. Review and future prospects for DNA barcoding methods in forensic palynology. Forensic Sci. Int. Genet. 2016, 21, 110–116. [Google Scholar] [CrossRef]
- Cowart, D.A.; Pinheiro, M.; Mouchel, O.; Maguer, M.; Grall, J.; Mine, J.; Arnaud-Haond, S. Metabarcoding is powerful yet still blind: A comparative analysis of morphological and molecular surveys of seagrass communities. PLoS ONE 2015, 10, e0117562. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, A.R.; Dittbern, M.N.; Johnston, E.T.; Conklin, K.Y. A metabarcoding comparison of windward and leeward airborne algal diversity across the Ko ‘olau mountain range on the island of O’ahu, Hawaii. J. Phycol. 2017, 53, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Djemiel, C.; Plassard, D.; Terrat, S.; Crouzet, O.; Sauze, J.; Mondy, S.; Nowak, V.; Wingate, L.; Ogée, J.; Maron, P.-A. µgreen-db: A reference database for the 23S rRNA gene of eukaryotic plastids and cyanobacteria. Sci. Rep. 2020, 10, 5915. [Google Scholar] [CrossRef] [PubMed]
- Lebret, K.; Tesson, S.V.; Kritzberg, E.S.; Tomas, C.; Rengefors, K. Phylogeography of the freshwater raphidophyte Gonyostomum semen confirms a recent expansion in northern Europe by a single haplotype. J. Phycol. 2015, 51, 768–781. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.I.; Vis, M.L. Stream algal biofilm community diversity along an acid mine drainage recovery gradient using multimarker metabarcoding. J. Phycol. 2020, 56, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Chappuis, E.; Terradas, M.; Cefalì, M.E.; Mariani, S.; Ballesteros, E. Vertical zonation is the main distribution pattern of littoral assemblages on rocky shores at a regional scale. Estuar. Coast. Shelf Sci. 2014, 147, 113–122. [Google Scholar] [CrossRef]
- Burkett, V.; Fernandez, L.; Nicholls, R.; Woodroffe, C. Climate change impacts on coastal biodiversity. In Climate Change and Biodiversity in the Americas; Fenech, A., MacIver, D., Dallmeier, F., Eds.; Environment Canada: Ottawa, ON, Canada, 2008; pp. 167–193. [Google Scholar]
- Valdivia, N.; Scrosati, R.A.; Molis, M.; Knox, A.S. Variation in community structure across vertical intertidal stress gradients: How does it compare with horizontal variation at different scales? PLoS ONE 2011, 6, e24062. [Google Scholar] [CrossRef]
- Veiga, P.; Rubal, M.; Vieira, R.; Arenas, F.; Sousa-Pinto, I. Spatial variability in intertidal macroalgal assemblages on the North Portuguese coast: Consistence between species and functional group approaches. Helgol. Mar. Res. 2013, 67, 191. [Google Scholar] [CrossRef]
- Kokabi, M.; Yousefzadi, M.; Razaghi, M.; Feghhi, M.A. Zonation patterns, composition and diversity of macroalgal communities in the eastern coasts of Qeshm Island, Persian Gulf, Iran. Mar. Biodivers. Rec. 2016, 9, 96. [Google Scholar] [CrossRef]
- Sherwood, A.R.; Presting, G.G. Universal primers amplify a 23S rDNA plastid marker in eukaryotic algae and cyanobacteria 1. J. Phycol. 2007, 43, 605–608. [Google Scholar] [CrossRef]
- Leliaert, F.; De Clerck, O.; Verbruggen, H.; Boedeker, C.; Coppejans, E. Molecular phylogeny of the Siphonocladales (Chlorophyta: Cladophorophyceae). Mol. Phylogenet. Evol. 2007, 44, 1237–1256. [Google Scholar] [CrossRef] [PubMed]
- Bombin, S.; Wysor, B.; Lopez-Bautista, J.M. Assessment of littoral algal diversity from the northern Gulf of Mexico using environmental DNA metabarcoding. J. Phycol. 2021, 57, 269–278. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Flyvbjerg, H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 2015, 31, 3476–3482. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016. bioRxiv:081257. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639. [Google Scholar] [CrossRef] [PubMed]
- Guiry, M.; Guiry, G. AlgaeBase. Available online: https://www.algaebase.org (accessed on 11 September 2021).
- Holovachov, O.; Haenel, Q.; Bourlat, S.J.; Jondelius, U. Taxonomy assignment approach determines the efficiency of identification of OTUs in marine nematodes. R. Soc. Open Sci. 2017, 4, 170315. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Barbera, P.; Kozlov, A.M.; Czech, L.; Morel, B.; Darriba, D.; Flouri, T.; Stamatakis, A. EPA-ng: Massively Parallel Evolutionary Placement of Genetic Sequences. Syst. Biol. 2019, 68, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Czech, L.; Barbera, P.; Stamatakis, A. Genesis and Gappa: Processing, analyzing and visualizing phylogenetic (placement) data. Bioinformatics 2020, 36, 3263–3265. [Google Scholar] [CrossRef] [PubMed]
- Tomas, C.R. Identifying Marine Phytoplankton; Elsevier: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Dhargalkar, V.; Kavlekar, D. Seaweeds—A Field Manual. 2004. Available online: https://drs.nio.res.in/drs/bitstream/handle/2264/96/Seaweeds-Manual.pdf?sequence=1&isAllowed=y (accessed on 18 September 2019).
- Woelkerling, W.J. Marine Plants of the Caribbean. A Field Guide from Florida to Brazil. Phycologia 1989, 28, 537. [Google Scholar] [CrossRef]
- Whittaker, R.H. Evolution and measurement of species diversity. Taxon 1972, 21, 213–251. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef]
- Chang, Q.; Luan, Y.; Sun, F. Variance adjusted weighted UniFrac: A powerful beta diversity measure for comparing communities based on phylogeny. BMC Bioinform. 2011, 12, 118. [Google Scholar] [CrossRef]
- Straub, D.; Blackwell, N.; Langarica-Fuentes, A.; Peltzer, A.; Nahnsen, S.; Kleindienst, S. Interpretations of Environmental Microbial Community Studies Are Biased by the Selected 16S rRNA (Gene) Amplicon Sequencing Pipeline. Front. Microbiol. 2020, 11, 550420. [Google Scholar] [CrossRef] [PubMed]
- Reitmeier, S.; Hitch, T.C.; Fikas, N.; Hausmann, B.; Ramer-Tait, A.E.; Neuhaus, K.; Berry, D.; Haller, D.; Lagkouvardos, I.; Clavel, T. Handling of spurious sequences affects the outcome of high-throughput 16S rRNA gene amplicon profiling. ISME Commun. 2021, 1, 31. [Google Scholar] [CrossRef]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Venables, W.; Ripley, B. Random and mixed effects. In Modern Applied Statistics with S; Springer: Berlin/Heidelberg, Germany, 2002; pp. 271–300. [Google Scholar]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]
- Paulson, J.N.; Pop, M.; Bravo, H.C. metagenomeSeq: Statistical analysis for sparse high-throughput sequencing. Bioconductor Package 2013, 1, 191. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Horton, T.; Kroh, A.; Ahyong, S.; Bailly, N.; Boyko, C.B.; Brandão, S.N.; Gofas, S.; Hooper, J.N.A.; Hernandez, F.; Holovachov, O.; et al. World Register of Marine Species (WoRMS). 2020. Available online: https://www.marinespecies.org/ (accessed on 11 September 2021).
- Fukuyama, J.; McMurdie, P.J.; Dethlefsen, L.; Relman, D.A.; Holmes, S. Comparisons of distance methods for combining covariates and abundances in microbiome studies. In Biocomputing 2012; World Scientific: Singapore, 2012; pp. 213–224. [Google Scholar]
- Fukuyama, J. Emphasis on the deep or shallow parts of the tree provides a new characterization of phylogenetic distances. Genome Biol. 2019, 20, 131. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.H.; Strauss, S.Y. More closely related species are more ecologically similar in an experimental test. Proc. Natl. Acad. Sci. USA 2011, 108, 5302–5307. [Google Scholar] [CrossRef]
- Godoy, B.S.; Camargos, L.M.; Lodi, S. When phylogeny and ecology meet: Modeling the occurrence of Trichoptera with environmental and phylogenetic data. Ecol. Evol. 2018, 8, 5313–5322. [Google Scholar] [CrossRef] [PubMed]
- MGnify. ADDOMEx Tier 3 Experiments: Mesocosm Si with Gulf of Mexico Coastal Waters. 2018. Available online: https://www.gbif.org/dataset/552346e7-47c0-425c-9fbd-cb0366759db1 (accessed on 10 May 2021).
- MGnify. Amplicon Sequencing of Tara Oceans DNA Samples Corresponding to Size Fractions for Protists. 2018. Available online: https://www.gbif.org/dataset/d596fccb-2319-42eb-b13b-986c932780ad (accessed on 10 May 2021).
- MGnify. Marine Water Column Samples Targeted Loci Environmental. 2019. Available online: https://www.gbif.org/dataset/51fd12e4-402a-4271-94f8-9d5727af1cda (accessed on 11 May 2021).
- MGnify. Temporal Effect of Plant Diversity and Oiling on Nitrogen Cycling in Marsh Sediments. 2019. Available online: https://www.gbif.org/dataset/93c02d72-d8ac-4396-8522-bd7c8dbbd62d (accessed on 11 May 2021).
- MGnify. Effects of Triclosan on Bacterial Community Composition in Natural Seawater Microcosms. 2019. Available online: https://www.gbif.org/dataset/2d312e17-bd09-45ec-834c-9a372a0c7e8e (accessed on 11 May 2021).
- Titlyanov, E.A.; Titlyanova, T.V.; Li, X.; Huang, H. Common Marine Algae of Hainan Island (Guidebook). In Coral Reef Marine Plants of Hainan Island; Titlyanov, E.A., Titlyanova, T.V., Li, X., Huang, H., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 75–228. [Google Scholar]
- Green, L.A.; Neefus, C.D. The effects of short-and long-term freezing on Porphyra umbilicalis Kützing (Bangiales, Rhodophyta) blade viability. J. Exp. Mar. Biol. Ecol. 2014, 461, 499–503. [Google Scholar] [CrossRef]
- Saunders, G.W.; Jackson, C.; Salomaki, E.D. Phylogenetic analyses of transcriptome data resolve familial assignments for genera of the red-algal Acrochaetiales-Palmariales Complex (Nemaliophycidae). Mol. Phylogenet Evol. 2018, 119, 151–159. [Google Scholar] [CrossRef]
- Rødde, R.S.H.; Vårum, K.M.; Larsen, B.A.; Myklestad, S.M. Seasonal and geographical variation in the chemical composition of the red alga Palmaria palmata (L.) Kuntze. Bot. Mar. 2004, 47, 125–133. [Google Scholar] [CrossRef]
- Stoeckle, B.C.; Beggel, S.; Kuehn, R.; Geist, J. Influence of stream characteristics and population size on downstream transport of freshwater mollusk environmental DNA. Freshw. Sci. 2021, 40, 191–201. [Google Scholar] [CrossRef]
- Deiner, K.; Altermatt, F. Transport distance of invertebrate environmental DNA in a natural river. PLoS ONE 2014, 9, e88786. [Google Scholar] [CrossRef]
- Jane, S.F.; Wilcox, T.M.; McKelvey, K.S.; Young, M.K.; Schwartz, M.K.; Lowe, W.H.; Letcher, B.H.; Whiteley, A.R. Distance, flow and PCR inhibition: E DNA dynamics in two headwater streams. Mol. Ecol. Resour. 2015, 15, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Wacker, S.; Fossøy, F.; Larsen, B.M.; Brandsegg, H.; Sivertsgård, R.; Karlsson, S. Downstream transport and seasonal variation in freshwater pearl mussel (Margaritifera margaritifera) eDNA concentration. Environ. DNA 2019, 1, 64–73. [Google Scholar] [CrossRef]
- Pinevich, A.; Velichko, N.; Ivanikova, N. Cyanobacteria of the genus prochlorothrix. Front. Microbiol. 2012, 3, 173. [Google Scholar] [CrossRef] [PubMed]
- Geiss, U.; Bergmann, I.; Blank, M.; Schumann, R.; Hagemann, M.; Schoor, A. Detection of Prochlorothrix in brackish waters by specific amplification of pcb genes. Appl. Environ. Microbiol. 2003, 69, 6243–6249. [Google Scholar] [CrossRef]
- Molina-Menor, E.; Tanner, K.; Vidal-Verdu, A.; Pereto, J.; Porcar, M. Microbial communities of the Mediterranean rocky shore: Ecology and biotechnological potential of the sea-land transition. Microb. Biotechnol. 2019, 12, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
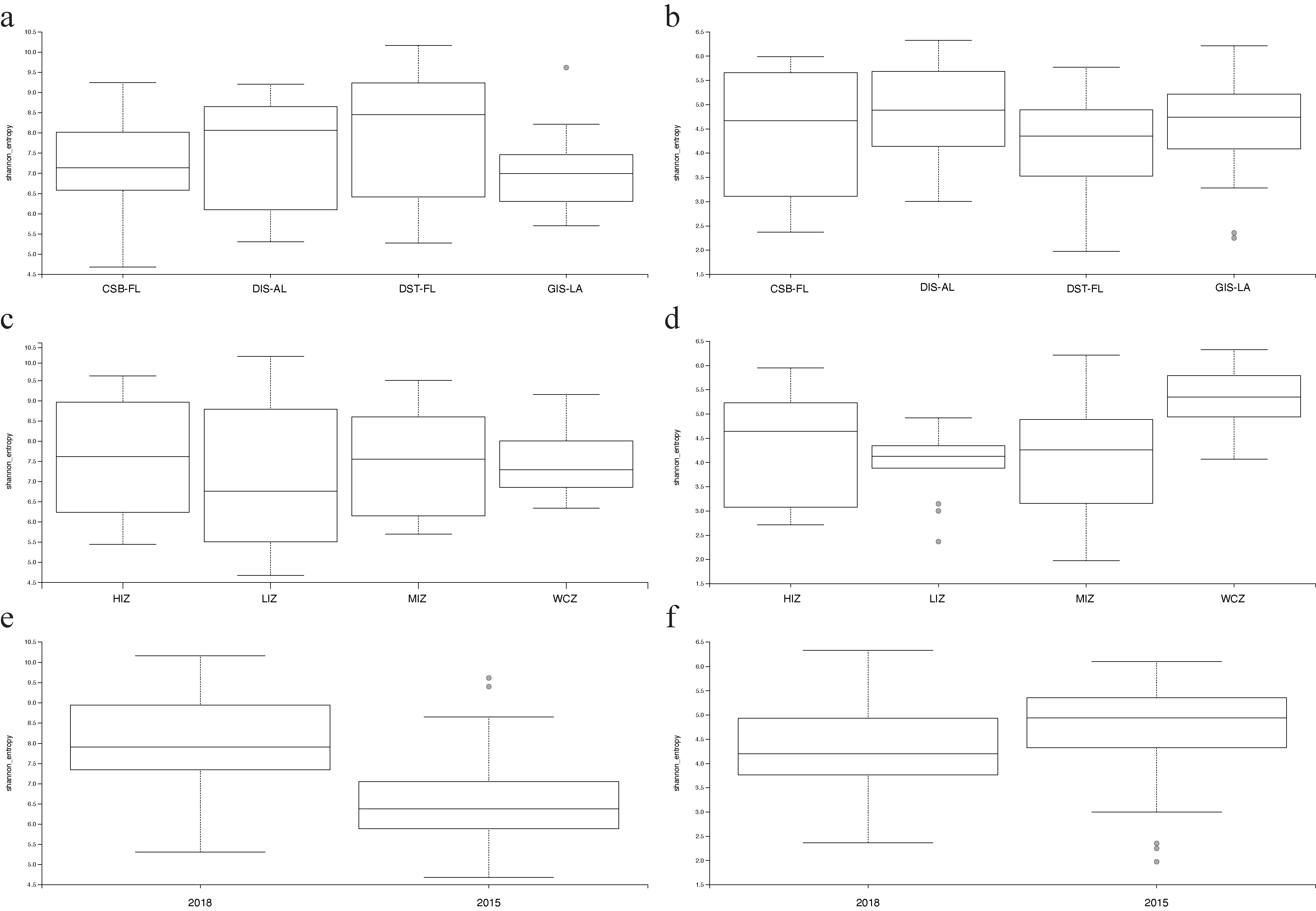

{kind=link}
{kind=link}
{kind=link}
| Samples | Shannon | Faith’s PD | BC | WU |
|---|---|---|---|---|
| UPA-algae | GL, ZN, SY *** | GL, ZN *, SY *** | GL ***, ZN *** | GL ***, ZN *** |
| UPA-all | GL, ZN, SY *** | GL, ZN **, SY | GL ***, ZN ***, | GL ***, ZN *** |
| LSU-algae | GL, ZN **, SY | GL, ZN **, SY | GL ***, ZN *** | GL ***, ZN *** |
| LSU-all | GL, ZN **, SY | GL, ZN **, SY | GL ***, ZN *** | GL ***, ZN *** |
| Markers | Algal Species Total | Native GoM | Marine/Brackish NIS | Inconclusive Status | Freshwater Species |
|---|---|---|---|---|---|
| UPA | 96 | 15 | 33 | 17 | 24 |
| LSU | 42 | 11 | 16 | 9 | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bombin, S.; Bombin, A.; Wysor, B.; Lopez-Bautista, J.M. Application of Environmental DNA Metabarcoding to Differentiate Algal Communities by Littoral Zonation and Detect Unreported Algal Species. Phycology 2024, 4, 605-620. https://doi.org/10.3390/phycology4040033
Bombin S, Bombin A, Wysor B, Lopez-Bautista JM. Application of Environmental DNA Metabarcoding to Differentiate Algal Communities by Littoral Zonation and Detect Unreported Algal Species. Phycology. 2024; 4(4):605-620. https://doi.org/10.3390/phycology4040033
Chicago/Turabian StyleBombin, Sergei, Andrei Bombin, Brian Wysor, and Juan M. Lopez-Bautista. 2024. "Application of Environmental DNA Metabarcoding to Differentiate Algal Communities by Littoral Zonation and Detect Unreported Algal Species" Phycology 4, no. 4: 605-620. https://doi.org/10.3390/phycology4040033
APA StyleBombin, S., Bombin, A., Wysor, B., & Lopez-Bautista, J. M. (2024). Application of Environmental DNA Metabarcoding to Differentiate Algal Communities by Littoral Zonation and Detect Unreported Algal Species. Phycology, 4(4), 605-620. https://doi.org/10.3390/phycology4040033