
Exploiting Synthetic Lethality of PRMT5 for Precision Treatment of MTAP-Deficient Glioblastoma


Abstract
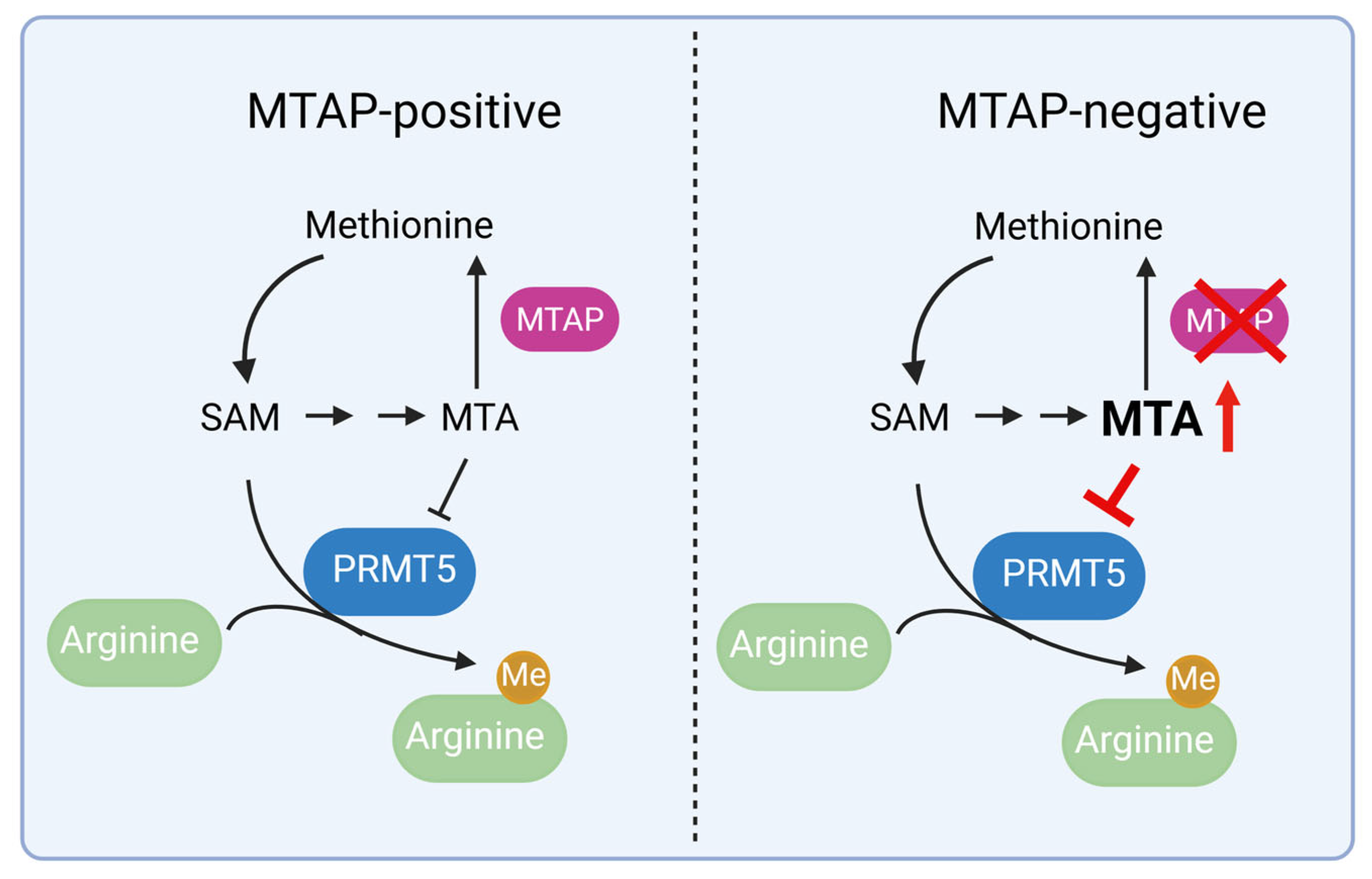
1. Introduction
2. Role of PRMT5 in GBM
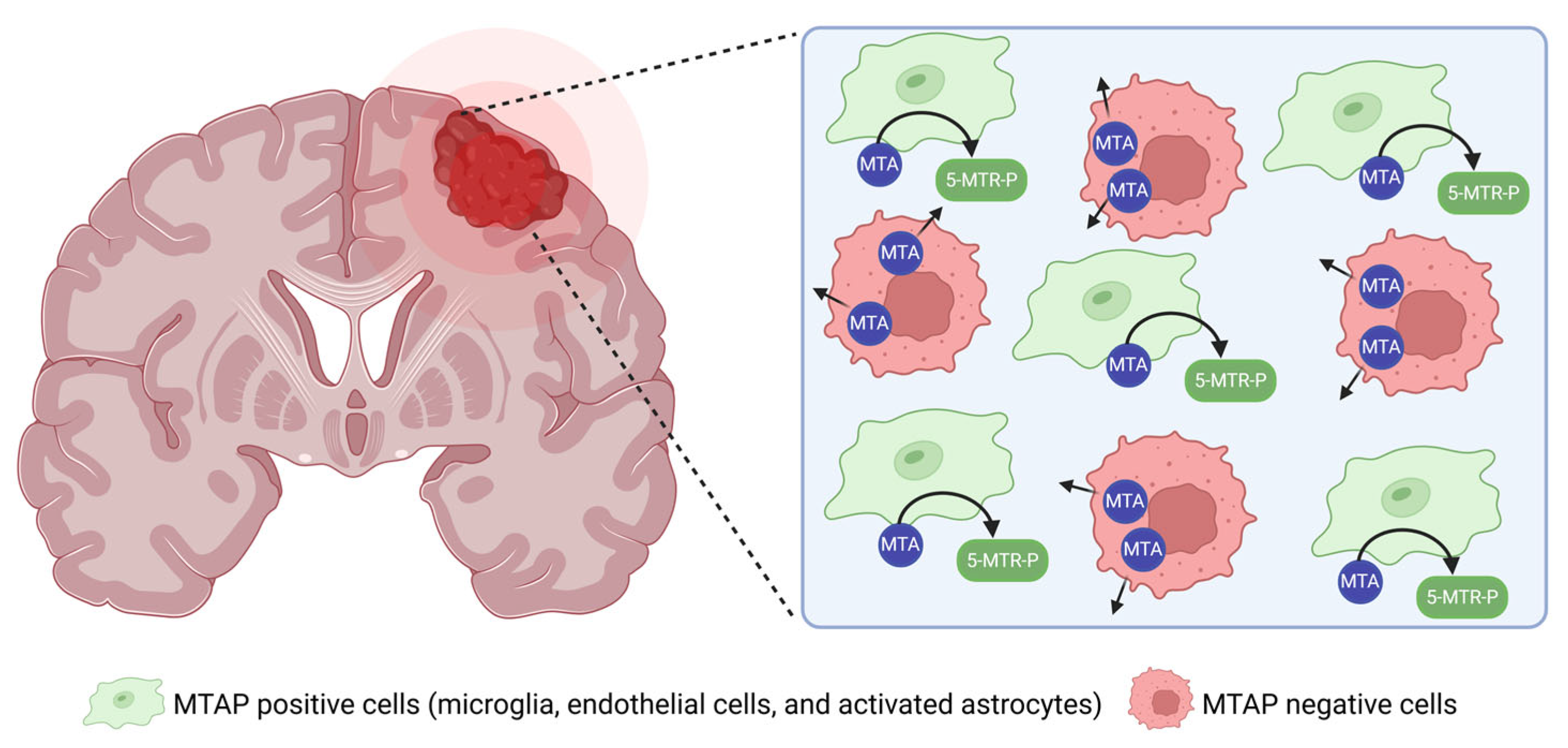
3. Preclinical and Clinical Studies of PRMT5 Inhibitors in GBM
3.1. First-Generation PRMT5-Targeted Therapies
3.1.1. GSK3326595 (GSK, Philadelphia, PA, USA)
3.1.2. JNJ-64619178 (Janssen Research & Development, LLC, New Brunswick, NJ, USA)
3.1.3. PF-06939999 (Pfizer, New York, NY, USA)
3.1.4. PRT811 (Prelude Therapeutics, Wilmington, DE, USA)
3.1.5. LLY-283 (Eli Lilly, Indianapolis, IN, USA)
3.1.6. CMP5
3.2. Second-Generation PRMT5-Targeted Therapies
3.2.1. MRTX1719 (BMS-986504-Bristol Myers Squibb, Princeton, NJ, USA)
3.2.2. AMG193 (Amgen, Thousand Oaks, CA, USA)
3.2.3. TNG908 and TNG462 (Tango Therapeutics, Boston, MA, USA)
4. Combination Strategies with PRMT5 Inhibitors in GBM Therapy
4.1. Combining PRMT5 Inhibition with Irradiation
4.2. Combining PRMT5 Inhibition with CDK4/6 Inhibitor
4.3. Combining PRMT5 Inhibition with MEK Inhibitor
4.4. Combining PRMT5 Inhibition with PTEN Deficiency in Cancer Therapy
4.5. Combining PRMT5 Inhibition with mTOR Inhibitor
4.6. Combining PRMT5 Inhibition with PP2A Inhibitor
4.7. Combining PRMT5 Inhibition with PD-1 Inhibitor
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Nguyen, T.T.T.; Greene, L.A.; Mnatsakanyan, H.; Badr, C.E. Revolutionizing Brain Tumor Care: Emerging Technologies and Strategies. Biomedicines 2024, 12, 1376. [Google Scholar] [CrossRef]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Moreno-Murciano, P.; Oriol-Caballo, M.; Lopez-Blanch, R.; Pineda, B.; Gutierrez-Arroyo, J.L.; Loras, A.; Gonzalez-Bonet, L.G.; Martinez-Cadenas, C.; Estrela, J.M.; et al. Glioblastoma Therapy: Past, Present and Future. Int. J. Mol. Sci. 2024, 25, 2529. [Google Scholar] [CrossRef]
- Teraiya, M.; Perreault, H.; Chen, V.C. An overview of glioblastoma multiforme and temozolomide resistance: Can LC-MS-based proteomics reveal the fundamental mechanism of temozolomide resistance? Front. Oncol. 2023, 13, 1166207. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Yuan, K.; Tao, J.; Qin, Y.; Li, Y.; Fu, J.; Li, Z.; Zhou, H.; Tang, Z.; Li, L.; et al. Glioblastoma multiforme: An updated overview of temozolomide resistance mechanisms and strategies to overcome resistance. Discov. Oncol. 2025, 16, 731. [Google Scholar] [CrossRef]
- Tsai, C.K.; Lin, C.Y.; Chang, Y.L.; Yang, F.C.; Chou, C.H.; Huang, Y.C.; Hueng, D.Y. Using novel oxidative phosphorylation inhibitors to attenuate drug resistance in human gliomas. EXCLI J. 2025, 24, 433–449. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Wu, Q.; Schapira, M.; Arrowsmith, C.H.; Barsyte-Lovejoy, D. Protein arginine methylation: From enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov. 2021, 20, 509–530. [Google Scholar] [CrossRef]
- Bray, C.; Balcells, C.; McNeish, I.A.; Keun, H.C. The potential and challenges of targeting MTAP-negative cancers beyond synthetic lethality. Front. Oncol. 2023, 13, 1264785. [Google Scholar] [CrossRef]
- Mavrakis, K.J.; McDonald, E.R., III; Schlabach, M.R.; Billy, E.; Hoffman, G.R.; deWeck, A.; Ruddy, D.A.; Venkatesan, K.; Yu, J.; McAllister, G.; et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 2016, 351, 1208–1213. [Google Scholar] [CrossRef]
- Woollard, W.J.; Kalaivani, N.P.; Jones, C.L.; Roper, C.; Tung, L.; Lee, J.J.; Thomas, B.R.; Tosi, I.; Ferreira, S.; Beyers, C.Z.; et al. Independent Loss of Methylthioadenosine Phosphorylase (MTAP) in Primary Cutaneous T-Cell Lymphoma. J. Invest. Dermatol. 2016, 136, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Behrmann, I.; Wallner, S.; Komyod, W.; Heinrich, P.C.; Schuierer, M.; Buettner, R.; Bosserhoff, A.K. Characterization of methylthioadenosin phosphorylase (MTAP) expression in malignant melanoma. Am. J. Pathol. 2003, 163, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Stopa, N.; Krebs, J.E.; Shechter, D. The PRMT5 arginine methyltransferase: Many roles in development, cancer and beyond. Cell Mol. Life Sci. 2015, 72, 2041–2059. [Google Scholar] [CrossRef]
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Li, B.; Wu, Y.; Wu, X.; Wang, Y. Targeting Arginine Methyltransferase PRMT5 for Cancer Therapy: Updated Progress and Novel Strategies. J. Med. Chem. 2023, 66, 8407–8427. [Google Scholar] [CrossRef]
- Han, X.; Li, R.; Zhang, W.; Yang, X.; Wheeler, C.G.; Friedman, G.K.; Province, P.; Ding, Q.; You, Z.; Fathallah-Shaykh, H.M.; et al. Expression of PRMT5 correlates with malignant grade in gliomas and plays a pivotal role in tumor growth in vitro. J. Neurooncol. 2014, 118, 61–72. [Google Scholar] [CrossRef]
- Yan, F.; Alinari, L.; Lustberg, M.E.; Martin, L.K.; Cordero-Nieves, H.M.; Banasavadi-Siddegowda, Y.; Virk, S.; Barnholtz-Sloan, J.; Bell, E.H.; Wojton, J.; et al. Genetic validation of the protein arginine methyltransferase PRMT5 as a candidate therapeutic target in glioblastoma. Cancer Res. 2014, 74, 1752–1765. [Google Scholar] [CrossRef]
- Banasavadi-Siddegowda, Y.K.; Welker, A.M.; An, M.; Yang, X.; Zhou, W.; Shi, G.; Imitola, J.; Li, C.; Hsu, S.; Wang, J.; et al. PRMT5 as a druggable target for glioblastoma therapy. Neuro Oncol. 2018, 20, 753–763. [Google Scholar] [CrossRef]
- Wolf, S.S. The protein arginine methyltransferase family: An update about function, new perspectives and the physiological role in humans. Cell Mol. Life Sci. 2009, 66, 2109–2121. [Google Scholar] [CrossRef]
- Lee, Y.H.; Stallcup, M.R. Minireview: Protein arginine methylation of nonhistone proteins in transcriptional regulation. Mol. Endocrinol. 2009, 23, 425–433. [Google Scholar] [CrossRef]
- Di Lorenzo, A.; Bedford, M.T. Histone arginine methylation. FEBS Lett. 2011, 585, 2024–2031. [Google Scholar] [CrossRef]
- Brehmer, D.; Beke, L.; Wu, T.; Millar, H.J.; Moy, C.; Sun, W.; Mannens, G.; Pande, V.; Boeckx, A.; van Heerde, E.; et al. Discovery and Pharmacological Characterization of JNJ-64619178, a Novel Small-Molecule Inhibitor of PRMT5 with Potent Antitumor Activity. Mol. Cancer Ther. 2021, 20, 2317–2328. [Google Scholar] [CrossRef] [PubMed]
- Wilczek, C.; Chitta, R.; Woo, E.; Shabanowitz, J.; Chait, B.T.; Hunt, D.F.; Shechter, D. Protein arginine methyltransferase Prmt5-Mep50 methylates histones H2A and H4 and the histone chaperone nucleoplasmin in Xenopus laevis eggs. J. Biol. Chem. 2011, 286, 42221–42231. [Google Scholar] [CrossRef] [PubMed]
- Braun, C.J.; Stanciu, M.; Boutz, P.L.; Patterson, J.C.; Calligaris, D.; Higuchi, F.; Neupane, R.; Fenoglio, S.; Cahill, D.P.; Wakimoto, H.; et al. Coordinated Splicing of Regulatory Detained Introns within Oncogenic Transcripts Creates an Exploitable Vulnerability in Malignant Glioma. Cancer Cell 2017, 32, 411–426.e411. [Google Scholar] [CrossRef]
- Bezzi, M.; Teo, S.X.; Muller, J.; Mok, W.C.; Sahu, S.K.; Vardy, L.A.; Bonday, Z.Q.; Guccione, E. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes. Dev. 2013, 27, 1903–1916. [Google Scholar] [CrossRef]
- Burgos, E.S.; Wilczek, C.; Onikubo, T.; Bonanno, J.B.; Jansong, J.; Reimer, U.; Shechter, D. Histone H2A and H4 N-terminal tails are positioned by the MEP50 WD repeat protein for efficient methylation by the PRMT5 arginine methyltransferase. J. Biol. Chem. 2015, 290, 9674–9689. [Google Scholar] [CrossRef] [PubMed]
- Migliori, V.; Muller, J.; Phalke, S.; Low, D.; Bezzi, M.; Mok, W.C.; Sahu, S.K.; Gunaratne, J.; Capasso, P.; Bassi, C.; et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat. Struct. Mol. Biol. 2012, 19, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Baiocchi, R.A.; Byrd, J.C.; Grever, M.R.; Jacob, S.T.; Sif, S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 2007, 26, 3558–3569. [Google Scholar] [CrossRef]
- Guccione, E.; Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 642–657. [Google Scholar] [CrossRef]
- Pawar, J.S.; Al-Amin, M.Y.; Hu, C.D. JNJ-64619178 radiosensitizes and suppresses fractionated ionizing radiation-induced neuroendocrine differentiation (NED) in prostate cancer. Front. Oncol. 2023, 13, 1126482. [Google Scholar] [CrossRef]
- Owens, J.L.; Beketova, E.; Liu, S.; Tinsley, S.L.; Asberry, A.M.; Deng, X.; Huang, J.; Li, C.; Wan, J.; Hu, C.D. PRMT5 Cooperates with pICln to Function as a Master Epigenetic Activator of DNA Double-Strand Break Repair Genes. iScience 2020, 23, 100750. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.L.; Sanchez-Bailon, M.P.; Chiang, K.; Reynolds, J.J.; Herrero-Ruiz, J.; Bandeiras, T.M.; Matias, P.M.; Maslen, S.L.; Skehel, J.M.; Stewart, G.S.; et al. PRMT5-Dependent Methylation of the TIP60 Coactivator RUVBL1 Is a Key Regulator of Homologous Recombination. Mol. Cell 2017, 65, 900–916. [Google Scholar] [CrossRef] [PubMed]
- Sachamitr, P.; Ho, J.C.; Ciamponi, F.E.; Ba-Alawi, W.; Coutinho, F.J.; Guilhamon, P.; Kushida, M.M.; Cavalli, F.M.G.; Lee, L.; Rastegar, N.; et al. PRMT5 inhibition disrupts splicing and stemness in glioblastoma. Nat. Commun. 2021, 12, 979. [Google Scholar] [CrossRef]
- Guderian, G.; Peter, C.; Wiesner, J.; Sickmann, A.; Schulze-Osthoff, K.; Fischer, U.; Grimmler, M. RioK1, a new interactor of protein arginine methyltransferase 5 (PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and substrate specificity. J. Biol. Chem. 2011, 286, 1976–1986. [Google Scholar] [CrossRef]
- Beketova, E.; Fang, S.; Owens, J.L.; Liu, S.; Chen, X.; Zhang, Q.; Asberry, A.M.; Deng, X.; Malola, J.; Huang, J.; et al. Protein Arginine Methyltransferase 5 Promotes pICln-Dependent Androgen Receptor Transcription in Castration-Resistant Prostate Cancer. Cancer Res. 2020, 80, 4904–4917. [Google Scholar] [CrossRef]
- McKinney, D.C.; McMillan, B.J.; Ranaghan, M.J.; Moroco, J.A.; Brousseau, M.; Mullin-Bernstein, Z.; O’Keefe, M.; McCarren, P.; Mesleh, M.F.; Mulvaney, K.M.; et al. Discovery of a First-in-Class Inhibitor of the PRMT5-Substrate Adaptor Interaction. J. Med. Chem. 2021, 64, 11148–11168. [Google Scholar] [CrossRef]
- Pal, S.; Sif, S. Interplay between chromatin remodelers and protein arginine methyltransferases. J. Cell Physiol. 2007, 213, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Hamard, P.J.; Santiago, G.E.; Liu, F.; Karl, D.L.; Martinez, C.; Man, N.; Mookhtiar, A.K.; Duffort, S.; Greenblatt, S.; Verdun, R.E.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep. 2018, 24, 2643–2657. [Google Scholar] [CrossRef]
- Koh, C.M.; Bezzi, M.; Guccione, E. The Where and the How of PRMT5. Curr. Mol. Biol. Rep. 2015, 1, 19–28. [Google Scholar] [CrossRef]
- Hu, M.; Chen, X. A review of the known MTA-cooperative PRMT5 inhibitors. RSC Adv. 2024, 14, 39653–39691. [Google Scholar] [CrossRef]
- Rodon, J.; Rodriguez, E.; Maitland, M.L.; Tsai, F.Y.; Socinski, M.A.; Berlin, J.D.; Thomas, J.S.; Al Baghdadi, T.; Wang, I.M.; Guo, C.; et al. A phase I study to evaluate the safety, pharmacokinetics, and pharmacodynamics of PF-06939999 (PRMT5 inhibitor) in patients with selected advanced or metastatic tumors with high incidence of splicing factor gene mutations. ESMO Open 2024, 9, 102961. [Google Scholar] [CrossRef]
- Hu, R.; Zhou, B.; Chen, Z.; Chen, S.; Chen, N.; Shen, L.; Xiao, H.; Zheng, Y. PRMT5 Inhibition Promotes PD-L1 Expression and Immuno-Resistance in Lung Cancer. Front. Immunol. 2021, 12, 722188. [Google Scholar] [CrossRef] [PubMed]
- Bonday, Z.Q.; Cortez, G.S.; Grogan, M.J.; Antonysamy, S.; Weichert, K.; Bocchinfuso, W.P.; Li, F.; Kennedy, S.; Li, B.; Mader, M.M.; et al. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med. Chem. Lett. 2018, 9, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Vieito, M.; Moreno, V.; Spreafico, A.; Brana, I.; Wang, J.S.; Preis, M.; Hernandez, T.; Genta, S.; Hansen, A.R.; Doger, B.; et al. Phase 1 Study of JNJ-64619178, a Protein Arginine Methyltransferase 5 Inhibitor, in Advanced Solid Tumors. Clin. Cancer Res. 2023, 29, 3592–3602. [Google Scholar] [CrossRef]
- Zhu, K.; Tao, H.; Song, J.L.; Jin, L.; Zhang, Y.; Liu, J.; Chen, Z.; Jiang, C.S.; Luo, C.; Zhang, H. Identification of 5-benzylidene-2-phenylthiazolones as potent PRMT5 inhibitors by virtual screening, structural optimization and biological evaluations. Bioorg Chem. 2018, 81, 289–298. [Google Scholar] [CrossRef]
- Siu, L.L.; Rasco, D.W.; Vinay, S.P.; Romano, P.M.; Menis, J.; Opdam, F.L.; Heinhuis, K.M.; Egger, J.L.; Gorman, S.A.; Parasrampuria, R.; et al. METEOR-1: A phase I study of GSK3326595, a first-in-class protein arginine methyltransferase 5 (PRMT5) inhibitor, in advanced solid tumours. Ann. Oncol. 2019, 30, v159. [Google Scholar] [CrossRef]
- Zhu, F.; Ryan, C.E.; Hackett, L.R.; Collins, M.C.; Chong, S.J.F.; Davids, M.S. Inhibition of PRMT5 Increases Sensitivity to BH3 Mimetics in Aggressive B Cell Malignancies. Blood 2022, 140, 8842–8843. [Google Scholar] [CrossRef]
- Monga, V.; Johanns, T.M.; Stupp, R.; Chandra, S.; Falchook, G.S.; Giglio, P.; Philipovskiy, A.; Alnahhas, I.; Babbar, N.; Sun, W.; et al. A phase 1 study of the protein arginine methyltransferase 5 (PRMT5) brain-penetrant inhibitor PRT811 in patients (pts) with recurrent high-grade glioma or uveal melanoma (UM). J. Clin. Oncol. 2023, 41, 3008. [Google Scholar] [CrossRef]
- Briggs, K.J.; Cottrell, K.M.; Tonini, M.R.; Tsai, A.; Zhang, M.; Whittington, D.A.; Zhang, W.; Lombardo, S.A.; Yoda, S.; Wilker, E.W.; et al. TNG908 is a brain-penetrant, MTA-cooperative PRMT5 inhibitor developed for the treatment of MTAP-deleted cancers. Transl. Oncol. 2025, 52, 102264. [Google Scholar] [CrossRef]
- Feustel, K.; Falchook, G.S. Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors in Oncology Clinical Trials: A review. J. Immunother. Precis. Oncol. 2022, 5, 58–67. [Google Scholar] [CrossRef]
- Mishra, V.; Singh, A.; Korzinkin, M.; Cheng, X.; Wing, C.; Sarkisova, V.; Koppayi, A.L.; Pogorelskaya, A.; Glushchenko, O.; Sundaresan, M.; et al. PRMT5 inhibition has a potent anti-tumor activity against adenoid cystic carcinoma of salivary glands. J. Exp. Clin. Cancer Res. 2025, 44, 11. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shao, X.; Zhao, X.; Ji, Y.; Liu, X.; Li, P.; Zhang, M.; Wang, Q. Targeting protein arginine methyltransferase 5 in cancers: Roles, inhibitors and mechanisms. Biomed. Pharmacother. 2021, 144, 112252. [Google Scholar] [CrossRef]
- Webb, L.M.; Amici, S.A.; Jablonski, K.A.; Savardekar, H.; Panfil, A.R.; Li, L.; Zhou, W.; Peine, K.; Karkhanis, V.; Bachelder, E.M.; et al. PRMT5-Selective Inhibitors Suppress Inflammatory T Cell Responses and Experimental Autoimmune Encephalomyelitis. J. Immunol. 2017, 198, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Jain, S.; Coulter, D.W.; Joshi, S.S.; Chaturvedi, N.K. PRMT5 as a Potential Therapeutic Target in MYC-Amplified Medulloblastoma. Cancers 2023, 15, 5855. [Google Scholar] [CrossRef]
- Marjon, K.; Cameron, M.J.; Quang, P.; Clasquin, M.F.; Mandley, E.; Kunii, K.; McVay, M.; Choe, S.; Kernytsky, A.; Gross, S.; et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016, 15, 574–587. [Google Scholar] [CrossRef]
- Fan, N.; Zhang, Y.; Zou, S. Methylthioadenosine phosphorylase deficiency in tumors: A compelling therapeutic target. Front. Cell Dev. Biol. 2023, 11, 1173356. [Google Scholar] [CrossRef]
- Cottrell, K.M.; Whittington, D.A.; Briggs, K.J.; Jahic, H.; Ali, J.A.; Amor, A.J.; Gotur, D.; Tonini, M.R.; Zhang, W.; Huang, A.; et al. MTA-Cooperative PRMT5 Inhibitors: Mechanism Switching Through Structure-Based Design. J. Med. Chem. 2025, 68, 4217–4236. [Google Scholar] [CrossRef] [PubMed]
- Bertino, J.R.; Waud, W.R.; Parker, W.B.; Lubin, M. Targeting tumors that lack methylthioadenosine phosphorylase (MTAP) activity: Current strategies. Cancer Biol. Ther. 2011, 11, 627–632. [Google Scholar] [CrossRef]
- Schneider, C.; Spielmann, V.; Braun, C.J.; Schneider, G. PRMT5 inhibitors: Therapeutic potential in pancreatic cancer. Transl. Oncol. 2025, 55, 102366. [Google Scholar] [CrossRef]
- Engstrom, L.D.; Aranda, R.; Waters, L.; Moya, K.; Bowcut, V.; Vegar, L.; Trinh, D.; Hebbert, A.; Smith, C.R.; Kulyk, S.; et al. MRTX1719 Is an MTA-Cooperative PRMT5 Inhibitor That Exhibits Synthetic Lethality in Preclinical Models and Patients with MTAP-Deleted Cancer. Cancer Discov. 2023, 13, 2412–2431. [Google Scholar] [CrossRef]
- Rodon, J.; Prenen, H.; Sacher, A.; Villalona-Calero, M.; Penel, N.; El Helali, A.; Rottey, S.; Yamamoto, N.; Ghiringhelli, F.; Goebeler, M.E.; et al. First-in-human study of AMG 193, an MTA-cooperative PRMT5 inhibitor, in patients with MTAP-deleted solid tumors: Results from phase I dose exploration. Ann. Oncol. 2024, 35, 1138–1147. [Google Scholar] [CrossRef]
- Belmontes, B.; Slemmons, K.K.; Su, C.; Liu, S.; Policheni, A.N.; Moriguchi, J.; Tan, H.; Xie, F.; Aiello, D.A.; Yang, Y.; et al. AMG 193, a Clinical Stage MTA-Cooperative PRMT5 Inhibitor, Drives Antitumor Activity Preclinically and in Patients with MTAP-Deleted Cancers. Cancer Discov. 2025, 15, 139–161. [Google Scholar] [CrossRef] [PubMed]
- Soeta, T.; Sugisawa, N.; Yamamura, A.; Tanaka, N.; Imoto, H.; Tsuchiya, T.; Aizawa, T.; Okamoto, K.; Kawamura, M.; Saijo, F.; et al. MRTX1719, an MTA-cooperative PRMT5 Inhibitor, Induces Cell Cycle Arrest and Synergizes With Oxaliplatin and Gemcitabine for Enhanced Anticancer Effects. Anticancer. Res. 2024, 44, 5231–5240. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, X.; Ma, R.; Zhang, X.; Ji, S.; Liu, Z.; Yang, G.; Wang, H.; Zhang, P.; Zhang, J.; et al. Inhibitory Effect of PRMT5/MTA Inhibitor on MTAP-Deficient Glioma May Be Influenced by Surrounding Normal Cells. Cancer Med. 2024, 13, e70526. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.R.; Aranda, R.; Bobinski, T.P.; Briere, D.M.; Burns, A.C.; Christensen, J.G.; Clarine, J.; Engstrom, L.D.; Gunn, R.J.; Ivetac, A.; et al. Fragment-Based Discovery of MRTX1719, a Synthetic Lethal Inhibitor of the PRMT5*MTA Complex for the Treatment of MTAP-Deleted Cancers. J. Med. Chem. 2022, 65, 1749–1766. [Google Scholar] [CrossRef] [PubMed]
- Barekatain, Y.; Ackroyd, J.J.; Yan, V.C.; Khadka, S.; Wang, L.; Chen, K.C.; Poral, A.H.; Tran, T.; Georgiou, D.K.; Arthur, K.; et al. Homozygous MTAP deletion in primary human glioblastoma is not associated with elevation of methylthioadenosine. Nat. Commun. 2021, 12, 4228. [Google Scholar] [CrossRef]
- Pettus, L.H.; Bourbeau, M.; Tamayo, N.A.; Amegadzie, A.; Beylkin, D.; Booker, S.K.; Butler, J.; Frohn, M.J.; Kaller, M.R.; Kohn, T.; et al. Discovery of AMG 193, an MTA-Cooperative PRMT5 Inhibitor for the Treatment of MTAP-Deleted Cancers. J. Med. Chem. 2025, 68, 6932–6954. [Google Scholar] [CrossRef]
- Ikushima, H.; Watanabe, K.; Shinozaki-Ushiku, A.; Oda, K.; Kage, H. Pan-cancer clinical and molecular landscape of MTAP deletion in nationwide and international comprehensive genomic data. ESMO Open 2025, 10, 104535. [Google Scholar] [CrossRef]
- Cottrell, K.M.; Briggs, K.J.; Tsai, A.; Tonini, M.R.; Whittington, D.A.; Gong, S.; Liang, C.; McCarren, P.; Zhang, M.; Zhang, W.; et al. Discovery of TNG462: A Highly Potent and Selective MTA-Cooperative PRMT5 Inhibitor to Target Cancers with MTAP Deletion. J. Med. Chem. 2025, 68, 5097–5119. [Google Scholar] [CrossRef]
- Cottrell, K.M.; Briggs, K.J.; Whittington, D.A.; Jahic, H.; Ali, J.A.; Davis, C.B.; Gong, S.; Gotur, D.; Gu, L.; McCarren, P.; et al. Discovery of TNG908: A Selective, Brain Penetrant, MTA-Cooperative PRMT5 Inhibitor That Is Synthetically Lethal with MTAP-Deleted Cancers. J. Med. Chem. 2024, 67, 6064–6080. [Google Scholar] [CrossRef]
- Briggs, K.; Tsai, A.; Zhang, M.; Tonini, M.; Haines, B.; Huang, A.; Cottrell, K. TNG462 is a potential best-in-class MTA-cooperative PRMT5 inhibitor for the treatment of peripheral MTAP-deleted solid tumors. Eur. J. Cancer 2022, 174, S84. [Google Scholar] [CrossRef]
- Che, Y.; Liu, Y.; Yao, Y.; Hill, H.A.; Li, Y.; Cai, Q.; Yan, F.; Jain, P.; Wang, W.; Rui, L.; et al. Exploiting PRMT5 as a target for combination therapy in mantle cell lymphoma characterized by frequent ATM and TP53 mutations. Blood Cancer J. 2023, 13, 27. [Google Scholar] [CrossRef]
- Lin, C.C.; Chang, T.C.; Wang, Y.; Guo, L.; Gao, Y.; Bikorimana, E.; Lemoff, A.; Fang, Y.V.; Zhang, H.; Zhang, Y.; et al. PRMT5 is an actionable therapeutic target in CDK4/6 inhibitor-resistant ER+/RB-deficient breast cancer. Nat. Commun. 2024, 15, 2287. [Google Scholar] [CrossRef]
- Brown-Burke, F.; Hwang, I.; Sloan, S.; Hinterschied, C.; Helmig-Mason, J.; Long, M.; Chan, W.K.; Prouty, A.; Chung, J.H.; Zhang, Y.; et al. PRMT5 inhibition drives therapeutic vulnerability to combination treatment with BCL-2 inhibition in mantle cell lymphoma. Blood Adv. 2023, 7, 6211–6224. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Kreuger, I.Z.M.; Slieker, R.C.; van Groningen, T.; van Doorn, R. Therapeutic Strategies for Targeting CDKN2A Loss in Melanoma. J. Invest. Dermatol. 2023, 143, 18–25.e11. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes. Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef]
- AbuHammad, S.; Cullinane, C.; Martin, C.; Bacolas, Z.; Ward, T.; Chen, H.; Slater, A.; Ardley, K.; Kirby, L.; Chan, K.T.; et al. Regulation of PRMT5-MDM4 axis is critical in the response to CDK4/6 inhibitors in melanoma. Proc. Natl. Acad. Sci. USA 2019, 116, 17990–18000. [Google Scholar] [CrossRef]
- Li, L.; Zhao, G.D.; Shi, Z.; Qi, L.L.; Zhou, L.Y.; Fu, Z.X. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol. Lett. 2016, 12, 3045–3050. [Google Scholar] [CrossRef]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: From mechanism to clinical studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef]
- Falchook, G.S.; Lewis, K.D.; Infante, J.R.; Gordon, M.S.; Vogelzang, N.J.; DeMarini, D.J.; Sun, P.; Moy, C.; Szabo, S.A.; Roadcap, L.T.; et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Fecher, L.A.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Greger, J.G.; Eastman, S.D.; Zhang, V.; Bleam, M.R.; Hughes, A.M.; Smitheman, K.N.; Dickerson, S.H.; Laquerre, S.G.; Liu, L.; Gilmer, T.M. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol. Cancer Ther. 2012, 11, 909–920. [Google Scholar] [CrossRef]
- Berberich, A.; Kessler, T.; Thome, C.M.; Pusch, S.; Hielscher, T.; Sahm, F.; Oezen, I.; Schmitt, L.M.; Ciprut, S.; Hucke, N.; et al. Targeting Resistance against the MDM2 Inhibitor RG7388 in Glioblastoma Cells by the MEK Inhibitor Trametinib. Clin. Cancer Res. 2019, 25, 253–265. [Google Scholar] [CrossRef]
- Schreck, K.C.; Guajardo, A.; Lin, D.D.M.; Eberhart, C.G.; Grossman, S.A. Concurrent BRAF/MEK Inhibitors in BRAF V600-Mutant High-Grade Primary Brain Tumors. J. Natl. Compr. Canc Netw. 2018, 16, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, D.T.; Dvir, A.; Lossos, A.; Tzuk-Shina, T.; Lior, T.; Limon, D.; Yust-Katz, S.; Lokiec, A.; Ram, Z.; Ross, J.S.; et al. Clinical utility and treatment outcome of comprehensive genomic profiling in high grade glioma patients. J. Neurooncol. 2016, 130, 211–219. [Google Scholar] [CrossRef]
- Banasavadi-Siddegowda, Y.K.; Namagiri, S.; Otani, Y.; Sur, H.; Rivas, S.; Bryant, J.P.; Shellbourn, A.; Rock, M.; Chowdhury, A.; Lewis, C.T.; et al. Targeting protein arginine methyltransferase 5 sensitizes glioblastoma to trametinib. Neurooncol. Adv. 2022, 4, vdac095. [Google Scholar] [CrossRef]
- Tan, W.; Gu, Z.; Shen, B.; Jiang, J.; Meng, Y.; Da, Z.; Liu, H.; Tao, T.; Cheng, C. PTEN/Akt-p27(kip1) Signaling Promote the BM-MSCs Senescence and Apoptosis in SLE Patients. J. Cell Biochem. 2015, 116, 1583–1594. [Google Scholar] [CrossRef]
- Lee, J.J.; Kim, B.C.; Park, M.J.; Lee, Y.S.; Kim, Y.N.; Lee, B.L.; Lee, J.S. PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionizing radiation. Cell Death Differ. 2011, 18, 666–677. [Google Scholar] [CrossRef]
- Banasavadi-Siddegowda, Y.K.; Russell, L.; Frair, E.; Karkhanis, V.A.; Relation, T.; Yoo, J.Y.; Zhang, J.; Sif, S.; Imitola, J.; Baiocchi, R.; et al. PRMT5-PTEN molecular pathway regulates senescence and self-renewal of primary glioblastoma neurosphere cells. Oncogene 2017, 36, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Cornu, M.; Albert, V.; Hall, M.N. mTOR in aging, metabolism, and cancer. Curr. Opin. Genet. Dev. 2013, 23, 53–62. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef]
- Holmes, B.; Benavides-Serrato, A.; Saunders, J.T.; Landon, K.A.; Schreck, A.J.; Nishimura, R.N.; Gera, J. The protein arginine methyltransferase PRMT5 confers therapeutic resistance to mTOR inhibition in glioblastoma. J. Neurooncol. 2019, 145, 11–22. [Google Scholar] [CrossRef]
- Otani, Y.; Sur, H.P.; Rachaiah, G.; Namagiri, S.; Chowdhury, A.; Lewis, C.T.; Shimizu, T.; Gangaplara, A.; Wang, X.; Vezina, A.; et al. Inhibiting protein phosphatase 2A increases the antitumor effect of protein arginine methyltransferase 5 inhibition in models of glioblastoma. Neuro Oncol. 2021, 23, 1481–1493. [Google Scholar] [CrossRef]
- Hong, C.S.; Ho, W.; Zhang, C.; Yang, C.; Elder, J.B.; Zhuang, Z. LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol. Ther. 2015, 16, 821–833. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Clinical Trial Identifier/Therapy | Study Name | Phase/Recruitment Status | Key Findings/Conclusions |
|---|---|---|---|
| NCT02783300/GSK3326595 | An Open-label, Dose Escalation Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics and Clinical Activity of GSK3326595 in Participants with Solid Tumors and Non-Hodgkin′s Lymphoma (Meteor 1) | Phase 1/completed | GSK3326595 was generally well-tolerated, with about 95% of patients experiencing treatment-related adverse events, most commonly fatigue, anemia, nausea, and alopecia [46]. |
| NCT03573310/JNJ-64619178 | A Study of JNJ-64619178, an Inhibitor of PRMT5 in Participants with Advanced Solid Tumors, NHL, and Lower Risk MDS | Phase 1/active, not recruiting | JNJ-64619178 showed manageable safety, early anti-tumor activity, and suitable dosing, supporting further evaluation in Phase 2 trials [22,47]. |
| NCT03854227/PF-06939999 | A Dose Escalation Study Of PF-06939999 In Participants with Advanced or Metastatic Solid Tumors | Phase 1/terminated | PF-06939999 was generally well-tolerated. Dose-limiting toxicities were observed in 17% of patients during the dose-escalation phase, including thrombocytopenia, anemia, and neutropenia [41]. |
| NCT04089449/PRT811 | A Study of PRT811 in Participants with Advanced Solid Tumors, CNS Lymphoma and Gliomas | Phase 1/completed | PRT811 demonstrated an acceptable safety profile [48]. |
| NCT06883747/BMS-986504 (MRTX1719) | Clinical Trial of BMS-986504 in Recurrent GBM Patients | Early Phase 1/recruiting | MRTX1719 is being evaluated for its pharmacokinetics (PK), safety, and tolerability in patients with recurrent glioblastoma who harbor MTAP gene deletions. |
| NCT05275478/TNG908 | Safety and Tolerability of TNG908 in Patients With MTAP-deleted Solid Tumors | Phase 1, 2/active, not recruiting | TNG908 is being evaluated in a phase 1/2 clinical trial to assess its safety, tolerability, and early signs of ant-tumor activity in patients with MTAP-deleted advanced or metastatic solid tumors [49]. |
| NCT05732831/TNG462 | Safety and Tolerability of TNG462 in Patients With MTAP-deleted Solid Tumors | Phase 1, 2/recruiting | TNG462 is being evaluated in a phase 1/2 clinical trial to assess its safety and tolerability as a single agent and in combination in patients with advanced or metastatic solid tumors harboring MTAP deletions. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.T.T.; Yi, E.; Badr, C.E. Exploiting Synthetic Lethality of PRMT5 for Precision Treatment of MTAP-Deficient Glioblastoma. Int. J. Transl. Med. 2025, 5, 27. https://doi.org/10.3390/ijtm5030027
Nguyen TTT, Yi E, Badr CE. Exploiting Synthetic Lethality of PRMT5 for Precision Treatment of MTAP-Deficient Glioblastoma. International Journal of Translational Medicine. 2025; 5(3):27. https://doi.org/10.3390/ijtm5030027
Chicago/Turabian StyleNguyen, Trang T. T., Eunhee Yi, and Christian E. Badr. 2025. "Exploiting Synthetic Lethality of PRMT5 for Precision Treatment of MTAP-Deficient Glioblastoma" International Journal of Translational Medicine 5, no. 3: 27. https://doi.org/10.3390/ijtm5030027
APA StyleNguyen, T. T. T., Yi, E., & Badr, C. E. (2025). Exploiting Synthetic Lethality of PRMT5 for Precision Treatment of MTAP-Deficient Glioblastoma. International Journal of Translational Medicine, 5(3), 27. https://doi.org/10.3390/ijtm5030027