
Fundamentals of Diatomic Molecular Spectroscopy


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Angular Momentum Commutators
3.1.1. Invariance for Unitary Transformations
3.1.2. Invariance for Time Reversal or Reversal of Motion
3.2. Diatomic Wave Function
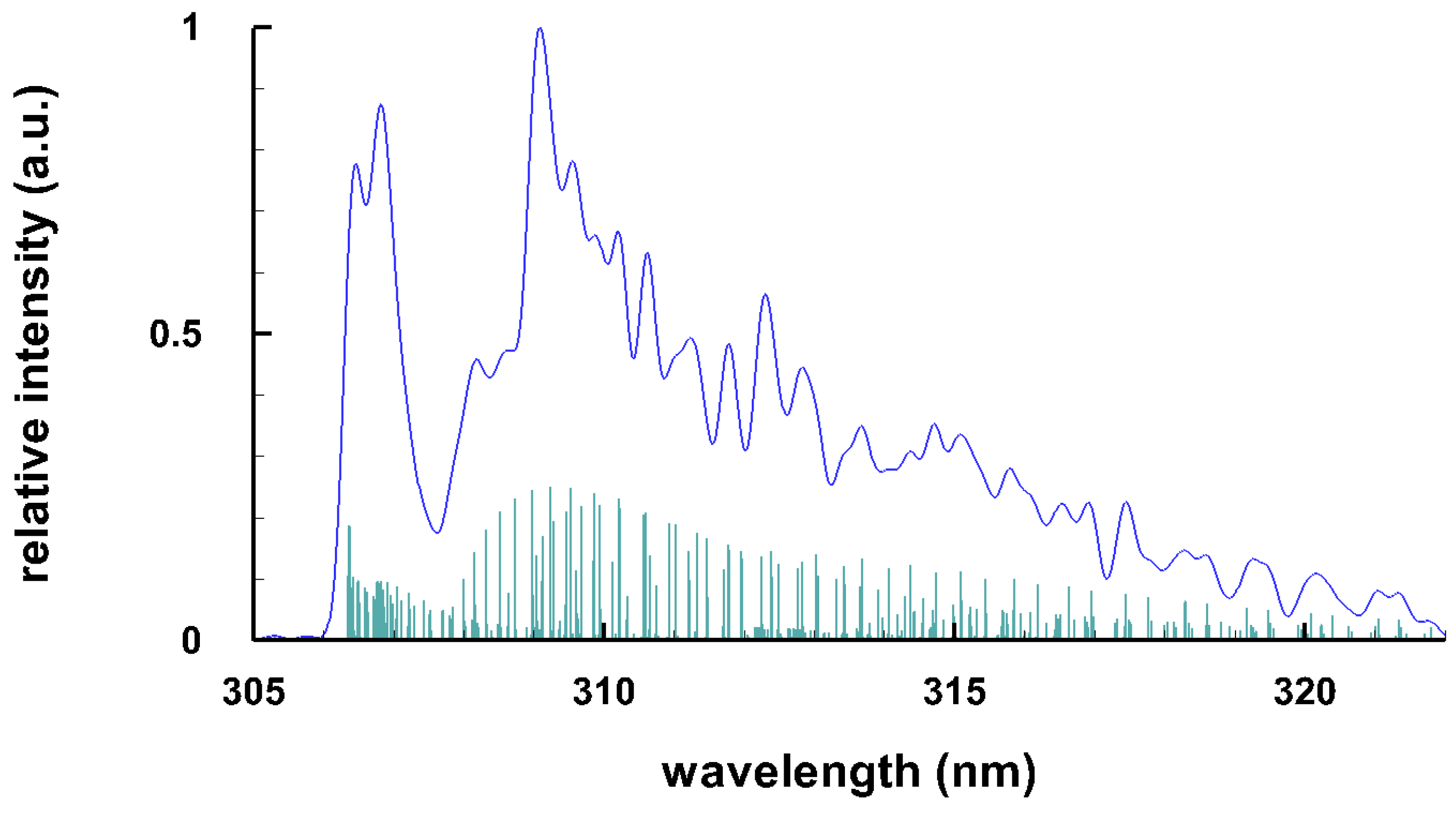
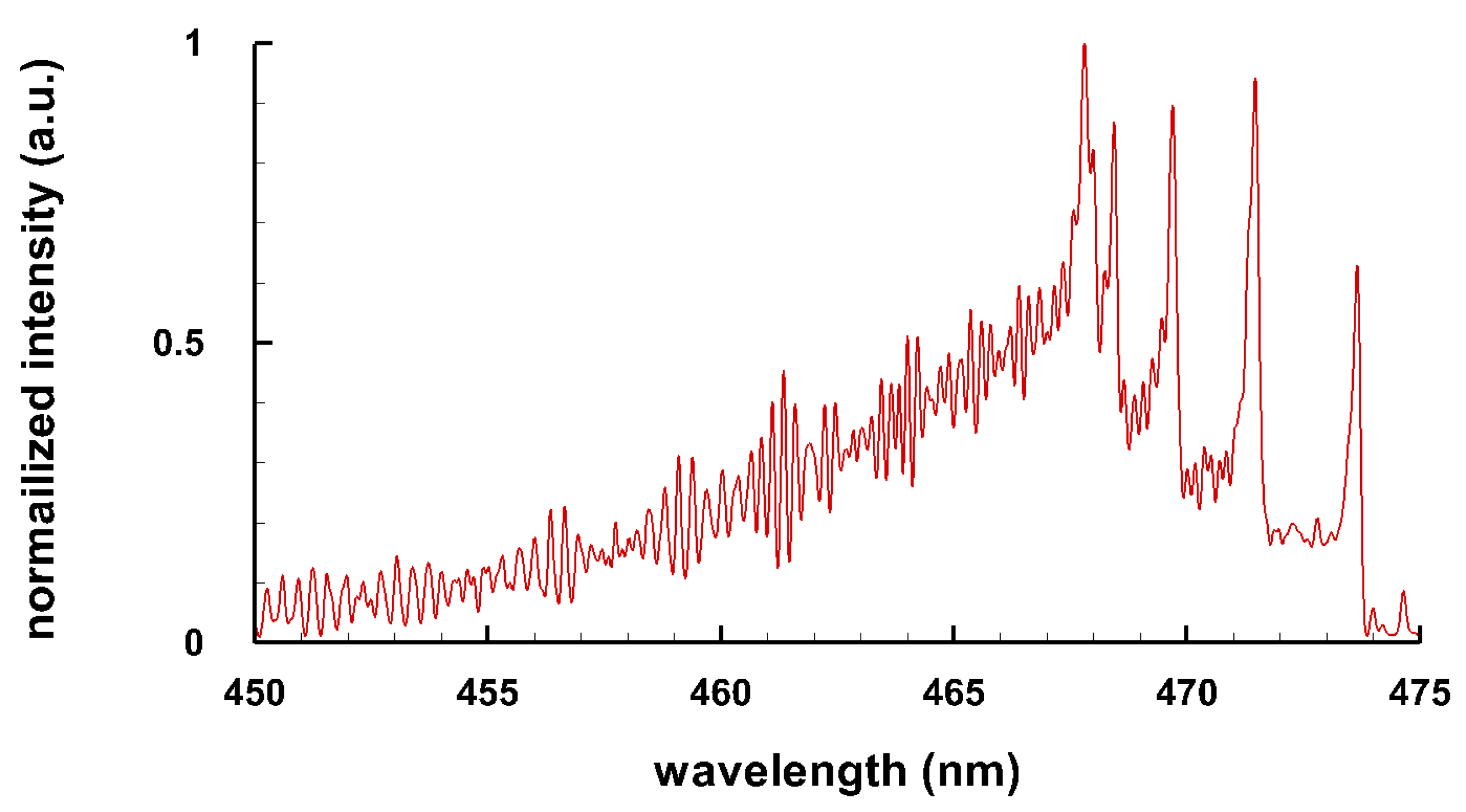
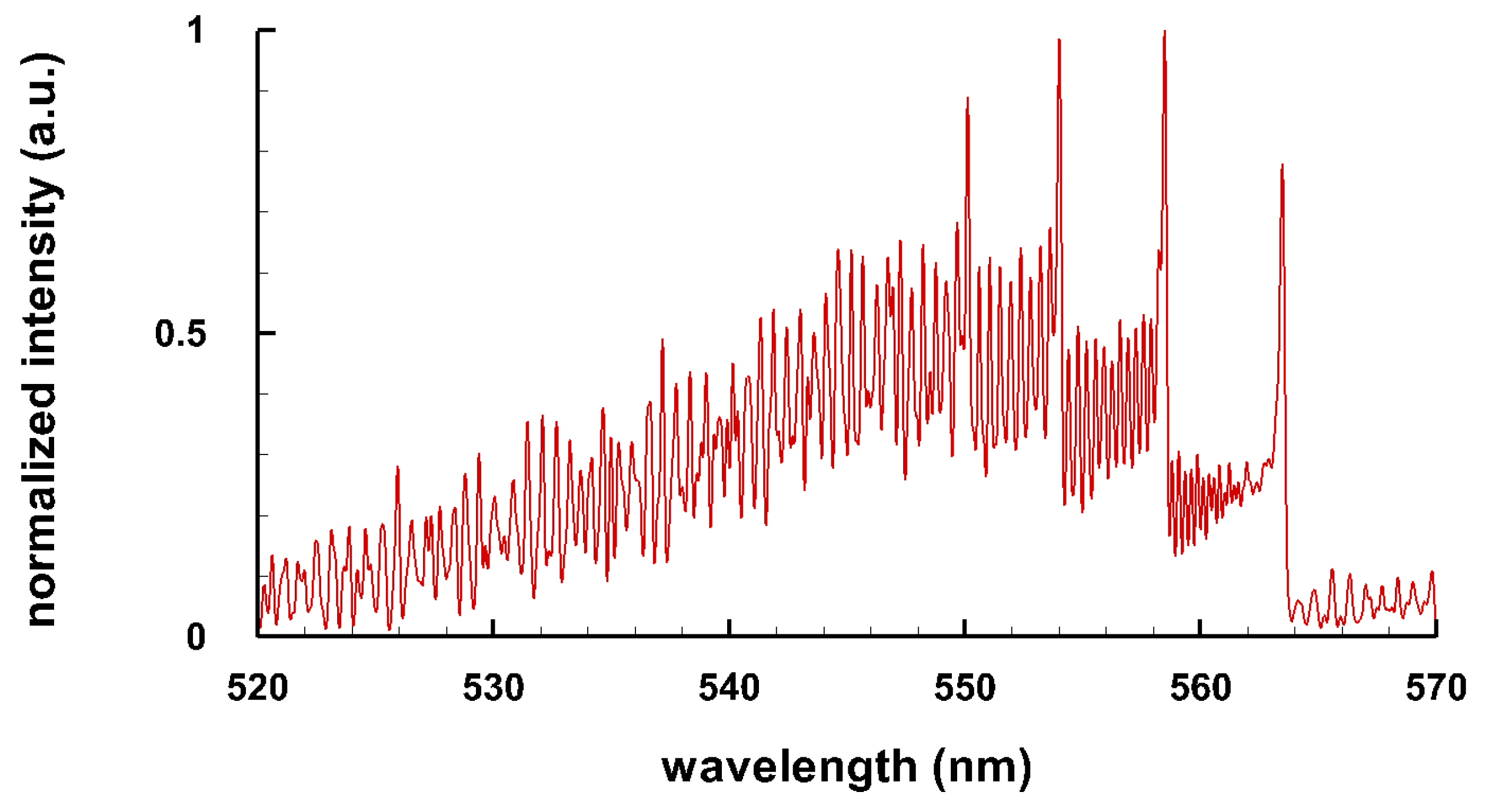
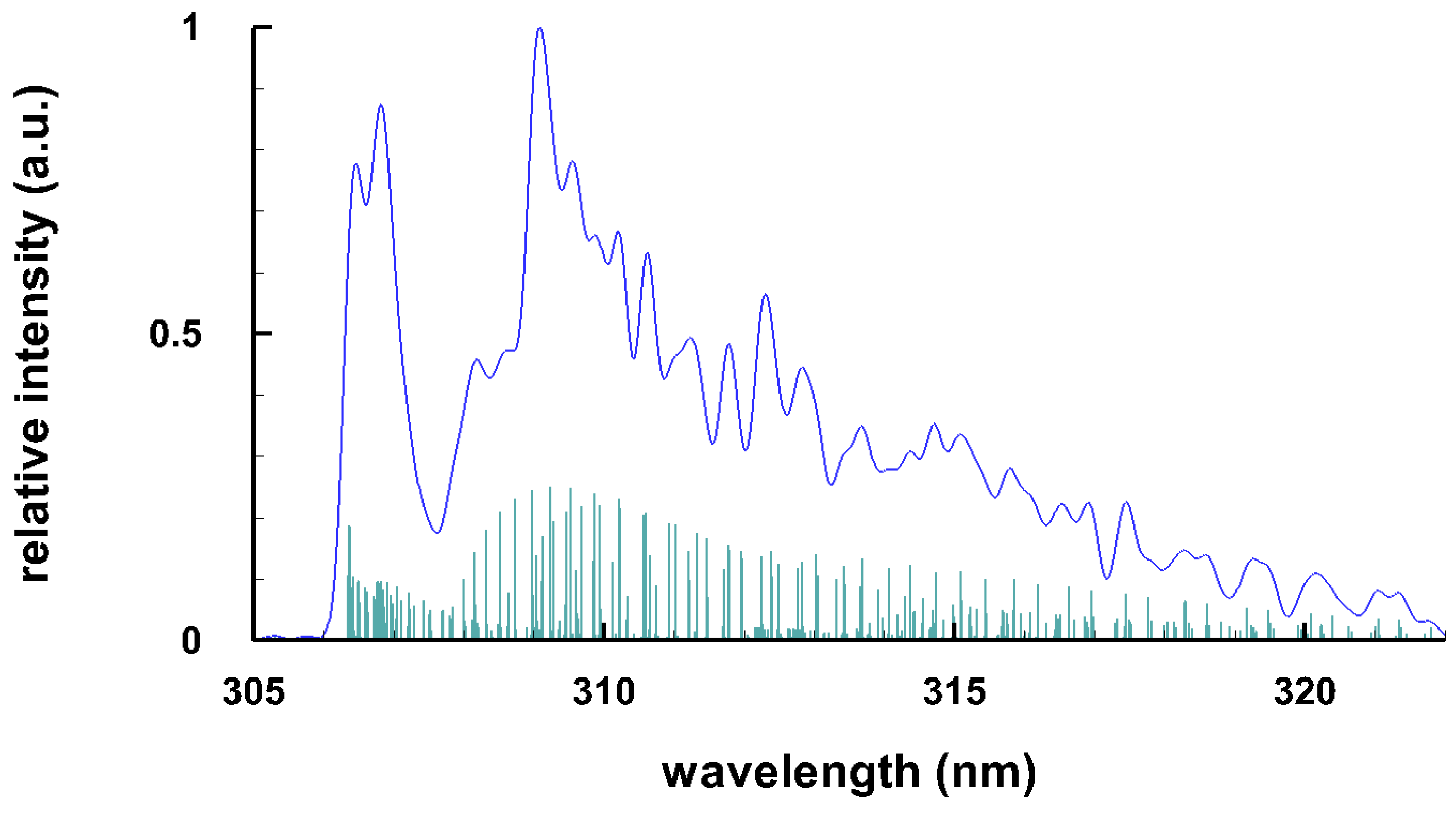
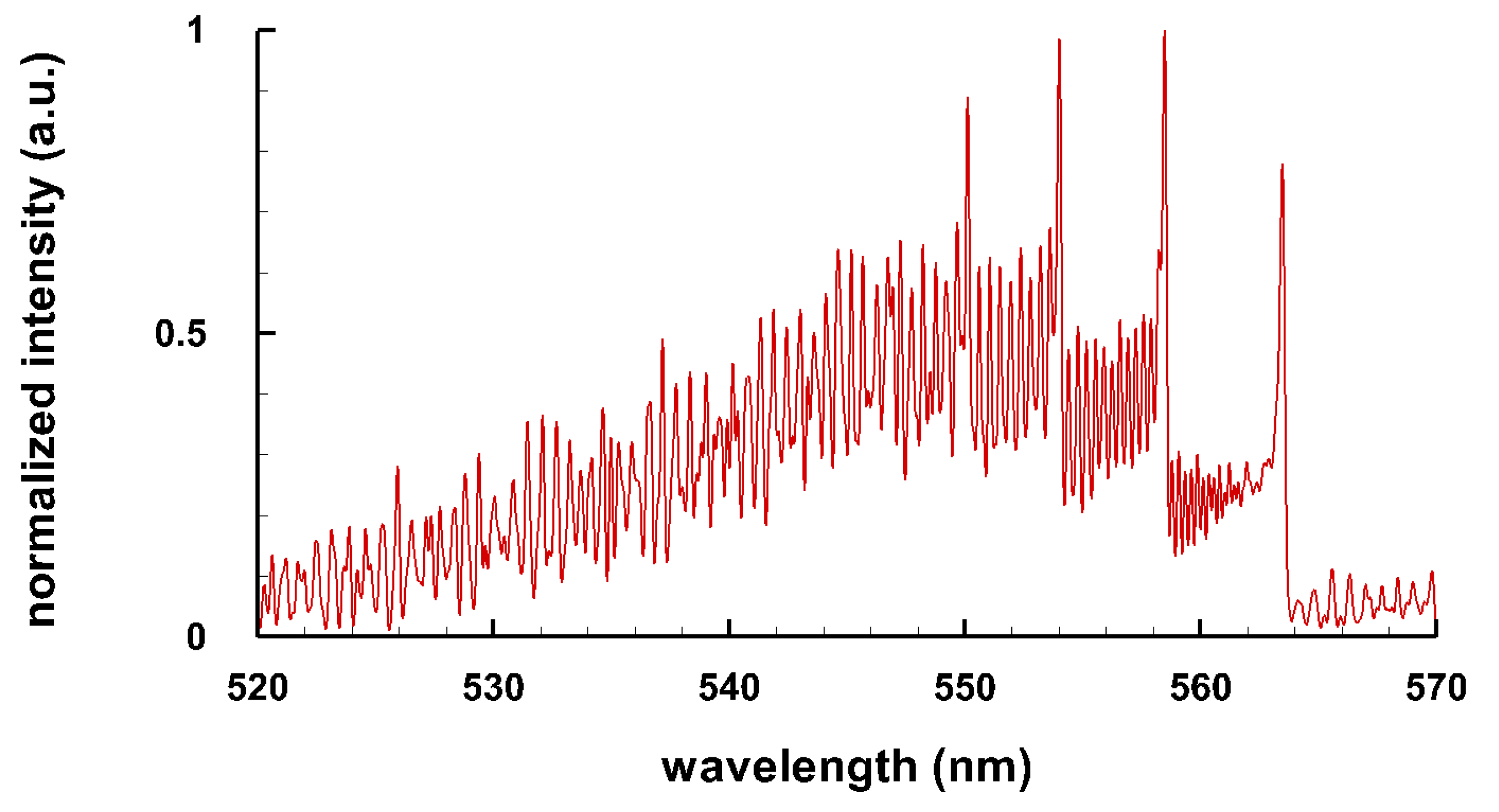
3.3. Selected Diatomic Spectra
4. Discussion and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AM | Angular Momentum |
| BESP | Boltzmann Equilibrium Spectrum Program |
| CM | Classical Mechanics |
| NMT | Nelder–Mead Temperature |
| PGOPHER | Program for simulating rotational, vibrational, and electronic spectra, or |
| “Program Gopher” | |
| QM | Quantum Mechanics |
| QMT | Quantum Mechanics Theory |
| RAM | Reversed Angular Momentum |
| WWE | Wigner–Witmer Eigenfunction |
References
- Klein, O. Zur Frage der Quantelung des asymmetrischen Kreisels. Z. Phys. 1929, 58, 730–734. [Google Scholar] [CrossRef]
- Van Vleck, J.H. The Coupling of Angular Momentum Vectors in Molecules. Rev. Mod. Phys. 1951, 23, 213–227. [Google Scholar] [CrossRef]
- Nöther, E. Invariante Variationsprobleme. Nachr. D. König. Gesellsch. D. Wiss. Göttingen 1918, 918, 235–257, Invariant variation problems. Transp. Theory Statist. Phys. 1971, 1, 183–207. [Google Scholar]
- Condon, E.U.; Shortley, G. The Theory of Atomic Spectra; Cambridge University Press: Cambridge, UK, 1953. [Google Scholar]
- Parigger, C.G.; Hornkohl, J.O. Quantum Mechanics of the Diatomic Molecule with Applications; IOP Publishing: Bristol, UK, 2020. [Google Scholar]
- Parigger, C.G.; Hornkohl, J.O. Diatomic Molecular Spectroscopy with Standard and Anomalous Commutators. Int. Rev. At. Mol. Phys. 2010, 1, 25–43. [Google Scholar]
- Dirac, P.A.M. The Elimination of Nodes in Quantum Mechanics. Proc. Roy. Soc. Lond. A 1926, 111, 281–305. [Google Scholar]
- Heisenberg, W.; Born, M.; Jordan, P. Zur Quantenmechanik II. Z. Phys. 1926, 35, 557–615. [Google Scholar]
- Popper, K. The Logic and Evolution of Scientific Theory. In All Life is Problem Solving; Routledge: London, UK, 1999; Chapter 1; pp. 3–22. [Google Scholar]
- Brown, J.; Carrington, A. Rotational Spectroscopy of Diatomic Molecules; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Lefebvre-Brion, H.; Field, R.W. The Spectra and Dynamics of Diatomic Molecules; Elsevier: Amsterdam, NL, USA, 2004. [Google Scholar]
- Bunker, P.R.; Jensen, P. Fundamentals of Molecular Spectroscopy; IOP Publishing: Bristol, UK, 2005. [Google Scholar]
- Gottfried, K. Quantum Mechanics; Addison-Wesley: Reading, UK, 1989. [Google Scholar]
- Baym, G. Lectures on Quantum Mechanics; Benjamin/Cummings: Reading, UK, 1969. [Google Scholar]
- Shore, B.W.; Menzel, D.H. Principles of Atomic Spectra; Addison-Wesley: Reading, UK, 1968. [Google Scholar]
- Judd, B.R. Angular Momentum Theory for Diatomic Molecules; Academic Press: New York, NY, USA, 1975. [Google Scholar]
- Mizushima, M. The Theory of Rotating Diatomic Molecules; John Wiley & Sons: New York, NY, USA, 1975. [Google Scholar]
- Kovacs, I. Rotational Structure in the Spectra of Diatomic Molecules; Elsevier: New York, NY, USA, 1969. [Google Scholar]
- Hougen, J.T. The Calculation of Rotational Energy Levels and Rotational Line Intensities in Diatomic Molecules; NBS Monograph 115; U.S. Government Printing Office: Washington, DC, USA, 1970.
- Kroto, H.W. Molecular Rotation Spectra; Dover: New York, NY, USA, 1992. [Google Scholar]
- Carrington, A.; Levy, D.H.; Miller, T.A. Electron Resonance of Gaseous Diatomic Molecules. Adv. Chem. Phys. 1970, 18, 149–248. [Google Scholar]
- Freed, K.F. Theory of the Hyperfine Structure of Molecules: Application to 3Π States of Diatomic Molecules Intermediate between Hund’s Cases (a) and (b). J. Chem. Phys. 1966, 45, 4214–4241. [Google Scholar] [CrossRef]
- Cohen-Tannoudji, C.; Diu, B.; Laloe, F. Quantum Mechanics, Volume 1: Basic Concepts, Tools, and Application, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2019. [Google Scholar]
- Cohen-Tannoudji, C.; Diu, B.; Laloe, F. Quantum Mechanics, Volume 2: Angular Momentum, Spin, and Approximation Methds, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2019. [Google Scholar]
- Arfken, G.B.; Weber, H.J.; Harris, F.E. Mathematical Methods for Physicists, A Comprehensive Guide, 7th ed.; Academic Press: New York, NY, USA, 2012. [Google Scholar]
- Western, C.M. PGOPHER: A program for simulating rotational, vibrational and electronic spectra. J. Quant. Spectrosc. Radiat. Transf. 2017, 186, 221–242. [Google Scholar] [CrossRef] [Green Version]
- Kunze, H.-J. Introduction to Plasma Spectroscopy; Springer: Heidelberg, Germany, 2009. [Google Scholar]
- Demtröder, W. Laser Spectroscopy 1: Basic Principles, 5th ed.; Springer: Heidelberg, Germany, 2014. [Google Scholar]
- Demtröder, W. Laser Spectroscopy 2: Experimental Techniques, 5th ed.; Springer: Heidelberg, Germany, 2015. [Google Scholar]
- Hertel, I.V.; Schulz, C.-P. Atoms, Molecules and Optical Physics 1, Atoms and Spectroscopy; Springer: Heidelberg, Germany, 2015. [Google Scholar]
- Hertel, I.V.; Schulz, C.-P. Atoms, Molecules and Optical Physics 2, Molecules and Photons—Spectroscopy and Collisions; Springer: Heidelberg, Germany, 2015. [Google Scholar]
- Cremers, D.E.; Radziemski, L.J. Handbook of Laser-Induced Breakdown Spectroscopy; Wiley: New York, NY, USA, 2006. [Google Scholar]
- Miziolek, A.W.; Palleschi, V.; Schechter, I. (Eds.) Laser Induced Breakdown Spectroscopy; Cambridge University Press: New York, NY, USA, 2006. [Google Scholar]
- Singh, J.P.; Thakur, S.N. (Eds.) Laser-Induced Breakdown Spectroscopy, 2nd ed.; Elsevier: New York, NY, USA, 2020. [Google Scholar]
- Tennyson, J.; Lodi, L.; McKemmish, L.K.; Yurchenko, S.N. The ab initio calculation of spectra of open shell diatomic molecules. arXiv 2016, arXiv:1605.02301v1. [Google Scholar]
- Davydov, A.S. Quantum Mechanics; Pergamon Press: Oxford, UK, 1965. [Google Scholar]
- Wigner, E.; Witmer, E.E. Über die Struktur der zweiatomigen Molekelspectren nach der Quantenmechanik. Z. Physik 1928, 51, 859–886. [Google Scholar] [CrossRef]
- Hettema, H. (Ed.) On the Structure of the Spectra of Two-Atomic Molecules According to Quantum Mechanics. In Quantum Chemistry: Classic Scientific Papers; World Scientific: Singapore, 2000; pp. 287–311. [Google Scholar]
- Bransden, B.H.; Joachain, C.J. Physics of Atoms and Molecules, 2nd ed.; Prentice Hall: Essex, UK, 2003. [Google Scholar]
- Parigger, C.G.; Woods, A.C.; Surmick, D.M.; Gautam, G.; Witte, M.J.; Hornkohl, J.O. Computation of diatomic molecular spectra for selected transitions of aluminum monoxide, cyanide, diatomic carbon, and titanium monoxide. Spectrochim. Acta Part B At. Spectrosc. 2015, 107, 132–138. [Google Scholar] [CrossRef]
- Parigger, C.G.; Helstern, C.M.; Jordan, B.S.; Surmick, D.M.; Splinter, R. Laser-Plasma Spectroscopy of Hydroxyl with Applications. Molecules 2020, 25, 988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parigger, C.G.; Surmick, D.M.; Helstern, C.M.; Gautam, G.; Bol’shakov, A.A.; Russo, R.E. Molecular laser-induced breakdown spectroscopy. In Laser-Induced Breakdown Spectroscopy, 2nd ed.; Singh, J.P., Thakur, S.N., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Chapter 7; pp. 167–209. [Google Scholar]
- Parigger, C.G.; Helstern, C.M.; Jordan, B.S.; Surmick, D.M.; Splinter, R. Laser-Plasma Spatiotemporal Cyanide Spectroscopy and Applications. Molecules 2020, 25, 615. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parigger, C.G. Fundamentals of Diatomic Molecular Spectroscopy. Foundations 2021, 1, 208-216. https://doi.org/10.3390/foundations1020016
Parigger CG. Fundamentals of Diatomic Molecular Spectroscopy. Foundations. 2021; 1(2):208-216. https://doi.org/10.3390/foundations1020016
Chicago/Turabian StyleParigger, Christian G. 2021. "Fundamentals of Diatomic Molecular Spectroscopy" Foundations 1, no. 2: 208-216. https://doi.org/10.3390/foundations1020016
APA StyleParigger, C. G. (2021). Fundamentals of Diatomic Molecular Spectroscopy. Foundations, 1(2), 208-216. https://doi.org/10.3390/foundations1020016