Simple Summary
Ruminants rely heavily on microorganisms for survival, and the diverse microbial populations in their gastrointestinal tracts are well documented. However, recent studies suggest that microbes may also exist in significant populations outside the gastrointestinal tract, possibly even in the blood. These may include both pathogenic and commensal organisms. Mycoplasma are small, cell wall–lacking bacteria known to be pathogenic, obligate parasites of cattle and other mammals. Hemoplasmas are a subset of Mycoplasma that depend on erythrocytes as hosts. Mycoplasma wenyonii is one such hemoplasma known to infect cattle. While prior research shows that hemoplasma infection is common in dairy cattle and may reduce milk production, its impact on beef cattle remains poorly understood. This study aimed to generate data on bacterial presence in the blood of apparently healthy cattle and assess M. wenyonii infection levels in Texas beef cattle. The results revealed a diverse array of bloodborne bacteria and a high infection rate of M. wenyonii. These findings highlight the issue of chronic Mycoplasma infection in beef cattle and expand our understanding of a possible bovine blood microbiome.
Abstract
Microbiomes have become an increasingly important field of study in the past decade, with data supporting microbial roles in disease control, metabolic efficiency, and more. Microbial DNA is detectable outside the digestive tract, including in blood. Bloodborne pathogens such as hemotrophic Mycoplasma are endemic in cattle. Hemoplasmas are associated with reduced male fertility and decreased milk production in dairy cattle, but their impact on beef cattle remains unclear. Strain variability, such as between the Massachusetts and INFAP01 strains of Mycoplasma wenyonii, may complicate detection. Coinfection with multiple species likely contributes to disease progression from latent to acute infection. To assess microbial DNA in blood and quantify erythrocytic M. wenyonii, blood was collected from 120 beef cattle in Erath County, Texas: 61 cows, 55 calves, and 4 bulls. DNA was extracted and used to prepare 16S rRNA V4 libraries and perform PCR. After rarefaction, ASVs were analyzed and separated into four groups: adult females (n = 61), adult males (n = 4), juvenile males (n = 27), and juvenile females (n = 28). Statistical analysis revealed differences in Actinobacteria by sex (p < 0.001) and higher Bartonella and Mycoplasma abundances in adults (p < 0.001). PCR revealed that M. wenyonii infection was more frequent in adult females (p = 0.006), suggesting age-related variation in infection.
1. Introduction
Bacterial infections are a serious cause of illness in cattle. Over the decades, much time and money has been spent on understanding the lifecycle and virulence factors of bacterial species that are known to cause serious disease. However, there are also rare bacteria responsible for chronic, latent disease that are more challenging to detect and therefore are in need of further study. Furthermore, within the context of latent infection, there is a confounding effect from the possibility of multiple concurrent infections with different species or multiple strains of a single species. A better understanding of such organisms and their complex relationships can lead to better detection and treatment, to allow for increased production outputs of mammalian hosts, as well as animal welfare outcomes.
One such under-studied bacterial pathogen is Mycoplasma wenyonii, a species of hemotrophic Mycoplasma that survives parasitically attached to the erythrocytes of cattle [1]. In healthy animals, the infection often causes few to no outward signs. Severe disease is possible, however, with reports of hemolytic anemia, pyrexia, and internal abscess, often resulting in natural death or euthanasia [2].
Due to the latent nature of the infection, little is known about how this bacterium survives, infects its host’s cells, and is transferred between individuals, as well as the true prevalence rate of infections. Latent hemoplasma infection has been established as a possible etiology of decreased milk production [3]. M. wenyonii infection is also associated with swollen teats, rough coats, decreased milk production, and reduced reproductive productivity in first-calf dairy heifers [4]. Male animals have been found to be aspermic during active infection [5].
Currently, two distinct strains of M. wenyonii have been described. The Massachusetts strain has been sequenced in full [6]. More recently, another strain dubbed INIFAP02 was discovered in cattle from Chihuahua, Mexico, and its genomic draft has been published [7]. A recent study has shown that older detection protocols based on the Massachusetts strain may not be successful at detecting the presence of the Mexican strain [8]. It is likely that other strains exist with unquantified pathogenicity that may or may not be detectable using current methods.
Additionally, there is the possibility of interaction with other pathogenic bacterial species. This includes the closely related Candidatus Mycoplasma haemobos, a more recently discovered hemotrophic Mycoplasma that is being shown to be similarly prevalent or even more abundant than M. wenyonii [9]. Other bloodborne pathogens can also present an infection risk for cattle, such as those belonging to genus Anaplasma, which are thought to be transferred by ticks [8]. Another recent study has shown members of Rickettsia, Babesia, and Borrelia to be present in cattle located in temperate regions [10]. Bartonella has also been widely reported in cattle herds, showing a preference for pregnant females [11]. These other genera of hemotrophic bacteria have confounded the reliability of Mycoplasma detection tools, and since most cannot currently be cultured in the laboratory, scientists and veterinarians must rely heavily on DNA-based screening tools.
There is room for greater exploration of the bovine blood microbiome. Recent studies in humans have shown that there may be a substantial endogenous blood microbiome, contradictory to the previously accepted belief that blood must be sterile, only experiencing transient invasion from migratory microbial visitors from other flora-rich sites in the body [12]. It has definitively been shown that there is a robust bovine-specific microbiome in the feces, ruminal fluid, and milk of cattle, but little is known concerning the microbial populations that may reside in blood. This is likely due to the inability to culture many bloodborne microbes.
For many decades, the standard method for discovering more about a bacterium has been by studying the species in culture. However, to date M. wenyonii and any of the related hemotrophic Mycoplasma species have not been successfully cultured [13]. For this reason, it is necessary to utilize alternative methods to investigate such species. One of the most effective ways to conduct a survey of a bacterial population is through the process of 16S rRNA gene sequencing. 16S rRNA sequencing involves sequencing the ribosomal RNA gene that codes for a variable region of the 16S ribosomal subunit. Within the 16S gene, there exist multiple regions of greater sequence variability, known as Highly Variable Regions, including the frequently targeted V4 region [14]. Upon PCR amplification and subsequent sequencing, 16S sequence data is used to group similar sequences together to form Operational Taxonomic Units (OTU), or alternatively Amplicon Sequence Variant (ASV) tables, which are then compared to established reference databases [15]. The level of similarity between the sequences indicates how the organisms that are present may be related taxonomically. This technology allows for a quick and efficient survey of the relative differences in microbial populations within and between different samples. However, there are limitations to the inferences that can be made about absolute numbers of microbes present. Also, the presence of 16S microbial DNA does not directly correlate to a living microbial presence in the sample tissue, because the DNA may remain in samples for some time following microbial cell death. An RNA-based approach is necessary to confirm that living microbes are present in samples, or methods that degrade DNA from non-living cells are required [16].
In this study, we have employed a 16S rRNA V4 sequencing approach, in conjunction with both digital polymerase chain reaction (dPCR), and quantitative PCR (qPCR), to survey the microbial population present in bovine blood and examine the variability of the M. wenyonii population therein. We hypothesized that bovine blood would contain diverse microbial populations, similar to the blood microbiomes found in other mammalian species, including pathogenic species, commensals, and obligate cellular parasites. We also hypothesized that multiple strains of M. wenyonii may be present within the samples. Third, we expected that cow, calf, and bull categories may have different levels of species abundance and different dominant strains of Mycoplasma. Finally, it is hypothesized that the INFAP01 strain of M. wenyonii from Mexico will be present in this Texas herd in differing proportions.
2. Materials and Methods
2.1. Sampling
Prior to the start of the study, blood was collected by a local veterinarian and veterinary staff from a single herd of beef cattle located in Erath County, Texas, during the course of routine veterinary care. Jugular arterial blood was taken from 61 adult females, 55 of their calves, and 4 adult bulls. Samples were collected in 10 mL ethylenediaminetetraacetic acid (EDTA) tubes to prevent coagulation. Upon receipt by the university, blood was separated into 200 µL aliquots, stored in 1.5 mL flip-cap tubes, and frozen at −20 °C until DNA extraction.
2.2. Extraction
DNA extraction was performed utilizing a QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to manufacturer instructions. The final 100 µL elution was stored frozen at −20 °C for later use.
2.3. 16S Library Preparation
16S rRNA V4 sequencing amplicons were created with Illumina-tagged universal prokaryotic primers 515F-Y 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGTGYCAGCMGCCGCGGTAA-3′ [17] and 806RB 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGGACTACNVGGGTWTCTAAT-3′ [18] according to the Illumina 16S Metagenomic Sequencing Library Preparation Guide and subsequently underwent index PCR for the application of identifying barcodes (Illumina, San Diego, CA, USA). To ensure the quality and quantity of the dsDNA, a Qubit (ThermoFisher Scientific, Waltham, MA, USA) fluorometric quantification dsDNA high sensitivity assay was performed before pooling the samples for sequencing. For size selection of amplicon products, a Pippin (Sage Science, Beverly, MA, USA) preparation protocol was performed to select for appropriately sized fragments in the 300–600 bp range. The samples were sent to the Texas A&M AgriLife Genomics and Bioinformatics Service (College Station, TX, USA) for 300 × 2 bp paired-end MiSeq sequencing using an Illumina v3 600-cycle sequencing kit (Illumina, San Diego, CA, USA).
2.4. Quantitative Real-Time PCR (qPCR)
The first qPCR target utilized the following primer and probe sequences, as reported by Meli and others [19], to target the 16S gene of M. wenyonii: forward 5′-CCACGTGAACGATGAAGGTCTT-3′, reverse 5′-GGCACATAGTTAGCTGTCACTTATTCAA-3′, and finally the probe 5′-Cy5-AGTACCATCAAGGCGCGCTCATTTCCTAG-TQ5-3′ [19]. For the second target, forward and reverse primers, as well as a TaqMan probe, were synthesized according to the template outlined by Persson Waller and others in 2023 [8] to specifically amplify targets similar to the strain of M. wenyonii recently reported in Mexico, INFIAP02, via the polC gene. The probe and primer sequences used were as follows: forward 5′-ATTTGAGCTTACCTCCGCCT-3′, reverse 5′-GAGGATGTCTTTTCCCGCCTAT-3′, and probe 5′-FAM-ACCTTAGAGGAAATTCAAGGCCT-BHQ1-3′ [8].
Standard curves were generated from plasmids for absolute quantification, and gradient PCR was performed to optimize the primer annealing temperature. Serial dilutions of the plasmids listed in Supplemental Table S1 were created to function as positive controls for both targets. The copy number for each plasmid was estimated by running the serial dilution set on both the qPCR instrument and the dPCR instrument. Absolute quantification values for the standards were generated through a dPCR run, allowing comparison of Cq values from the qPCR instrument to estimate the copy number present in each dilution.
Primer concentrations were 400 nM each, forward and reverse, with probes at a 200 nM concentration. Kapa Robust 5X Master Mix (Roche, Basel, Switzerland) was used in a 10 µL reaction with 1 µL template DNA. A Bio-Rad (Hercules, CA, USA) CFX384 Touch real-time PCR detection instrument was used for amplification and fluorescence detection. The PCR protocol included an initial 3 min denaturation step at 95 °C, followed by 40 cycles of 10 s at 95 °C for denaturation and 30 s at 55 °C for annealing. After PCR, the data were transferred to Microsoft Excel 365 MSO Version 2504 (Redmond, WA, USA) Excel for analysis.
2.5. Digital Polymerase Chain Reaction (dPCR)
For dPCR, samples were thawed immediately before preparation and then vortexed and centrifuged briefly to ensure that all contents were adequately mixed. A master solution was then created for each sample reaction that included 23.5 µL molecular water, 7 µL Roche (Roche, Basel, Switzerland) Digital LightCycler® Master Mix 5×, 3.5 µL primer/probe working solution, and finally 1 µL template DNA for a 35 µL total reaction volume, resulting in a final concentration of 100 nM for the probe and 250 nM for each primer. The reagents were mixed in 1.5 mL centrifuge tubes that remained on ice during preparation. All 35 µL reaction mixes were loaded into eight-well Roche Digital LightCycler® Universal Nanowell Plates. The dPCR protocol consisted of incubation for 5 min at 50 °C, initial denaturation for 3 min at 95 °C, and finally 40 cycles of 10 s at 95 °C and 30 s at 60 °C. The initial results were then visualized with Roche Digital LightCycler® Development Software version 1.1 to identify positive nanowell clustering and perform absolute quantification.
2.6. Sequence Pipeline and Statistical Analysis
The raw fastq sequence file was converted into an ASV table using a 64-bit version of Usearch 11.0.6 [20]. Based on average read quality, raw reads were length-trimmed using the fastx_truncate command to 220 bp for the forward reads and 110 bp for the reverse reads before merging with the fastq_mergepairs command. Amplicons lacking both forward and reverse primers were discarded, and primers were trimmed from amplicons using the search_pcr2 command. The resulting full-length primer-trimmed amplicons were quality-filtered to remove any sequences with one or more Ns and any sequences with more than 0.5 expected errors using the fastq_filter command. Quality-filtered sequences were dereplicated with the fastx_uniques command. ASVs were picked as unique zero-radius OTUs, and chimeras were filtered with the unoise3 command. An ASV table was created using the otutab command, and taxonomy was assigned using the sintax command with the Greengenes 13.8 database [21]. Data was rarified to 1000 sequences per sample prior to community analyses, and the taxonomy was edited with a Linux stream editor (sed) command in Ubuntu version 22.04.5 before import into MicrobiomeAnalyst Version 2.0.
MicrobiomeAnalyst Version 2.0 was used to create histograms representing prokaryotic alpha diversity, a Mycoplasma ASV heatmap, and a correlation network among the top three blood pathogen-associated genera [22]. To illustrate alpha diversity, histograms were generated using both Chao1 and Shannon indices, with a Mann–Whitney test performed to determine significance between adult and juvenile populations, and Kruskal–Wallis ANOVA was conducted to determine significance between groups of cows, calves, and bulls. A heatmap was generated to show abundance differences only between Mycoplasma-associated ASVs, with all other ASVs dropped for that purpose. A correlation network was created at the ASV level for genera known for blood infection, with a Spearman ranked correlation test having a correlation threshold of 0.40, a significance threshold < 0.05, and 100 permutations.
To visualize differences in the blood parasite microbiome, PCOa plots were created using PRIMER7 software (Primer-e, Quest Research Limited, Auckland, NZ, USA). Plots used a Bray–Curtis similarity matrix as input created from the rarefied, square root-transformed ASV sequence counts from only the families Anaplasmataceae, Bartonellaceae, and Mycoplasmataceae (other ASV sequences were removed from the rarefied ASV tables). PRIMER7 version 7.0.17 was also used to conduct PERMANOVAs comparing both bull-cow-calf groups and male-female groups of samples.
Mycoplasma ASVs were aligned with the MegAlign Pro module in DNASTAR software version 17.6.2.9 (DNASTAR, Madison, WI, USA) to identify SNP locations within the amplicons. An NCBI Nucleotide BLAST+ Version 2.16.0 search against the non-redundant database was used to confirm likely identification of Mycoplasma ASVs.
The phylogenetic tree was generated in DNASTAR MegAlign Pro using an alignment of 16S fragments assembled through Mauve, and the tree itself was compiled with RAxML maximum likelihood estimation and 100 bootstrap permutations. Published genomes from M. wenyonii Massachusetts (CP003703), M. wenyonii INIFAP02 (QKVO00000000), C. m. haemobos (EF460765), and M. pneumoniae 89,925 (CP179160) were used as comparison points to illustrate distances.
Differential abundance analysis was conducted using QIIME 1.9 [23]. Prior to statistical comparisons, ASVs with fewer than 10 sequence counts were filtered from the raw ASV table using the filter_otus_from_otu_table.py script. The ASV table was then normalized by cumulative sum scaling using the normalize_table.py script [24]. Differential abundance between groups of samples was then compared using the differential_abundance.py script using the DESeq2_nbinom flag [25] to produce log fold change values between categorical groups. Relative abundance was visualized through the creation of graphs in Microsoft Excel (Microsoft, Remond, WA, USA).
Statistical analyses were performed in QIIME 1.9 and R Studio version 4.3.1. Differences in sample means for ordinal variables were generated through Kruskal–Wallis ANOVA and post hoc analysis by Wilcoxon test with Bonferroni adjustment for multiple tests. For categorical variable correlation analysis, bootstrapped Spearman rank correlation was utilized with 100 permutations. The results were visualized, and the images were generated, in MicrobiomeAnalyst software Version 2.0.
Frequency of infection across groups was statistically analyzed for both dPCR and qPCR data using a chi-square test. Mean cycles of quantification were analyzed using Kruskal–Wallis nonparametric ANOVA for relative differences across groups.
3. Results
The DNA sequencing run produced a total of 5,545,803 paired-end sequences. A total of 19 blood samples produced an insufficient number of DNA sequences, including 13 calves, 4 adult cows, and 1 bull, and were eliminated from analysis after rarefaction.
Prokaryotic diversity (Figure 1A,B) was compared between adults and juveniles, as well as among the groups of cows, calves, and bulls. Adult animals had significantly greater Chao1 species richness when compared to juveniles by Mann–Whitney test (p < 0.0001). Bulls had the greatest species richness compared to both cows (p < 0.003) and calves (p < 0.002) via Kruskal–Wallis ANOVA. Cows also demonstrated greater species richness when compared to calves (p < 0.0001).
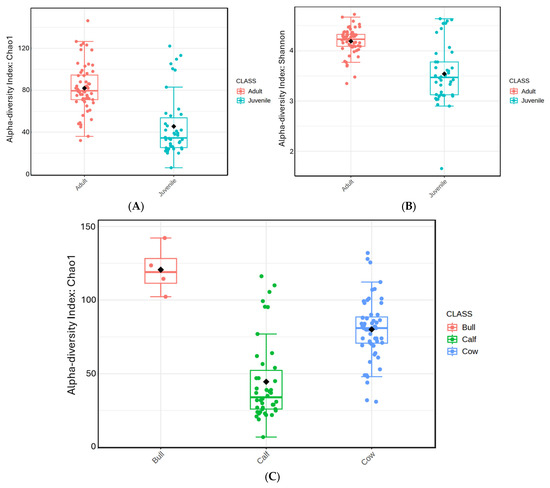
Figure 1.
Box and whisker plot of prokaryotic richness and diversity in blood samples of adult and juvenile animals. (A,C) depict prokaryotic richness and diversity by age and by herd status, respectively, according to the Chao1 index. (B) presents prokaryotic diversity by age according to the Shannon index.
An evaluation of beta diversity among the top three blood-associated genera revealed that cows and bulls clustered separately, with calves overlapping with both groups, eventually moving closer to adult clusters as their age in days increased (Figure 2A). An additional PCoA revealed (Figure 2B) how the relative abundance of the top three blood-associated genera changed within individuals across clusters, with Mycoplasma and Bartonella sequence counts increasing as animal age increased.
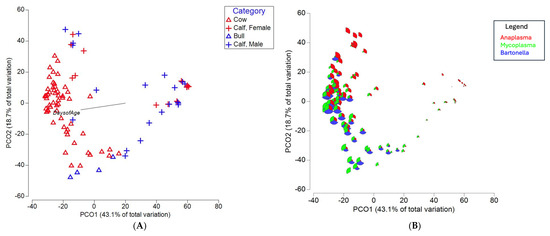
Figure 2.
PCoA plots of known blood pathogens generated from a Bray–Curtis similarity matrix. (A) Clustering of bulls, cows, and calves delineated by sex, with a vector included that indicates increasing age in days. (B) The same PCoA plot in panel A, with samples shown in a bubble plot with proportions of Anaplasma in red, Mycoplasma in green, and Bartonella in blue.
To analyze the relative abundance of the top eight phyla, animals were split into groups of adult females, adult males, female calves, and male calves (Figure 3). Tenericutes (the phylum containing the genus Mycoplasma) abundance was lower in calves than in adults of either sex. When compared to adult females, calves had a significant −2.2 log fold change (p = 0.0003). Compared to adult males, calves tended to have a 4 log fold decrease (p = 0.055). Males had a greater presence of Acidobacteria, with a 3.8 log fold increase over females (p < 0.0001). Adult males (bulls) had the greatest number of Acidobacteria overall, with an 8.2 log fold change over adult female cows (p < 0.0001). Nitrospirae and Firmicutes abundances were significantly greater in bulls, with 6.42 log fold change (p = 0.008) and a 2 log fold change (p < 0.0001), respectively.
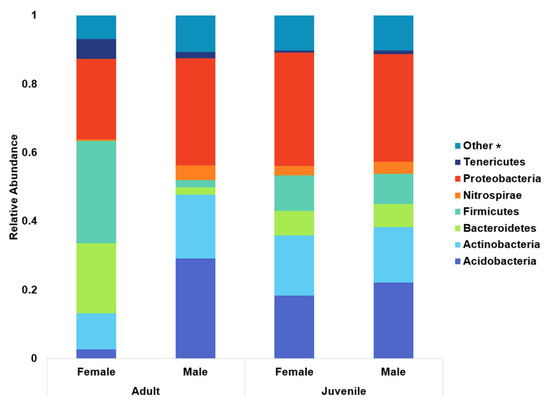
Figure 3.
Relative abundance of the top eight prokaryotic phyla broken down into adult females, adult males, female calves, and male calves. The genus Mycoplasma belongs to phylum Tenericutes, while Anaplasma and Bartonella both belong to Proteobacteria. * Constituent phyla of the “other” category are available in the Supplemental Materials.
The relative abundance of blood parasite families was also separated into groups of calves, cows, and bulls (Figure 4). There was a notable scarcity of Anaplasmataceae in adult male bulls, with a 10.5 log fold reduction when compared to females (p < 0.0001) and a 10.7 log fold reduction compared to calves (p < 0.0001). Adults of both sexes were more similar when compared to calves, with juveniles of both sexes having less of both Bartonellaceae (p < 0.0001) and Mycoplasmataceae (p < 0.0001) compared to their adult counterparts.
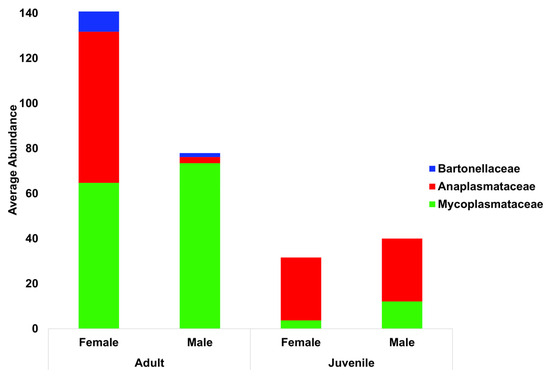
Figure 4.
Blood parasite total abundance at the family level, with animals grouped by both life stage and sex. Bartonellaceae are indicated in blue, Anaplasmataceae in red, and Mycoplasmataceae in green.
Phylogenic comparison of the ASVs identified in this project revealed observable differences between the ASVs found here and published strains of both M. wenyonii and M. c. haemobos (Figure 5).
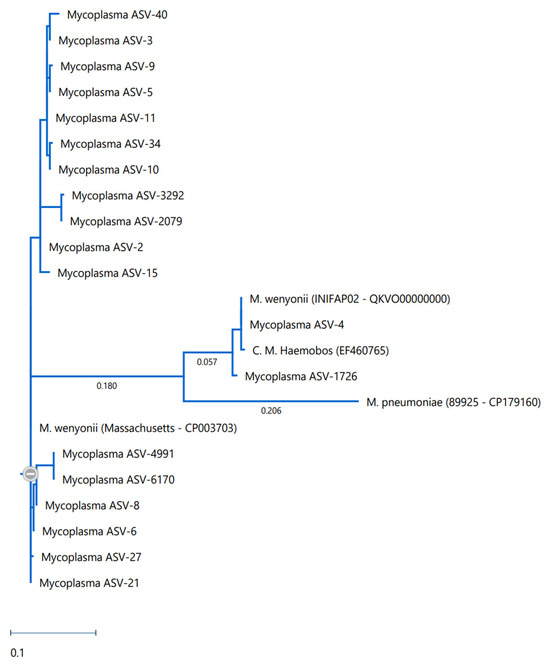
Figure 5.
Phylogenic comparison of known M. wenyonii strains (Massachusetts and INIFAP02) and M. pneumoniae, alongside the C. m. haemobos and M. wenyonii ASVs identified in this study. The circled minus sign represents the collapse point of the tree as generated by the software.
A total of 19 unique ASV clusters were identified after rarefaction that likely belong to genus Mycoplasma. Sequence fragments were not long enough to identify with greater specificity than genus. Mycoplasma ASV differences were illustrated with a heatmap image based on differences generated from Kruskal–Wallis ANOVA (Figure 6). For bulls, ASV 2, 5, 8, 27, and 40 had the most significant differences from cows and calves, while ASV 15 and 2079 stood out distinctly as only appearing in adult females. Calves had fewer Mycoplasma and Bartonella ASVs and sequence counts than bulls or cows.
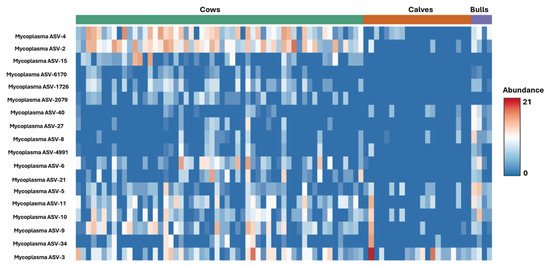
Figure 6.
Heatmap of Mycoplasma ASV distribution across bulls, cows, and calves.
A blood parasite correlation network at the ASV level showed that most Anaplasma ASVs clustered with each other, apart from both Mycoplasma and Bartonella ASVs, which tended to cluster together (Figure 7).
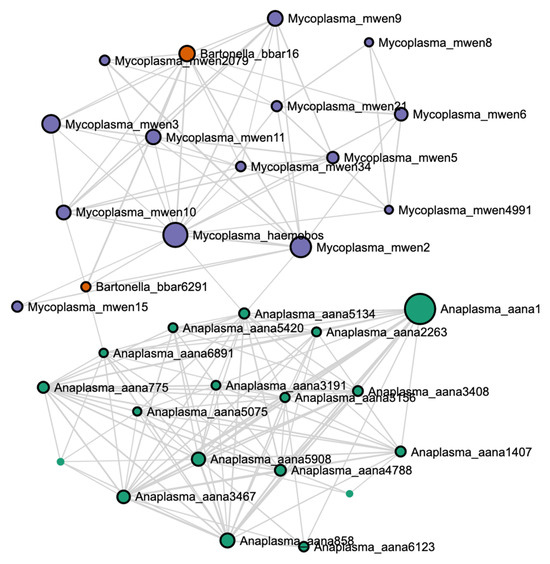
Figure 7.
Blood parasite correlation network at the ASV level created with a Spearman ranked correlation test with a correlation threshold set at 0.40, significance threshold at 0.05, and 100 bootstrap permutations.
When comparing the average CQ values among groups for the 16S qPCR assay (Table 1), there were significant differences across groups (Kruskal–Wallis χ2 = 10.319, df = 2, p = 0.006), with cows being significantly different from calves (p = 0.027). There was a tendency toward differences in copy number between cows and calves for the 16S dPCR assay (Kruskal–Wallis chi-squared = 5.6091, df = 2, p = 0.06054; adjusted Dunn’s p-value = 0.05874288). Statistical differences in bacterial load for the PolC assay were not possible between qPCR and dPCR when comparing the bull, cow, and calf groups due to low numbers of positive qPCR samples. Overall, the group had a higher infection rate when tested broadly for M. wenyonii infection, with only a few animals testing positive for the INFAP02 strain.

Table 1.
Comparison of the number of infected animals using either the polC (INIFAP02 strain specific) targeted probe or the 16S probe (all M. wenyonii), as well as comparison between the numbers of positive animals using dPCR vs. qPCR. The average copy number (ACN) is included for all assays and animal groups. Values that are different from each other across rows are represented by superscripts.
4. Discussion
The blood microbiome is an emerging area of study, from humans to cattle. Traditionally, blood is considered a sterile environment, and yet, recent studies have shown that bacteria are present throughout most tissues, including blood [26]. When sampling blood, there are many possible routes of contamination during the process. Even with best sampling practices, there is still the possibility of having a plug of dermis from the needle puncture included in the blood sample. Beyond the issue of sampling contamination, there is also room for debate regarding the permanence of such DNA-detected microbial populations. Currently, there is no agreement as to whether these bacteria represent active populations or simply dormant visitors that are being dealt with by the host immune system. Recent studies have shown that resuscitation and subsequent culture of bacteria extracted from the blood of healthy humans is possible [12]. However, a great deal more evidence is necessary to challenge the prevailing understanding of the sterility of blood. Since the organisms being detected in this study cannot be grown in culture, this could account for their presence being detected only through DNA-based diagnostics.
A recent study conducted in South Africa found a large presence of Firmicutes, Bacteriodetes, Proteobacteria, and Actinobacteria in bovine blood from randomly selected, apparently healthy cattle through 16S rRNA sequencing [27]. Additionally, a 2021 study by Scarsella and others examined the microbiota of feces, milk, and blood in dairy cattle to determine if differences existed between healthy cattle and those with mastitis [28]. That study reported similar relative proportions of bacterial phyla from blood samples, with the authors considering the bacteria present as likely to be dormant. Both studies suggest similar findings to the current study, where a large fraction of the microbial DNA detected in blood belongs to known blood pathogens, but not all. With the methods used in this study, it is impossible to determine if the DNA detected through 16S surveying belonged to living organisms.
As ruminants, cattle have robust microbial populations within their rumen that are used to generate fermentation end-products such as microbial cell proteins and VFAs that are necessary for the animal’s metabolism [29]. According to previous studies, Bacteroidetes and Firmicutes are the most abundant phyla found in the bovine rumen [30]. This is notable in the context of the current study, as, outside of the phyla responsible for blood pathogens, Bacteroidetes and Firmicutes were some of the most abundant phyla found here. Other standout phyla included Acidobacteria and Actinobacteria, which were differentially abundant in males and females and of which many species thrive in lower pH environments [31,32]. Notably, the current study observed a greater abundance of Acidobacteria in adult males than in females or juveniles of either sex. This shift in the microbiome indicated a potential difference in blood pH between bulls and cows. Since blood pH was not directly measured in the study, we have no direct confirmation regarding blood pH differences that may explain the differential abundance of Acidobacteria. There is currently no data supporting pH differences between healthy cattle of different sexes, with pH being highly regulated through homeostatic mechanisms [33]. It is possible that differences in diet and subsequent volatile fatty acid (VFA) concentration could play a role in explaining the differences in phyla abundance across groups. Dietary factors will influence ruminal microbial communities and have subsequent impacts on blood parameters such as pH downstream, particularly in animals experiencing physiological imbalances [34]. Additionally, the rumen environment is one that develops over the course of an animal’s life. After birth, young ruminants very quickly acquire necessary cellulolytic bacteria, but the relative proportions of anaerobes and aerobic bacteria change as the animal develops [29]. The information that was available with the samples included only identification tag number, date of birth, and sex of the animals, but dietary information was not within the scope of the current study.
Many genera are known to be responsible for bacteremia, including the genera found in this study (Anaplasma, Mycoplasma, and Bartonella). These intra-erythrocytic parasites have frequently been found in seemingly healthy cattle [27,35]. However, they have also been implicated in severe disease [36]. The existence of multiple concurrent infections may contribute a valuable piece of the puzzle to understanding the relationship between latent infection and acute disease. An outbreak in Switzerland among dairy cattle of fatal hemolytic anemia led to an investigation that uncovered as many as five blood parasites present within the acutely ill animals [36]. While Anaplasma marginale was considered to be the key player in the outbreak, Mycoplasma were implicated in the expansion of the disease [36]. A 2021 study investigating the connection between Mycoplasma and mastitis found similar results—Mycoplasma populations were greater in cows with mastitis, yet Mycoplasma was not detected in milk samples, suggesting that Mycoplasma may impact host immune response but is not directly responsible for mastitis [28]. A 2022 study in sheep noticed a correlation between Mycoplasma fermentans and the foot-rot-causing pathogen Dichelobacter nodosus. The study was able to determine that M. fermentans-infected skin cells were unable to produce a satisfactory inflammatory response when exposed to D. nodosus, leading to the conclusion that M. fermentans infection leads to susceptibility to other pathogens [37]. These studies highlight the necessity of understanding how pathogenic species interact with one another. When viewing the correlation network in the current study, it can be seen that Anaplasma and Mycoplasma appeared largely uncorrelated to one another. This may reflect a lack of coordinated interaction and could explain why the herd appeared visually healthy despite having a large presence of erythrocytic infectious agents. It was also noted that relative abundance of Anaplasma was greatest in calves, while Mycoplasma numbers were greatest in bulls. This could be related to cumulative exposure events, or could possibly be related to pH differences, as discussed previously. Further investigation is warranted to investigate mechanisms that might explain differences in blood parasite abundance within a single cattle herd.
In addition to the coinfection model contributing to the development of acute disease from latent infection, bacterial load also appears to play a role. The Mycoplasma wenyonii bacterial load was greater in herds that experienced fatal anemia outbreaks than in those that had not [19]. A study of 22 German cattle herds by Ade and others similarly revealed a robust bacterial load of hemotrophic Mycoplasma. Of the 19 samples that identified positively for M. wenyonii in the German study, they had an average bacterial load of 4.29 × 105 bacteria/mL blood [9]. These estimates are similar to the findings of the current study, which place the burden of infection at a similar order of magnitude.
Mode of transmission is yet another grey area when it comes to hemotrophic Mycoplasma. Blood-sucking arthropods have long been considered the most likely suspect, but currently non-vector-specific blood and bodily fluid contact is considered the primary post-natal source of infectious transmission [1]. It is possible that higher contact with both blood and bodily fluid from other infected animals could explain the greater relative quantity of M. wenyonii in the adult animals in this study, as breeding animals do come into greater contact with such sources of infection during sexual reproduction.
The possibility of placental transfer of hemotrophic Mycoplasma has not been fully investigated. It appears that placental transfer may be possible, but not overwhelming. A 2011 study found that 10.5% of calves born to hemoplasma-infected dams were born infected, as tested through precolostral blood samples taken immediately after birth [38]. This may explain the lower relative abundance of Mycoplasma in calves compared to adult female cows found in this study. Conversely, it has been noted that Anaplasma presence is higher in cattle under three years of age [39]. This supports the findings of this study that Anaplasma was more abundant in calves than in adults. This differential abundance across age groups may also help to explain the disparate clustering seen in this study.
Both dPCR and qPCR were used for this study to facilitate comparison between more traditional qPCR techniques and the newer dPCR technology. From this study, it was evident that dPCR was much more effective at detecting low copy number targets and is likely to be used in future studies of blood pathogens.
Of the M. wenyonii DNA detected in this study, there appears to be considerable diversity within the population. Upon phylogenetic comparison, it was seen that many of the M. wenyonii ASVs detected were not identical to published strains. Despite hypothesizing that the INIFAP02 strain, which was originally described from samples associated with Chihuahua, Mexico [7], would be more abundant in Texas due to geographic proximity, this was not found to be the case. The PCR results did show that the INIFAP02 strain was present but was not the primary agent responsible for M. wenyonii infection in this herd. Additionally, the phylogenetic analyses also support this, showing that the ASVs generated from this study were more closely related to the Massachusetts strain than the INIFAP02 strain.
5. Conclusions
The findings from this study demonstrate that there is a large presence of bacterial DNA in bovine blood; however, future studies will be necessary to determine if this DNA represents stable populations or even living organisms, as DNA presence may not indicate living cells. Furthermore, these data suggest that multiple erythrocytic organisms may be present in seemingly healthy herds. Finally, the dPCR data from this study shows that M. wenyonii is detectable in Texas cattle, including examples of the INFAP02 strain, and that infection burden may differ across age groups.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ruminants5030045/s1, Table S1: Sequences of plasmids used in qPCR for absolute quantification; Table S2: Phyla contained in category “Other” in Figure 3.
Author Contributions
Conceptualization, C.L.R., J.A.B., J.L.S., K.B.W., B.D.L. and N.A.B.; methodology, J.A.B. and J.L.S.; software, J.A.B.; validation, J.A.B., K.T. and N.A.B.; formal analysis, J.A.B., K.T. and N.A.B.; investigation, K.T. and N.A.B.; resources, B.D.L., C.L.R. and J.A.B.; data curation, J.A.B., J.L.S. and N.A.B.; writing—original draft preparation, N.A.B.; writing—review and editing, C.L.R., J.A.B., J.L.S. and K.B.W.; visualization, J.A.B. and N.A.B.; supervision, B.D.L., C.L.R. and J.A.B.; project administration, C.L.R. and K.B.W.; funding acquisition, B.D.L., C.L.R. and J.A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. Internal funding was provided by the Tarleton State University REID President’s Excellence in Research Scholars (PERS) Grant.
Institutional Review Board Statement
The animal material used in this research was collected under the supervision of trained veterinarians prior to the start of this study, and not for the purpose of this research. The analysis for this research study was carried out because the material was already available. Because this research did not direct the manipulation of animals or have any effect on the well-being of animals, it did not require approval of the Tarleton State University Institutional Animal Care and Use Committee.
Data Availability Statement
Sequence data is available from NCBI SRA under project number PRJNA 1314281.
Acknowledgments
The authors would like to extend their sincere thanks to Larry Reid, DVM and his clinic for providing samples and insight into producer concerns. Finally, we would like to thank all student workers, graduate students, and technicians at Texas A&M AgriLife and Tarleton State University who assisted with their time and expertise, with special thanks to Caroly Leija, Maggie Robison, and Harley Scott.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Strugnell, B.; McAuliffe, L. Mycoplasma wenyonii infection in cattle. Practice 2012, 34, 146–154. [Google Scholar] [CrossRef]
- Gladden, N.; Haining, H.; Henderson, L.; Marchesi, F.; Graham, L.; McDonald, M.; Murdoch, F.R.; Bruguera Sala, A.; Orr, J.; Ellis, K. A Case Report of Mycoplasma Wenyonii Associated Immune-Mediated Haemolytic Anaemia in a Dairy Cow. Ir. Vet. J. 2016, 69, 1. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, M.; Yamakawa, K.; Aoki, T.; Matsumoto, K.; Ishii, M.; Inokuma, H. Effect of Chronic Hemoplasma Infection on Cattle Productivity. J. Vet. Med. Sci. 2013, 75, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Thrall, M.A.; Smith, J.L.; Salman, M.D.; Ching, S.V.; Collins, J.K. Eperythrozoon Wenyonii Infection in Dairy Cattle. J. Am. Vet. Med. Assoc. 1990, 196, 1244–1250. [Google Scholar] [CrossRef]
- Montes, A.J.; Wolfe, D.F.; Welles, E.G.; Tyler, J.W.; Tepe, E. Infertility Associated with Eperythrozoon Wenyonii Infection in a Bull. J. Am. Vet. Med. Assoc. 1994, 204, 261–263. [Google Scholar] [CrossRef]
- dos Santos, A.P.; Guimaraes, A.M.S.; do Nascimento, N.C.; SanMiguel, P.J.; Messick, J.B. Complete Genome Sequence of Mycoplasma Wenyonii Strain Massachusetts. J. Bacteriol. 2012, 194, 5458–5459. [Google Scholar] [CrossRef][Green Version]
- Quiroz-Castañeda, R.E.; Martínez-Ocampo, F.; Dantán-González, E. Draft Genome Sequence of Mycoplasma Wenyonii, a Second Hemotropic Mycoplasma Species Identified in Mexican Bovine Cattle. Microbiol. Resour. Announc. 2018, 7, e00875-18. [Google Scholar] [CrossRef]
- Persson Waller, K.; Dahlgren, K.; Grandi, G.; Holding, M.L.; Näslund, K.; Omazic, A.; Sprong, H.; Ullman, K.; Leijon, M. A Disease Outbreak in Beef Cattle Associated with Anaplasma and Mycoplasma Infections. Animals 2023, 13, 286. [Google Scholar] [CrossRef]
- Ade, J.; Niethammer, F.; Schade, B.; Schilling, T.; Hoelzle, K.; Hoelzle, L.E. Quantitative Analysis of Mycoplasma Wenyonii and ‘Candidatus Mycoplasma Haemobos” Infections in Cattle Using Novel gapN-Based Realtime PCR Assays. Vet. Microbiol. 2018, 220, 1–6. [Google Scholar] [CrossRef]
- Adjadj, N.R.; Cargnel, M.; Ribbens, S.; Quinet, C.; Malandrin, L.; Mignon, B.; Mori, M. Prevalence of Anaplasma Phagocytophilum, Borrelia Burgdorferi Sensu Lato, Rickettsia Spp. and Babesia Spp. in Cattle Serum and Questing Ticks from Belgium. Ticks Tick-Borne Dis. 2023, 14, 102146. [Google Scholar] [CrossRef]
- Dahmani, M.; Sambou, M.; Scandola, P.; Raoult, D.; Fenollar, F.; Mediannikov, O. Bartonella Bovis and Candidatus Bartonella Davousti in Cattle from Senegal. Comp. Immunol. Microbiol. Infect. Dis. 2017, 50, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Panaiotov, S.; Filevski, G.; Equestre, M.; Nikolova, E.; Kalfin, R. Cultural Isolation and Characteristics of the Blood Microbiome of Healthy Individuals. Adv. Microbiol. 2018, 8, 406–421. [Google Scholar] [CrossRef]
- Hemotropic Mycoplasma Infections in Animals—Circulatory System. Available online: https://www.merckvetmanual.com/circulatory-system/blood-parasites/hemotropic-mycoplasma-infections-in-animals (accessed on 8 February 2023).
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A Detailed Analysis of 16S Ribosomal RNA Gene Segments for the Diagnosis of Pathogenic Bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA Gene Sequencing for Species and Strain-Level Microbiome Analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, E.U.; Carroll, K.; Speshock, J.; Brady, J. A New Technique for Use in Culturing Prokaryotes Comprising the Mouse Intestinal Microbiome. Adv. Microbiol. 2023, 13, 119–147. [Google Scholar] [CrossRef]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef]
- Meli, M.L.; Willi, B.; Dreher, U.M.; Cattori, V.; Knubben-Schweizer, G.; Nuss, K.; Braun, U.; Lutz, H.; Hofmann-Lehmann, R. Identification, Molecular Characterization, and Occurrence of Two Bovine Hemoplasma Species in Swiss Cattle and Development of Real-Time TaqMan Quantitative PCR Assays for Diagnosis of Bovine Hemoplasma Infections. J. Clin. Microbiol. 2010, 48, 3563–3568. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R. Hugenholtz An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Lu, Y.; Zhou, G.; Ewald, J.; Pang, Z.; Shiri, T.; Xia, J. MicrobiomeAnalyst 2.0: Comprehensive Statistical, Functional and Integrative Analysis of Microbiome Data. Nucleic Acids Res. 2023, 51, W310–W318. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Castillo, D.J.; Rifkin, R.F.; Cowan, D.A.; Potgieter, M. The Healthy Human Blood Microbiome: Fact or Fiction? Front. Cell. Infect. Microbiol. 2019, 9, 148. [Google Scholar] [CrossRef]
- Khoza, B.; Byaruhanga, C.; Makgabo, S.M.; Nyangiwe, N.; Mnisi, T.; Nxumalo, S.; Oosthuizen, M.C.; Mnisi, Z.T. Tick Distribution and a cComparative Analysis of Bovine Blood Microbiome in Two Provinces of South Africa Using 16S rRNA PacBio Approach. Front. Trop. Dis. 2024, 5, 1399364. [Google Scholar] [CrossRef]
- Scarsella, E.; Zecconi, A.; Cintio, M.; Stefanon, B. Characterization of Microbiome on Feces, Blood and Milk in Dairy Cows with Different Milk Leucocyte Pattern. Animals 2021, 11, 1463. [Google Scholar] [CrossRef]
- Mizrahi, I. Rumen Symbioses. In The Prokaryotes: Prokaryotic Biology and Symbiotic Associations; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 533–544. ISBN 978-3-642-30194-0. [Google Scholar]
- Ahmad, A.A.; Zhang, J.; Liang, Z.; Du, M.; Yang, Y.; Zheng, J.; Yan, P.; Long, R.; Tong, B.; Han, J.; et al. Age-Dependent Variations in Rumen Bacterial Community of Mongolian Cattle from Weaning to Adulthood. BMC Microbiol. 2022, 22, 213. [Google Scholar] [CrossRef]
- Anandan, R.; Dharumadurai, D.; Manogaran, G.P.; Anandan, R.; Dharumadurai, D.; Manogaran, G.P. An Introduction to Actinobacteria. In Actinobacteria—Basics and Biotechnological Applications; IntechOpen: London, UK, 2016; ISBN 978-953-51-2248-7. [Google Scholar]
- Kalam, S.; Basu, A.; Ahmad, I.; Sayyed, R.Z.; El-Enshasy, H.A.; Dailin, D.J.; Suriani, N.L. Recent Understanding of Soil Acidobacteria and Their Ecological Significance: A Critical Review. Front. Microbiol. 2020, 11, 580024. [Google Scholar] [CrossRef]
- Hamm, L.L.; Nakhoul, N.; Hering-Smith, K.S. Acid-Base Homeostasis. Clin. J. Am. Soc. Nephrol. 2015, 10, 2232. [Google Scholar] [CrossRef]
- Morgante, M.; Gianesella, M.; Casella, S.; Ravarotto, L.; Stelletta, C.; Giudice, E. Blood Gas Analyses, Ruminal and Blood pH, Urine and Faecal pH in Dairy Cows during Subacute Ruminal Acidosis. Comp. Clin. Pathol. 2009, 18, 229–232. [Google Scholar] [CrossRef]
- Nouvel, L.X.; Hygonenq, M.-C.; Catays, G.; Martinelli, E.; Le Page, P.; Collin, É.; Inokuma, H.; Schelcher, F.; Citti, C.; Maillard, R. First Detection of Mycoplasma Wenyonii in France: Identification, Evaluation of the Clinical Impact and Development of a New Specific Detection Assay. Comp. Immunol. Microbiol. Infect. Dis. 2019, 63, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Hofmann-Lehmann, R.; Meli, M.L.; Dreher, U.M.; Gönczi, E.; Deplazes, P.; Braun, U.; Engels, M.; Schüpbach, J.; Jörger, K.; Thoma, R.; et al. Concurrent Infections with Vector-Borne Pathogens Associated with Fatal Hemolytic Anemia in a Cattle Herd in Switzerland. J. Clin. Microbiol. 2004, 42, 3775–3780. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, A.M.; Baumbach, C.-M.; Michler, J.K.; Pickwell, N.D.; Staley, C.E.; Franklin, J.M.; Wattegedera, S.R.; Entrican, G.; Tötemeyer, S. Natural Mycoplasma Infection Reduces Expression of Pro-Inflammatory Cytokines in Response to Ovine Footrot Pathogens. Animals 2022, 12, 3235. [Google Scholar] [CrossRef] [PubMed]
- Hornok, S.; Micsutka, A.; Meli, M.L.; Lutz, H.; Hofmann-Lehmann, R. Molecular Investigation of Transplacental and Vector-Borne Transmission of Bovine Haemoplasmas. Vet. Microbiol. 2011, 152, 411–414. [Google Scholar] [CrossRef]
- Boularias, G.; Azzag, N.; Gandoin, C.; Bouillin, C.; Chomel, B.; Haddad, N.; Boulouis, H.-J. Bovines Harbor a Diverse Array of Vector-Borne Pathogens in Northeast Algeria. Pathogens 2020, 9, 883. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).