
Early Neural Changes as Underlying Pathophysiological Mechanism in Diabetic Retinopathy

 ,
,  ,
, 
{kind=link}
Abstract
:1. Diabetic Retinopathy as a World-Wide Burden on Health Systems
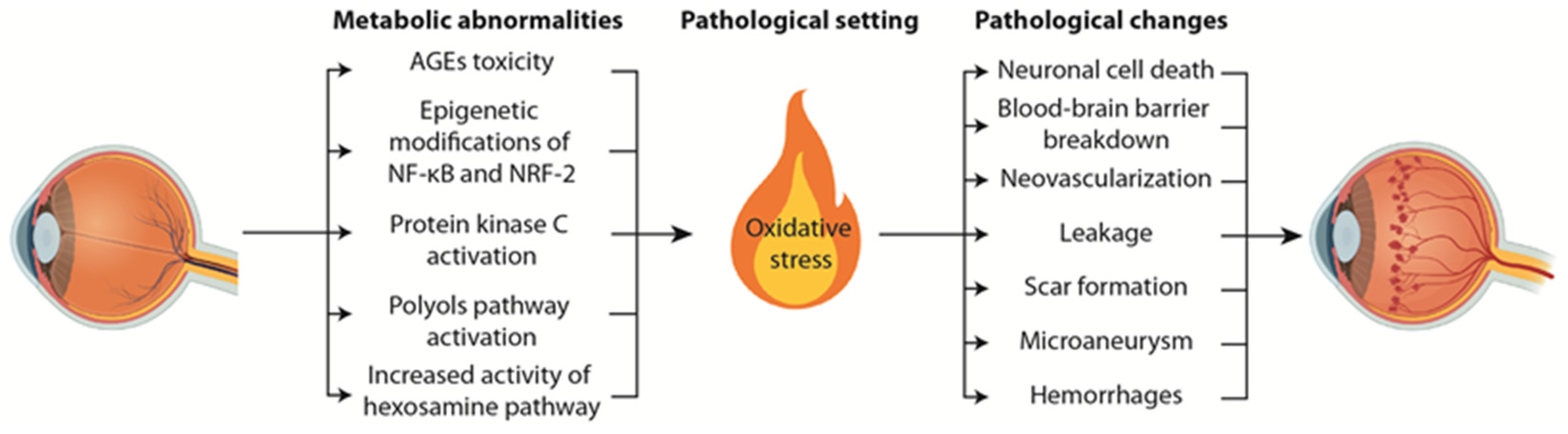
2. Pathophysiological Mechanisms Underlying Diabetic Retinopathy
3. Early Neural Alterations Are Associated to DR Pathological Mechanisms
4. In Vivo and In Vitro Models for the Study of DR
5. Novel Therapeutic Targets in DR
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Romero-Aroca, P.; Baget-Bernaldiz, M.; Pareja-Rios, A.; Lopez-Galvez, M.; Navarro-Gil, R.; Verges, R. Diabetic Macular Edema Pathophysiology: Vasogenic versus Inflammatory. J. Diabetes Res. 2016, 2016, 2156273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Floch, J.P.; Doucet, J.; Bauduceau, B.; Verny, C. Retinopathy, nephropathy, peripheral neuropathy and geriatric scale scores in elderly people with Type 2 diabetes. Diabet. Med. A J. Br. Diabet. Assoc. 2014, 31, 107–111. [Google Scholar] [CrossRef]
- Bourne, R.R.; Stevens, G.A.; White, R.A.; Smith, J.L.; Flaxman, S.R.; Price, H.; Jonas, J.B.; Keeffe, J.; Leasher, J.; Naidoo, K.; et al. Causes of vision loss worldwide, 1990-2010: A systematic analysis. Lancet Glob. Health 2013, 1, e339–e349. [Google Scholar] [CrossRef] [Green Version]
- Yau, J.W.; Rogers, S.L.; Kawasaki, R.; Lamoureux, E.L.; Kowalski, J.W.; Bek, T.; Chen, S.J.; Dekker, J.M.; Fletcher, A.; Grauslund, J.; et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 2012, 35, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Kempen, J.H.; O’Colmain, B.J.; Leske, M.C.; Haffner, S.M.; Klein, R.; Moss, S.E.; Taylor, H.R.; Hamman, R.F. The prevalence of diabetic retinopathy among adults in the United States. Arch. Ophthalmol. 2004, 122, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Q.; Welchowski, T.; Schmid, M.; Letow, J.; Wolpers, C.; Pascual-Camps, I.; Holz, F.G.; Finger, R.P. Prevalence, incidence and future projection of diabetic eye disease in Europe: A systematic review and meta-analysis. Eur. J. Epidemiol. 2020, 35, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Rema, M.; Premkumar, S.; Anitha, B.; Deepa, R.; Pradeepa, R.; Mohan, V. Prevalence of diabetic retinopathy in urban India: The Chennai Urban Rural Epidemiology Study (CURES) eye study, I. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2328–2333. [Google Scholar] [CrossRef] [Green Version]
- Raman, R.; Rani, P.K.; Reddi Rachepalle, S.; Gnanamoorthy, P.; Uthra, S.; Kumaramanickavel, G.; Sharma, T. Prevalence of diabetic retinopathy in India: Sankara Nethralaya Diabetic Retinopathy Epidemiology and Molecular Genetics Study report 2. Ophthalmology 2009, 116, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Klein, B.E.; Moss, S.E.; Davis, M.D.; DeMets, D.L. The Wisconsin epidemiologic study of diabetic retinopathy. II. Prevalence and risk of diabetic retinopathy when age at diagnosis is less than 30 years. Arch. Ophthalmol. 1984, 102, 520–526. [Google Scholar] [CrossRef]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Raymond, N.T.; Varadhan, L.; Reynold, D.R.; Bush, K.; Sankaranarayanan, S.; Bellary, S.; Barnett, A.H.; Kumar, S.; O’Hare, J.P. Higher prevalence of retinopathy in diabetic patients of South Asian ethnicity compared with white Europeans in the community: A cross-sectional study. Diabetes Care 2009, 32, 410–415. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Zhang, D.; Ren, Q.; Su, X.; Sun, Z. Prevalence and risk factors of diabetic retinopathy in diabetic patients: A community based cross-sectional study. Medicine 2020, 99, e19236. [Google Scholar] [CrossRef]
- Knowler, W.C.; Barrett-Connor, E.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [CrossRef]
- Song, P.; Yu, J.; Chan, K.Y.; Theodoratou, E.; Rudan, I. Prevalence, risk factors and burden of diabetic retinopathy in China: A systematic review and meta-analysis. J. Glob. Health 2018, 8, 010803. [Google Scholar] [CrossRef]
- Hammes, H.P. Diabetic retinopathy: Hyperglycaemia, oxidative stress and beyond. Diabetologia 2018, 61, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Spencer, B.G.; Estevez, J.J.; Liu, E.; Craig, J.E.; Finnie, J.W. Pericytes, inflammation, and diabetic retinopathy. Inflammopharmacology 2020, 28, 697–709. [Google Scholar] [CrossRef]
- Eshaq, R.S.; Aldalati, A.M.Z.; Alexander, J.S.; Harris, N.R. Diabetic retinopathy: Breaking the barrier. Pathophysiol. Off. J. Int. Soc. Pathophysiol. 2017, 24, 229–241. [Google Scholar] [CrossRef]
- Yang, X.; Yu, X.W.; Zhang, D.D.; Fan, Z.G. Blood-retinal barrier as a converging pivot in understanding the initiation and development of retinal diseases. Chin. Med. J. 2020, 133, 2586–2594. [Google Scholar] [CrossRef]
- Vinores, S.A. Breakdown of the Blood–Retinal Barrier. Encycl. Eye 2010, 216–222. [Google Scholar] [CrossRef]
- Omri, S.; Behar-Cohen, F.; Rothschild, P.-R.; Gélizé, E.; Jonet, L.; Jeanny, J.C.; Omri, B.; Crisanti, P. PKCζ mediates breakdown of outer blood-retinal barriers in diabetic retinopathy. PLoS ONE 2013, 8, e81600. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Gandhi, J.K.; Zhong, X.; Wei, Y.; Gong, J.; Duh, E.J.; Vinores, S.A. TNFalpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1336–1344. [Google Scholar] [CrossRef] [Green Version]
- Lechner, J.; O’Leary, O.E.; Stitt, A.W. The pathology associated with diabetic retinopathy. Vis. Res. 2017, 139, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, C.P.; Ferris, F.L., 3rd; Klein, R.E.; Lee, P.P.; Agardh, C.D.; Davis, M.; Dills, D.; Kampik, A.; Pararajasegaram, R.; Verdaguer, J.T. Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology 2003, 110, 1677–1682. [Google Scholar] [CrossRef]
- Cilenšek, I.; Mankoč, S.; Globočnik Petrovič, M.; Petrovič, D. The 4a/4a genotype of the VNTR polymorphism for endothelial nitric oxide synthase (eNOS) gene predicts risk for proliferative diabetic retinopathy in Slovenian patients (Caucasians) with type 2 diabetes mellitus. Mol. Biol. Rep. 2012, 39, 7061–7067. [Google Scholar] [CrossRef]
- Jafarzadeh, F.; Javanbakht, A.; Bakhtar, N.; Dalvand, A.; Shabani, M.; Mehrabinejad, M.M. Association between diabetic retinopathy and polymorphisms of cytokine genes: A systematic review and meta-analysis. Int. Ophthalmol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Balasubbu, S.; Sundaresan, P.; Rajendran, A.; Ramasamy, K.; Govindarajan, G.; Perumalsamy, N.; Hejtmancik, J.F. Association analysis of nine candidate gene polymorphisms in Indian patients with type 2 diabetic retinopathy. BMC Med. Genet. 2010, 11, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampton, B.M.; Schwartz, S.G.; Brantley, M.A., Jr.; Flynn, H.W., Jr. Update on genetics and diabetic retinopathy. Clin. Ophthalmol. 2015, 9, 2175–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson-Berka, J.L.; Rana, I.; Armani, R.; Agrotis, A. Reactive oxygen species, Nox and angiotensin II in angiogenesis: Implications for retinopathy. Clin. Sci. 2013, 124, 597–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, J.V.; Kuffova, L.; Delibegovic, M. The Role of Inflammation in Diabetic Retinopathy. Front. Immunol. 2020, 11, 583687. [Google Scholar] [CrossRef]
- Rübsam, A.; Parikh, S.; Fort, P.E. Role of Inflammation in Diabetic Retinopathy. Int. J. Mol. Sci. 2018, 19, 942. [Google Scholar] [CrossRef] [Green Version]
- Ucgun, N.I.; Zeki-Fikret, C.; Yildirim, Z. Inflammation and diabetic retinopathy. Mol. Vis. 2020, 26, 718–721. [Google Scholar] [PubMed]
- Hong, F.; Yang, D.Y.; Li, L.; Zheng, Y.F.; Wang, X.J.; Guo, S.R.N.; Jiang, S.; Zhu, D.; Tao, Y. Relationship Between Aqueous Humor Levels of Cytokines and Axial Length in Patients With Diabetic Retinopathy. Asia-Pac. J. Ophthalmol. 2020, 9, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Cao, D.; Yu, H.; Hu, Y.; He, M.; Yang, D.; Zhuang, X.; Zhang, L. Comprehensive analysis of vitreous humor chemokines in type 2 diabetic patients with and without diabetic retinopathy. Acta Diabetol. 2019, 56, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, S.; Xia, X. Role of intravitreal inflammatory cytokines and angiogenic factors in proliferative diabetic retinopathy. Curr. Eye Res. 2012, 37, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Dagher, Z.; Park, Y.S.; Asnaghi, V.; Hoehn, T.; Gerhardinger, C.; Lorenzi, M. Studies of rat and human retinas predict a role for the polyol pathway in human diabetic retinopathy. Diabetes 2004, 53, 2404–2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef]
- Uchiki, T.; Weikel, K.A.; Jiao, W.; Shang, F.; Caceres, A.; Pawlak, D.; Handa, J.T.; Brownlee, M.; Nagaraj, R.; Taylor, A. Glycation-altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging, and age-related disease (in nondiabetics). Aging Cell 2012, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowan, S.; Jiang, S.; Korem, T.; Szymanski, J.; Chang, M.L.; Szelog, J.; Cassalman, C.; Dasuri, K.; McGuire, C.; Nagai, R.; et al. Involvement of a gut-retina axis in protection against dietary glycemia-induced age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2017, 114, E4472–E4481. [Google Scholar] [CrossRef] [Green Version]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic targeting of advanced glycation end-products in age-related diseases. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 3631–3643. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W. Advanced glycation: An important pathological event in diabetic and age related ocular disease. Br. J. Ophthalmol. 2001, 85, 746–753. [Google Scholar] [CrossRef]
- Stitt, A.W.; Li, Y.M.; Gardiner, T.A.; Bucala, R.; Archer, D.B.; Vlassara, H. Advanced glycation end products (AGEs) co-localize with AGE receptors in the retinal vasculature of diabetic and of AGE-infused rats. Am. J. Pathol. 1997, 150, 523–531. [Google Scholar]
- Chen, M.; Curtis, T.M.; Stitt, A.W. Advanced glycation end products and diabetic retinopathy. Curr. Med. Chem. 2013, 20, 3234–3240. [Google Scholar] [CrossRef] [PubMed]
- Aragonès, G.; Dasuri, K.; Olukorede, O.; Francisco, S.G.; Renneburg, C.; Kumsta, C.; Hansen, M.; Kageyama, S.; Komatsu, M.; Rowan, S.; et al. Autophagic receptor p62 protects against glycation-derived toxicity and enhances viability. Aging Cell 2020, 19, e13257. [Google Scholar] [CrossRef] [PubMed]
- Aragonès, G.; Rowan, S.; Francisco, S.G.; Yang, W.; Weinberg, J.; Taylor, A.; Bejarano, E. Glyoxalase System as a Therapeutic Target against Diabetic Retinopathy. Antioxidants 2020, 9, 1062. [Google Scholar] [CrossRef] [PubMed]
- Queisser, M.A.; Yao, D.; Geisler, S.; Hammes, H.P.; Lochnit, G.; Schleicher, E.D.; Brownlee, M.; Preissner, K.T. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes 2010, 59, 670–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safi, S.Z.; Qvist, R.; Kumar, S.; Batumalaie, K.; Ismail, I.S. Molecular mechanisms of diabetic retinopathy, general preventive strategies, and novel therapeutic targets. BioMed Res. Int. 2014, 2014, 801269. [Google Scholar] [CrossRef] [Green Version]
- Tezel, G.; Luo, C.; Yang, X. Accelerated aging in glaucoma: Immunohistochemical assessment of advanced glycation end products in the human retina and optic nerve head. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Inagaki, Y.; Takenaka, K.; Jinnouchi, Y.; Yoshida, Y.; Matsuura, T.; Narama, I.; Motomiya, Y.; et al. Pigment epithelium-derived factor inhibits advanced glycation end product-induced retinal vascular hyperpermeability by blocking reactive oxygen species-mediated vascular endothelial growth factor expression. J. Biol. Chem. 2006, 281, 20213–20220. [Google Scholar] [CrossRef] [Green Version]
- Sorbinil Retinopathy Triail Research Group. A Randomized Trial of Sorbinil, an Aldose Reductase Inhibitor, in Diabetic Retinopathy. Arch. Ophthalmol. 1990, 108, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengellér, Z.; Szabó, C.; Brownlee, M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 2003, 112, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Barber, A.J.; Antonetti, D.A.; LaNoue, K.F.; Robinson, K.A.; Buse, M.G.; Gardner, T.W. Excessive hexosamines block the neuroprotective effect of insulin and induce apoptosis in retinal neurons. J. Biol. Chem. 2001, 276, 43748–43755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idris, I.; Gray, S.; Donnelly, R. Protein kinase C activation: Isozyme-specific effects on metabolism and cardiovascular complications in diabetes. Diabetologia 2001, 44, 659–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiello, L.P.; Clermont, A.; Arora, V.; Davis, M.D.; Sheetz, M.J.; Bursell, S.E. Inhibition of PKC beta by oral administration of ruboxistaurin is well tolerated and ameliorates diabetes-induced retinal hemodynamic abnormalities in patients. Investig. Ophthalmol. Vis. Sci. 2006, 47, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Xia, P.; Inoguchi, T.; Kern, T.S.; Engerman, R.L.; Oates, P.J.; King, G.L. Characterization of the mechanism for the chronic activation of diacylglycerol-protein kinase C pathway in diabetes and hypergalactosemia. Diabetes 1994, 43, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Aiello, L.P.; Vignati, L.; Sheetz, M.J.; Zhi, X.; Girach, A.; Davis, M.D.; Wolka, A.M.; Shahri, N.; Milton, R.C. Oral protein kinase c β inhibition using ruboxistaurin: Efficacy, safety, and causes of vision loss among 813 patients (1,392 eyes) with diabetic retinopathy in the Protein Kinase C β Inhibitor-Diabetic Retinopathy Study and the Protein Kinase C β Inhibitor-Diabetic Retinopathy Study 2. Retina 2011, 31, 2084–2094. [Google Scholar] [CrossRef]
- Kowluru, R.A. Retinopathy in a Diet-Induced Type 2 Diabetic Rat Model and Role of Epigenetic Modifications. Diabetes 2020, 69, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Karmakar, A.; Ganesan, S.K. Targeting epigenetic modifications as a potential therapeutic option for diabetic retinopathy. J. Cell. Physiol. 2020, 235, 1933–1947. [Google Scholar] [CrossRef] [PubMed]
- Sui, A.; Chen, X.; Demetriades, A.M.; Shen, J.; Cai, Y.; Yao, Y.; Yao, Y.; Zhu, Y.; Shen, X.; Xie, B. Inhibiting NF-κB Signaling Activation Reduces Retinal Neovascularization by Promoting a Polarization Shift in Macrophages. Investig. Ophthalmol. Vis. Sci. 2020, 61, 4. [Google Scholar] [CrossRef]
- Miller, W.P.; Sunilkumar, S.; Giordano, J.F.; Toro, A.L.; Barber, A.J.; Dennis, M.D. The stress response protein REDD1 promotes diabetes-induced oxidative stress in the retina by Keap1-independent Nrf2 degradation. J. Biol. Chem. 2020, 295, 7350–7361. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.; Hogg, R.E.; Chakravarthy, U. Antioxidants and diabetic retinopathy. Curr. Diabetes Rep. 2013, 13, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.; Lonn, E.M.; Yi, Q.; Gerstein, H.C.; Hoogwerf, B.J.; Pogue, J.; Bosch, J.; Dagenais, G.R.; Yusuf, S. Effects of vitamin E on cardiovascular outcomes in people with mild-to-moderate renal insufficiency: Results of the HOPE study. Kidney Int. 2004, 65, 1375–1380. [Google Scholar] [CrossRef] [Green Version]
- Hammes, H.P.; Lin, J.; Renner, O.; Shani, M.; Lundqvist, A.; Betsholtz, C.; Brownlee, M.; Deutsch, U. Pericytes and the pathogenesis of diabetic retinopathy. Diabetes 2002, 51, 3107–3112. [Google Scholar] [CrossRef] [Green Version]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, E.H.; van Dijk, H.W.; Jiao, C.; Kok, P.H.; Jeong, W.; Demirkaya, N.; Garmager, A.; Wit, F.; Kucukevcilioglu, M.; van Velthoven, M.E.; et al. Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc. Natl. Acad. Sci. USA 2016, 113, E2655–E2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simó, R.; Stitt, A.W.; Gardner, T.W. Neurodegeneration in diabetic retinopathy: Does it really matter? Diabetologia 2018, 61, 1902–1912. [Google Scholar] [CrossRef] [Green Version]
- Lynch, S.K.; Abràmoff, M.D. Diabetic retinopathy is a neurodegenerative disorder. Vis. Res. 2017, 139, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; McKay, G.J.; Jenkins, A.; Gardiner, T.A.; Lyons, T.J.; Hammes, H.P.; Simó, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Kato, K.; Hamada, Y.; Nakayama, M.; Chaya, S.; Nakashima, E.; Naruse, K.; Kasuya, Y.; Mizubayashi, R.; Miwa, K.; et al. A protein kinase C-beta-selective inhibitor ameliorates neural dysfunction in streptozotocin-induced diabetic rats. Diabetes 1999, 48, 2090–2095. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, M.; Wickremasinghe, S.; Osborne, A.; Van Wijngaarden, P.; Martin, K.R. Diabetic retinopathy: A complex pathophysiology requiring novel therapeutic strategies. Expert Opin. Biol. Ther. 2018, 18, 1257–1270. [Google Scholar] [CrossRef]
- Trento, M.; Durando, O.; Lavecchia, S.; Charrier, L.; Cavallo, F.; Costa, M.A.; Hernández, C.; Simó, R.; Porta, M. Vision related quality of life in patients with type 2 diabetes in the EUROCONDOR trial. Endocrine 2017, 57, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.K.; Lo, A.C. Animal models of diabetic retinopathy: Summary and comparison. J. Diabetes Res. 2013, 2013, 106594. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, J.H.; Yu, Y.S.; Cho, C.S.; Kim, K.W. Blockade of angiotensin II attenuates VEGF-mediated blood-retinal barrier breakdown in diabetic retinopathy. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2009, 29, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.; Palinski, W.; Schmid-Schönbein, G.W. Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am. J. Pathol. 1991, 139, 81–100. [Google Scholar]
- Kern, T.S.; Tang, J.; Mizutani, M.; Kowluru, R.A.; Nagaraj, R.H.; Romeo, G.; Podesta, F.; Lorenzi, M. Response of capillary cell death to aminoguanidine predicts the development of retinopathy: Comparison of diabetes and galactosemia. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3972–3978. [Google Scholar]
- Barber, A.J.; Antonetti, D.A.; Kern, T.S.; Reiter, C.E.; Soans, R.S.; Krady, J.K.; Levison, S.W.; Gardner, T.W.; Bronson, S.K. The Ins2Akita mouse as a model of early retinal complications in diabetes. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2210–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, S.G.; Boden, J.P.; Biecker, E.; Reichen, J.; Rothen, B. Endothelin antagonism prevents diabetic retinopathy in NOD mice: A potential role of the angiogenic factor adrenomedullin. Exp. Biol. Med. 2006, 231, 1101–1105. [Google Scholar]
- Midena, E.; Segato, T.; Radin, S.; di Giorgio, G.; Meneghini, F.; Piermarocchi, S.; Belloni, A.S. Studies on the retina of the diabetic db/db mouse. I. Endothelial cell-pericyte ratio. Ophthalmic Res. 1989, 21, 106–111. [Google Scholar] [CrossRef]
- Sima, A.A.; Chakrabarti, S.; Garcia-Salinas, R.; Basu, P.K. The BB-rat--an authentic model of human diabetic retinopathy. Curr. Eye Res. 1985, 4, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, I.A.; Miyamura, N.; Amemiya, T. Vascular architecture of degenerated retina in WBN/Kob rats: Corrosion cast and electron microscopic study. Ophthalmic Res. 1999, 31, 367–377. [Google Scholar] [CrossRef]
- Yang, Y.S.; Danis, R.P.; Peterson, R.G.; Dolan, P.L.; Wu, Y.Q. Acarbose partially inhibits microvascular retinopathy in the Zucker Diabetic Fatty rat (ZDF/Gmi-fa). J. Ocul. Pharmacol. Ther. Off. J. Assoc. Ocul. Pharmacol. Ther. 2000, 16, 471–479. [Google Scholar] [CrossRef]
- Miyamoto, K.; Ogura, Y.; Nishiwaki, H.; Matsuda, N.; Honda, Y.; Kato, S.; Ishida, H.; Seino, Y. Evaluation of retinal microcirculatory alterations in the Goto-Kakizaki rat. A spontaneous model of non-insulin-dependent diabetes. Investig. Ophthalmol. Vis. Sci. 1996, 37, 898–905. [Google Scholar]
- Grossniklaus, H.E.; Kang, S.J.; Berglin, L. Animal models of choroidal and retinal neovascularization. Prog. Retin. Eye Res. 2010, 29, 500–519. [Google Scholar] [CrossRef] [Green Version]
- Miranda, M.; Muriach, M.; Johnsen, S.; Bosch-Morell, F.; Araiz, J.; Romá, J.; Romero, F.J. [Oxidative stress in a model for experimental diabetic retinopathy: Treatment with antioxidants]. Arch. Soc. Esp. Oftalmol. 2004, 79, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Hernández, C.; García-Ramírez, M.; Corraliza, L.; Fernández-Carneado, J.; Farrera-Sinfreu, J.; Ponsati, B.; González-Rodríguez, A.; Valverde, A.M.; Simó, R. Topical administration of somatostatin prevents retinal neurodegeneration in experimental diabetes. Diabetes 2013, 62, 2569–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnal, E.; Miranda, M.; Johnsen-Soriano, S.; Alvarez-Nölting, R.; Díaz-Llopis, M.; Araiz, J.; Cervera, E.; Bosch-Morell, F.; Romero, F.J. Beneficial effect of docosahexanoic acid and lutein on retinal structural, metabolic, and functional abnormalities in diabetic rats. Curr. Eye Res. 2009, 34, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Hombrebueno, J.R.; Chen, M.; Penalva, R.G.; Xu, H. Loss of synaptic connectivity, particularly in second order neurons is a key feature of diabetic retinal neuropathy in the Ins2Akita mouse. PLoS ONE 2014, 9, e97970. [Google Scholar] [CrossRef]
- Sheskey, S.R.; Antonetti, D.A.; Rentería, R.C.; Lin, C.M. Correlation of Retinal Structure and Visual Function Assessments in Mouse Diabetes Models. Investig. Ophthalmol. Vis. Sci. 2021, 62, 20. [Google Scholar] [CrossRef]
- Yang, Q.; Xu, Y.; Xie, P.; Cheng, H.; Song, Q.; Su, T.; Yuan, S.; Liu, Q. Retinal Neurodegeneration in db/db Mice at the Early Period of Diabetes. J. Ophthalmol. 2015, 2015, 757412. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, P.; Corraliza, L.; Villena, J.A.; Carvalho, A.R.; Garcia-Arumí, J.; Ramos, D.; Ruberte, J.; Simó, R.; Hernández, C. The db/db mouse: A useful model for the study of diabetic retinal neurodegeneration. PLoS ONE 2014, 9, e97302. [Google Scholar] [CrossRef] [Green Version]
- Mi, X.S.; Yuan, T.F.; Ding, Y.; Zhong, J.X.; So, K.F. Choosing preclinical study models of diabetic retinopathy: Key problems for consideration. Drug Des. Dev. Ther. 2014, 8, 2311–2319. [Google Scholar] [CrossRef] [Green Version]
- Valdés, J.; Trachsel-Moncho, L.; Sahaboglu, A.; Trifunović, D.; Miranda, M.; Ueffing, M.; Paquet-Durand, F.; Schmachtenberg, O. Organotypic retinal explant cultures as in vitro alternative for diabetic retinopathy studies. Altex 2016, 33, 459–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnichels, S.; Paquet-Durand, F.; Löscher, M.; Tsai, T.; Hurst, J.; Joachim, S.C.; Klettner, A. Retina in a dish: Cell cultures, retinal explants and animal models for common diseases of the retina. Prog. Retin. Eye Res. 2021, 81, 100880. [Google Scholar] [CrossRef]
- Gardiner, T.A.; Anderson, H.R.; Stitt, A.W. Inhibition of advanced glycation end-products protects against retinal capillary base-ment membrane expansion during long-term diabetes. J. Pathol. 2003, 201, 328–333. [Google Scholar] [CrossRef]
- Schlotterer, A.; Kolibabka, M.; Lin, J.; Acunman, K.; Dietrich, N.; Sticht, C.; Fleming, T.; Nawroth, P.; Hammes, H.P. Methylglyoxal induces retinopathy-type lesions in the absence of hyperglycemia: Studies in a rat model. FASEB J. 2019, 33, 4141–4153. [Google Scholar] [CrossRef] [Green Version]
- Karachalias, N.; Babaei-Jadidi, R.; Ahmed, N.; Thornalley, P.J. Accumulation of fructosyl-lysine and advanced glycation end products in the kidney, retina and peripheral nerve of streptozotocin-induced diabetic rats. Biochem. Soc. Trans. 2003, 31, 1423–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiba, T.; Inoguchi, T.; Sportsman, J.R.; Heath, W.F.; Bursell, S.; King, G.L. Correlation of diacylglycerol level and protein kinase C activity in rat retina to retinal circulation. Am. J. Physiol. 1993, 265, E783–E793. [Google Scholar] [CrossRef]
- Saxena, R.; Singh, D.; Saklani, R.; Gupta, S.K. Clinical biomarkers and molecular basis for optimized treatment of diabetic retinopathy: Current status and future prospects. Eye Brain 2016, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A.; Engerman, R.L.; Kern, T.S. Abnormalities of retinal metabolism in diabetes or experimental galactosemia VIII. Prevention by aminoguanidine. Curr. Eye Res. 2000, 21, 814–819. [Google Scholar] [CrossRef]
- Bosch, R.R.; Janssen, S.W.; Span, P.N.; Olthaar, A.; van Emst-de Vries, S.E.; Willems, P.H.; Martens, J.M.G.; Hermus, A.R.; Sweep, C.C. Exploring levels of hexosamine biosynthesis pathway intermediates and protein kinase C isoforms in muscle and fat tissue of Zucker Diabetic Fatty rats. Endocrine 2003, 20, 247–252. [Google Scholar] [CrossRef]
- Villa, M.; Parravano, M.; Micheli, A.; Gaddini, L.; Matteucci, A.; Mallozzi, C.; Facchiano, F.; Malchiodi-Albedi, F.; Pricci, F. A quick, simple method for detecting circulating fluorescent advanced glycation end-products: Correlation with in vitro and in vivo non-enzymatic glycation. Metab. Clin. Exp. 2017, 71, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Saltsman, K.A.; Ohashi, H.; King, G.L. Activation of protein kinase C by elevation of glucose concentration: Proposal for a mechanism in the development of diabetic vascular complications. Proc. Natl. Acad. Sci. USA 1989, 86, 5141–5145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, T.W.; Chew, E.Y. Future opportunities in diabetic retinopathy research. Curr. Opin. Endocrinol. Diabetes Obes. 2016, 23, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bressler, N.M.; Varma, R.; Suñer, I.J.; Dolan, C.M.; Ward, J.; Ehrlich, J.S.; Colman, S.; Turpcu, A. Vision-related function after ranibizumab treatment for diabetic macular edema: Results from RIDE and RISE. Ophthalmology 2014, 121, 2461–2472. [Google Scholar] [CrossRef] [PubMed]
- Abramoff, M.D.; Fort, P.E.; Han, I.C.; Jayasundera, K.T.; Sohn, E.H.; Gardner, T.W. Approach for a Clinically Useful Comprehensive Classification of Vascular and Neural Aspects of Diabetic Retinal Disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.J. A new view of diabetic retinopathy: A neurodegenerative disease of the eye. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 283–290. [Google Scholar] [CrossRef]
- Stem, M.S.; Gardner, T.W. Neurodegeneration in the pathogenesis of diabetic retinopathy: Molecular mechanisms and therapeutic implications. Curr. Med. Chem. 2013, 20, 3241–3250. [Google Scholar] [CrossRef] [Green Version]
- Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E and beta carotene for age-related cataract and vision loss: AREDS report no. 9. Arch. Ophthalmol. 2001, 119, 1439–1452. [Google Scholar] [CrossRef]
- Sala-Vila, A.; Díaz-López, A.; Valls-Pedret, C.; Cofán, M.; García-Layana, A.; Lamuela-Raventós, R.M.; Castañer, O.; Zanon-Moreno, V.; Martinez-Gonzalez, M.A.; Toledo, E.; et al. Dietary Marine ω-3 Fatty Acids and Incident Sight-Threatening Retinopathy in Middle-Aged and Older Individuals With Type 2 Diabetes: Prospective Investigation From the PREDIMED Trial. JAMA Ophthalmol. 2016, 134, 1142–1149. [Google Scholar] [CrossRef]
- Ribeiro, M.L.; Seres, A.I.; Carneiro, A.M.; Stur, M.; Zourdani, A.; Caillon, P.; Cunha-Vaz, J.G. Effect of calcium dobesilate on progression of early diabetic retinopathy: A randomised double-blind study. Graefe’s Arch. Clin. Exp. Ophthalmol. 2006, 244, 1591–1600. [Google Scholar] [CrossRef]
- Lanthony, P.; Cosson, J.P. [The course of color vision in early diabetic retinopathy treated with Ginkgo biloba extract. A preliminary double-blind versus placebo study]. J. Fr. D’ophtalmologie 1988, 11, 671–674. [Google Scholar]
- Huang, S.Y.; Jeng, C.; Kao, S.C.; Yu, J.J.; Liu, D.Z. Improved haemorrheological properties by Ginkgo biloba extract (Egb 761) in type 2 diabetes mellitus complicated with retinopathy. Clin. Nutr. 2004, 23, 615–621. [Google Scholar] [CrossRef]
- Alfonso-Muñoz, E.A.; Burggraaf-Sánchez de Las Matas, R.; Mataix Boronat, J.; Molina Martín, J.C.; Desco, C. Role of Oral Antioxidant Supplementation in the Current Management of Diabetic Retinopathy. Int. J. Mol. Sci. 2021, 22, 4020. [Google Scholar] [CrossRef]
- Eggers, E.D.; Carreon, T.A. The effects of early diabetes on inner retinal neurons. Vis. Neurosci. 2020, 37, E006. [Google Scholar] [CrossRef]
- Lv, J.; Bao, S.; Liu, T.; Wei, L.; Wang, D.; Ye, W.; Wang, N.; Song, S.; Li, J.; Chudhary, M.; et al. Sulforaphane delays diabetes-induced retinal photoreceptor cell degeneration. Cell Tissue Res. 2020, 382, 477–486. [Google Scholar] [CrossRef]
- Smit-McBride, Z.; Morse, L.S. MicroRNA and diabetic retinopathy-biomarkers and novel therapeutics. Ann. Transl. Med. 2021, 9, 1280. [Google Scholar] [CrossRef]
- Silva, V.A.; Polesskaya, A.; Sousa, T.A.; Corrêa, V.M.; André, N.D.; Reis, R.I.; Kettelhut, I.C.; Harel-Bellan, A.; De Lucca, F.L. Expression and cellular localization of microRNA-29b and RAX, an activator of the RNA-dependent protein kinase (PKR), in the retina of streptozotocin-induced diabetic rats. Mol. Vis. 2011, 17, 2228–2240. [Google Scholar]
- Lin, F.L.; Wang, P.Y.; Chuang, Y.F.; Wang, J.H.; Wong, V.H.Y.; Bui, B.V.; Liu, G.S. Gene Therapy Intervention in Neovascular Eye Disease: A Recent Update. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 2120–2138. [Google Scholar] [CrossRef]
- Wang, J.H.; Roberts, G.E.; Liu, G.S. Updates on Gene Therapy for Diabetic Retinopathy. Curr. Diabetes Rep. 2020, 20, 22. [Google Scholar] [CrossRef]
- Prea, S.M.; Chan, E.C.; Dusting, G.J.; Vingrys, A.J.; Bui, B.V.; Liu, G.S. Gene Therapy with Endogenous Inhibitors of Angiogenesis for Neovascular Age-Related Macular Degeneration: Beyond Anti-VEGF Therapy. J. Ophthalmol. 2015, 2015, 201726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Das, S.K.; Passi, S.F.; Uehara, H.; Bohner, A.; Chen, M.; Tiem, M.; Archer, B.; Ambati, B.K. AAV2 delivery of Flt23k intraceptors inhibits murine choroidal neovascularization. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 226–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, P.E.; Freeman, S.C.; Janot, A.C. Novel stem cell and gene therapy in diabetic retinopathy, age related macular degeneration, and retinitis pigmentosa. Int. J. Retin. Vitr. 2019, 5, 7. [Google Scholar] [CrossRef]
- Igarashi, T.; Miyake, K.; Kato, K.; Watanabe, A.; Ishizaki, M.; Ohara, K.; Shimada, T. Lentivirus-mediated expression of angiostatin efficiently inhibits neovascularization in a murine proliferative retinopathy model. Gene Ther. 2003, 10, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, J.W.; Mistry, A.; De Alwis, M.; Paleolog, E.; Baker, A.; Thrasher, A.J.; Ali, R.R. Inhibition of retinal neovascularisation by gene transfer of soluble VEGF receptor sFlt-1. Gene Ther. 2002, 9, 320–326. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.K.; Shen, W.Y.; Brankov, M.; Lai, C.M.; Constable, I.J.; Rakoczy, P.E. Potential long-term inhibition of ocular neovascularisation by recombinant adeno-associated virus-mediated secretion gene therapy. Gene Ther. 2002, 9, 804–813. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Ingham, S.A.; Harkins, K.A.; Do, D.V.; Nguyen, Q.D. The role of pharmacogenetics and advances in gene therapy in the treatment of diabetic retinopathy. Pharmacogenomics 2016, 17, 309–320. [Google Scholar] [CrossRef]
- Mao, X.B.; Cheng, Y.H.; Xu, Y.Y. miR-204-5p promotes diabetic retinopathy development via downregulation of microtubule-associated protein 1 light chain 3. Exp. Ther. Med. 2019, 17, 2945–2952. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Chang, Z.P.; Ren, R.T.; Wei, S.H.; Zhou, H.F.; Chen, X.F.; Hou, B.K.; Jin, X.; Zhang, M.N. Protective Effects of Adeno-associated Virus Mediated Brain-derived Neurotrophic Factor Expression on Retinal Ganglion Cells in Diabetic Rats. Cell. Mol. Neurobiol. 2012, 32, 467–475. [Google Scholar] [CrossRef]
- Verma, A.; Shan, Z.; Lei, B.; Yuan, L.; Liu, X.; Nakagawa, T.; Grant, M.B.; Lewin, A.S.; Hauswirth, W.W.; Raizada, M.K.; et al. ACE2 and Ang-(1-7) confer protection against development of diabetic retinopathy. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 28–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- First CRISPR therapy dosed. Nat. Biotechnol. 2020, 38, 382. [CrossRef] [Green Version]
- Ruan, G.X.; Barry, E.; Yu, D.; Lukason, M.; Cheng, S.H.; Scaria, A. CRISPR/Cas9-Mediated Genome Editing as a Therapeutic Approach for Leber Congenital Amaurosis 10. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronk, S.M.; Kelly-Goss, M.R.; Ray, H.C.; Mendel, T.A.; Hoehn, K.L.; Bruce, A.C.; Dey, B.K.; Guendel, A.M.; Tavakol, D.N.; Herman, I.M.; et al. Adipose-derived stem cells from diabetic mice show impaired vascular stabilization in a murine model of diabetic retinopathy. Stem Cells Transl. Med. 2015, 4, 459–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajashekhar, G.; Ramadan, A.; Abburi, C.; Callaghan, B.; Traktuev, D.O.; Evans-Molina, C.; Maturi, R.; Harris, A.; Kern, T.S.; March, K.L. Regenerative therapeutic potential of adipose stromal cells in early stage diabetic retinopathy. PLoS ONE 2014, 9, e84671. [Google Scholar] [CrossRef]
- Ezquer, M.; Urzua, C.A.; Montecino, S.; Leal, K.; Conget, P.; Ezquer, F. Intravitreal administration of multipotent mesenchymal stromal cells triggers a cytoprotective microenvironment in the retina of diabetic mice. Stem Cell Res. Ther. 2016, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Caballero, S.; Hazra, S.; Bhatwadekar, A.; Li Calzi, S.; Paradiso, L.J.; Miller, L.P.; Chang, L.J.; Kern, T.S.; Grant, M.B. Circulating mononuclear progenitor cells: Differential roles for subpopulations in repair of retinal vascular injury. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3000–3009. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wang, Y.; Kong, J.; Dong, M.; Duan, H.; Chen, S. Therapeutic efficacy of neural stem cells originating from umbilical cord-derived mesenchymal stem cells in diabetic retinopathy. Sci. Rep. 2017, 7, 408. [Google Scholar] [CrossRef]
- Yu, S.; Cheng, Y.; Zhang, L.; Yin, Y.; Xue, J.; Li, B.; Gong, Z.; Gao, J.; Mu, Y. Treatment with adipose tissue-derived mesenchymal stem cells exerts anti-diabetic effects, improves long-term complications, and attenuates inflammation in type 2 diabetic rats. Stem Cell Res. Ther. 2019, 10, 333. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cantó, A.; Martínez, J.; Perini-Villanueva, G.; Miranda, M.; Bejarano, E. Early Neural Changes as Underlying Pathophysiological Mechanism in Diabetic Retinopathy. Int. J. Transl. Med. 2022, 2, 1-16. https://doi.org/10.3390/ijtm2010001
Cantó A, Martínez J, Perini-Villanueva G, Miranda M, Bejarano E. Early Neural Changes as Underlying Pathophysiological Mechanism in Diabetic Retinopathy. International Journal of Translational Medicine. 2022; 2(1):1-16. https://doi.org/10.3390/ijtm2010001
Chicago/Turabian StyleCantó, Antolín, Javier Martínez, Giuliana Perini-Villanueva, María Miranda, and Eloy Bejarano. 2022. "Early Neural Changes as Underlying Pathophysiological Mechanism in Diabetic Retinopathy" International Journal of Translational Medicine 2, no. 1: 1-16. https://doi.org/10.3390/ijtm2010001
APA StyleCantó, A., Martínez, J., Perini-Villanueva, G., Miranda, M., & Bejarano, E. (2022). Early Neural Changes as Underlying Pathophysiological Mechanism in Diabetic Retinopathy. International Journal of Translational Medicine, 2(1), 1-16. https://doi.org/10.3390/ijtm2010001