
Pericyte and Vascular Smooth Muscle Death in Diabetic Retinopathy Involves Autophagy



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
3. Results
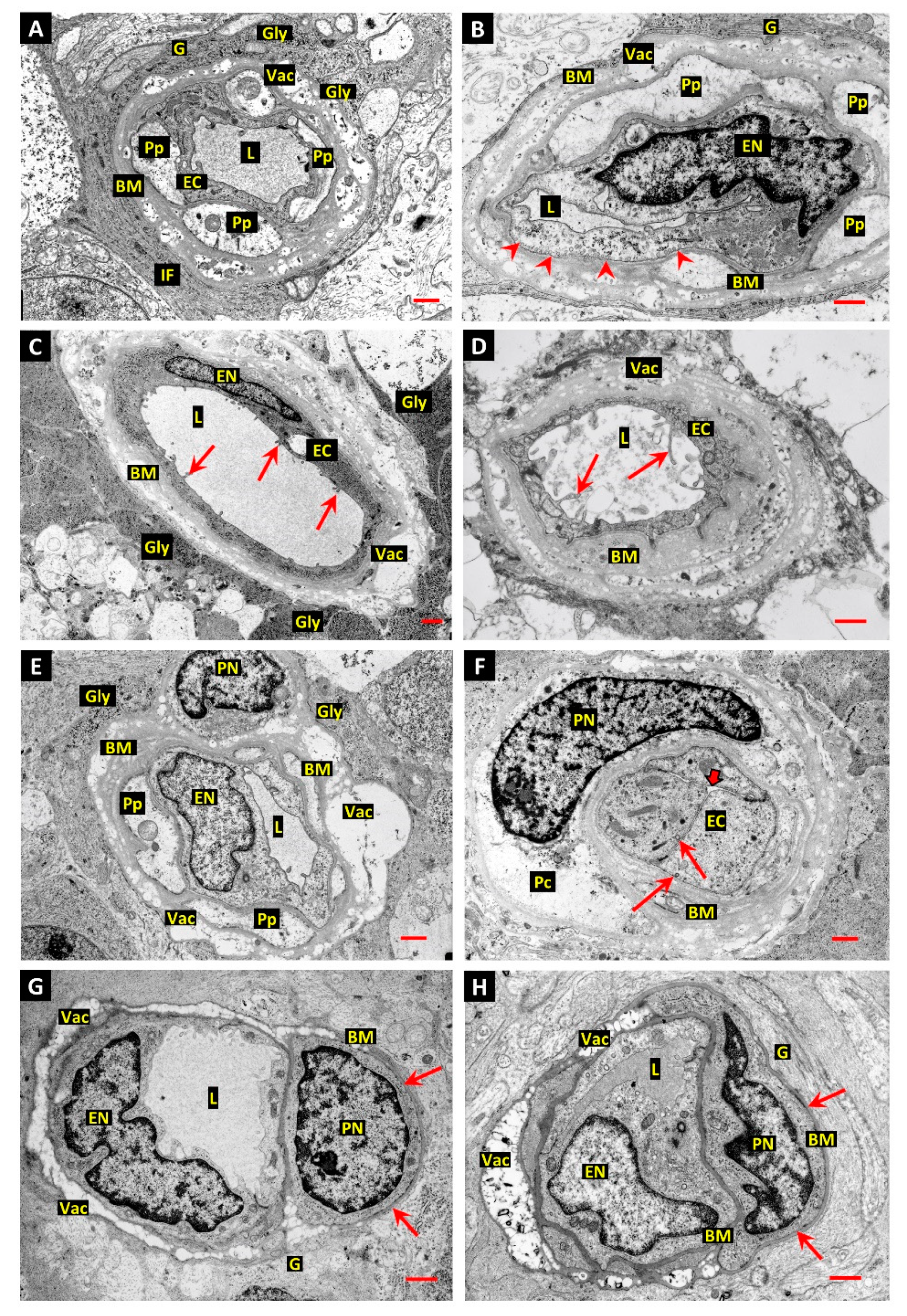
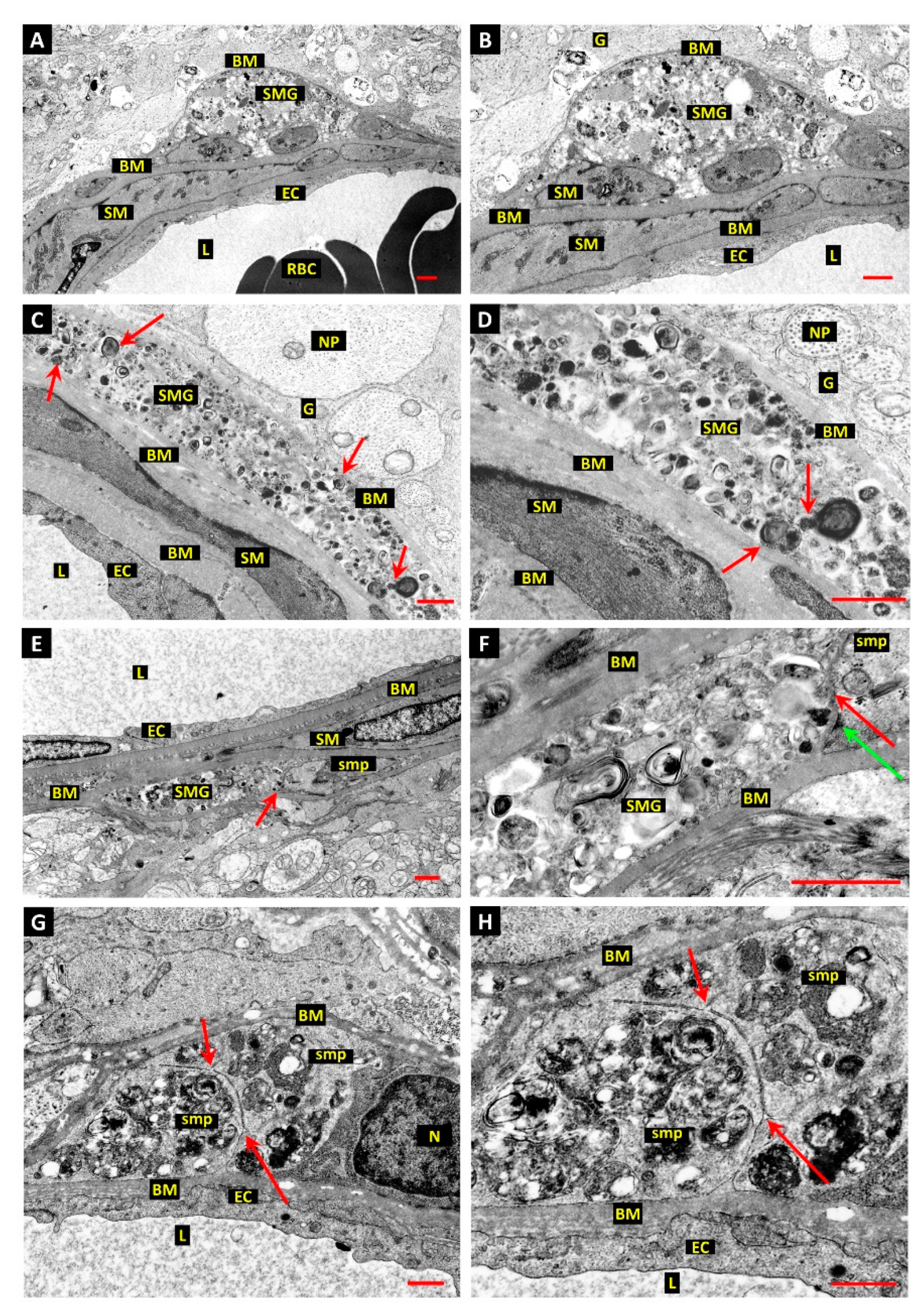
3.1. Differentiation of Post-Mortem (PM) Change from Diabetes-Related Mural Cell Death
3.2. Evidence of Mural Cell Autophagy in Diabetic Retinas
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; McKay, G.; Jenkins, A.; Gardiner, T.A.; Lyons, T.J.; Hammes, H.-P.; Simó, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef]
- Cogan, D.G.; Toussaint, D.; Kuwabara, T. Retinal Vascular Patterns. IV. Diabetic Retinopathy. Arch. Ophthalmol. 1961, 66, 366–378. [Google Scholar] [CrossRef]
- Gardiner, T.A.; Stitt, A.W.; Anderson, H.R.; Archer, D.B. Selective Loss of Vascular Smooth Muscle Cells in the Retinal Microcirculation of Diabetic Dogs. Br. J. Ophthalmol. 1994, 78, 54–60. [Google Scholar] [CrossRef]
- Gardiner, T.A.; Archer, D.B.; Curtis, T.M.; Stitt, A.W. Arteriolar Involvement in the Microvascular Lesions of Diabetic Retinopathy: Implications for Pathogenesis. Microcirculation 2007, 14, 25–38. [Google Scholar] [CrossRef]
- Tawata, M.; Ohtaka, M.; Hosaka, Y.; Onaya, T. Aldose Reductase mRNA Expression and Its Activity Are Induced by Glucose in Fetal Rat Aortic Smooth Muscle (A10) Cells. Life Sci. 1992, 51, 719–726. [Google Scholar] [CrossRef]
- Morrison, A.D. Effect of inhibition of polyol pathway activity on aortic smooth muscle metabolism. Clin. Investig. Med. 1990, 13, 119–122. [Google Scholar]
- Hohman, T.C.; Nishimura, C.; Robison, W.G., Jr. Aldose reductase and polyol in cultured pericytes of human retinal capillaries. Exp. Eye Res. 1989, 48, 55–60. [Google Scholar] [CrossRef]
- Stitt, A.W.; Li, Y.M.; Gardiner, T.A.; Bucala, R.; Archer, D.B.; Vlassara, H. Advanced glycation end products (AGEs) co-localize with AGE receptors in the retinal vasculature of diabetic and of AGE-infused rats. Am. J. Pathol. 1997, 150, 523–531. [Google Scholar]
- Fu, D.; Wu, M.; Zhang, J.; Du, M.; Yang, S.; Hammad, S.M.; Wilson, K.; Chen, J.; Lyons, T.J. Mechanisms of modified LDL-induced pericyte loss and retinal injury in diabetic retinopathy. Diabetologia 2012, 55, 3128–3140. [Google Scholar] [CrossRef]
- Martinet, W.; De Bie, M.; Schrijvers, D.M.; De Meyer, G.R.; Herman, A.G.; Kockx, M.M. 7-ketocholesterol induces protein ubiquitination, myelin figure formation, and light chain 3 processing in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2296–2301. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Yu, J.Y.; Yang, S.; Wu, M.; Hammad, S.M.; Connell, A.R.; Du, M.; Chen, J.; Lyons, T.J. Survival or death: A dual role for autophagy in stress-induced pericyte loss in diabetic retinopathy. Diabetologia 2016, 59, 2251–2261. [Google Scholar] [CrossRef]
- Martinet, W.; De Meyer, G.R. Autophagy in atherosclerosis. Curr. Atheroscler. Rep. 2008, 10, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson-Berka, J.L.; Babic, S.; De Gooyer, T.; Stitt, A.W.; Jaworski, K.; Ong, L.G.; Kelly, D.J.; Gilbert, R.E. Inhibition of platelet-derived growth factor promotes pericyte loss and angiogenesis in ischemic retinopathy. Am. J. Pathol. 2004, 164, 1263–1273. [Google Scholar] [CrossRef]
- Stitt, A.W.; Hughes, S.J.; Canning, P.; Lynch, O.; Cox, O.; Frizzell, N.; Thorpe, S.R.; Cotter, T.G.; Curtis, T.M.; Gardiner, T.A. Substrates modified by advanced glycation end-products cause dysfunction and death in retinal pericytes by reducing survival signals mediated by platelet-derived growth factor. Diabetologia 2004, 47, 1735–1746. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.P.; Lin, J.; Renner, O.; Shani, M.; Lundqvist, A.; Betsholtz, C.; Brownlee, M.; Deutsch, U. Pericytes and the pathogenesis of diabetic retinopathy. Diabetes 2002, 51, 3107–3112. [Google Scholar] [CrossRef]
- Bennett, M.R.; Evan, G.I.; Schwartz, S.M. Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. J. Clin. Investig. 1995, 95, 2266–2274. [Google Scholar] [CrossRef]
- Geraldes, P.; Hiraoka-Yamamoto, J.; Matsumoto, M.; Clermont, A.; Leitges, M.; Marette, A.; Aiello, L.P.; Kern, T.S.; King, G.L. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat. Med. 2009, 15, 1298–1306. [Google Scholar] [CrossRef]
- Cox, O.T.; Simpson, D.A.; Stitt, A.W.; Gardiner, T.A. Sources of PDGF expression in murine retina and the effect of short-term diabetes. Mol. Vis. 2003, 9, 665–672. [Google Scholar]
- Gerhardinger, C.; McClure, K.D.; Romeo, G.; Podesta, F.; Lorenzi, M. IGF-I mRNA and signaling in the diabetic retina. Diabetes 2001, 50, 175–183. [Google Scholar] [CrossRef][Green Version]
- Mizutani, M.; Kern, T.S.; Lorenzi, M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J. Clin. Investig. 1996, 97, 2883–2890. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Gardiner, T.A.; Archer, D.B. A morphologic and autoradiographic study of cell death and regeneration in the retinal microvasculature of normal and diabetic rats. Am. J. Ophthalmol. 1985, 100, 51–60. [Google Scholar] [CrossRef]
- Curtis, T.M.; Gardiner, T.A.; Stitt, A.W. Microvascular lesions of diabetic retinopathy: Clues towards understanding pathogenesis? Eye 2009, 23, 1496–1508. [Google Scholar] [CrossRef] [PubMed]
- Forrester, J.V.; Kuffova, L.; Delibegovic, M. The Role of Inflammation in Diabetic Retinopathy. Front. Immunol. 2020, 11, 583687. [Google Scholar] [CrossRef]
- Li, X.; Weber, N.C.; Cohn, D.M.; Hollmann, M.W.; DeVries, J.H.; Hermanides, J.; Preckel, B. Effects of Hyperglycemia and Diabetes Mellitus on Coagulation and Hemostasis. J. Clin. Med. 2021, 10, 2419. [Google Scholar] [CrossRef]
- Joussen, A.M.; Poulaki, V.; Le, M.L.; Koizumi, K.; Esser, C.; Janicki, H.; Schraermeyer, U.; Kociok, N.; Fauser, S.; Kirchhof, B.; et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004, 18, 1450–1452. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Flusberg, D.A.; Sorger, P.K. Surviving apoptosis: Life-death signaling in single cells. Trends Cell Biol. 2015, 25, 446–458. [Google Scholar] [CrossRef]
- Simpson, D.A.; Murphy, G.M.; Bhaduri, T.; Gardiner, T.A.; Archer, D.B.; Stitt, A.W. Expression of the VEGF gene family during retinal vaso-obliteration and hypoxia. Biochem. Biophys. Res. Commun. 1999, 262, 333–340. [Google Scholar] [CrossRef]
- Stitt, A.W.; Simpson, D.A.; Boocock, C.; Gardiner, T.A.; Murphy, G.M.; Archer, D.B. Expression of vascular endothelial growth factor (VEGF) and its receptors is regulated in eyes with intra-ocular tumours. J. Pathol. 1998, 186, 306–312. [Google Scholar] [CrossRef]
- Gerhardinger, C.; Brown, L.F.; Roy, S.; Mizutani, M.; Zucker, C.L.; Lorenzi, M. Expression of vascular endothelial growth factor in the human retina and in nonproliferative diabetic retinopathy. Am. J. Pathol. 1998, 152, 1453–1462. [Google Scholar] [PubMed]
- Stone, J.; Itin, A.; Alon, T.; Pe’er, J.; Gnessin, H.; Chan-Ling, T.; Keshet, E. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J. Neurosci. 1995, 15, 4738–4747. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef]
- Sanchez-Wandelmer, J.; Ktistakis, N.T.; Reggiori, F. ERES: Sites for autophagosome biogenesis and maturation? J. Cell Sci. 2015, 128, 185–192. [Google Scholar] [CrossRef]
- Bockler, S.; Westermann, B. Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev. Cell. 2014, 28, 450–458. [Google Scholar] [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Engerman, R.L.; Kern, T.S. Retinopathy in animal models of diabetes. Diabetes Metab. Rev. 1995, 11, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Engerman, R.L.; Bloodworth, J.M., Jr. Experimental Diabetic Retinopathy in Dogs. Arch. Ophthalmol. 1965, 73, 205–210. [Google Scholar] [CrossRef]
- Kroemer, G.; El-Deiry, W.S.; Golstein, P.; Peter, M.E.; Vaux, D.; Vandenabeele, P.; Zhivotovsky, B.; Blagosklonny, M.V.; Malorni, W.; Knight, R.A.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005, 12 (Suppl. 2), 1463–1467. [Google Scholar] [CrossRef]
- Anderson, H.R.; Stitt, A.W.; Gardiner, T.A.; Lloyd, S.J.; Archer, D.B. Induction of alloxan/streptozotocin diabetes in dogs: A revised experimental technique. Lab. Anim. 1993, 27, 281–285. [Google Scholar] [CrossRef]
- Ishikawa, T. Fine structure of retinal vessels in man and the macaque monkey. Investig. Ophthalmol. 1963, 2, 1–15. [Google Scholar]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef]
- Tanaka, K.; Matsuda, N. Proteostasis and neurodegeneration: The roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta 2014, 1843, 197–204. [Google Scholar] [CrossRef]
- Nishida, K.; Yamaguchi, O.; Otsu, K. Crosstalk between autophagy and apoptosis in heart disease. Circ. Res. 2008, 103, 343–351. [Google Scholar] [CrossRef]
- Denton, D.; Xu, T.; Kumar, S. Autophagy as a pro-death pathway. Immunol. Cell Biol. 2015, 93, 35–42. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Bialik, S.; Dasari, S.K.; Kimchi, A. Autophagy-dependent cell death—Where, how and why a cell eats itself to death. J. Cell Sci. 2018, 131, jcs215152. [Google Scholar] [CrossRef] [PubMed]
- Tinari, A.; Giammarioli, A.M.; Manganelli, V.; Ciarlo, L.; Malorni, W. Analyzing morphological and ultrastructural features in cell death. Methods Enzymol. 2008, 442, 1–26. [Google Scholar] [PubMed]
- Lane, J.D.; Allan, V.J.; Woodman, P.G. Active relocation of chromatin and endoplasmic reticulum into blebs in late apoptotic cells. J. Cell Sci. 2005, 118 Pt 17, 4059–4071. [Google Scholar] [CrossRef] [PubMed]
- Plomp, P.J.; Gordon, P.B.; Meijer, A.J.; Hoyvik, H.; Seglen, P.O. Energy dependence of different steps in the autophagic-lysosomal pathway. J. Biol. Chem. 1989, 264, 6699–6704. [Google Scholar] [CrossRef]
- Schutter, M.; Giavalisco, P.; Brodesser, S.; Graef, M. Local Fatty Acid Channeling into Phospholipid Synthesis Drives Phagophore Expansion during Autophagy. Cell 2020, 180, 135–149.e14. [Google Scholar] [CrossRef] [PubMed]
- Andrejeva, G.; Gowan, S.; Lin, G.; Wong Te Fong, A.C.L.; Shamsaei, E.; Parkes, H.G.; Mui, J.; Raynaud, F.; Asad, Y.; Vizcay-Barrena, G.; et al. De novo phosphatidylcholine synthesis is required for autophagosome membrane formation and maintenance during autophagy. Autophagy 2020, 16, 1044–1060. [Google Scholar] [CrossRef]
- Mills, J.C.; Stone, N.L.; Erhardt, J.; Pittman, R.N. Apoptotic membrane blebbing is regulated by myosin light chain phosphorylation. J. Cell Biol. 1998, 140, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Inbal, B.; Bialik, S.; Sabanay, I.; Shani, G.; Kimchi, A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 2002, 157, 455–468. [Google Scholar] [CrossRef]
- Levin-Salomon, V.; Bialik, S.; Kimchi, A. DAP-kinase and autophagy. Apoptosis 2014, 19, 346–356. [Google Scholar] [CrossRef]
- Gozuacik, D.; Bialik, S.; Raveh, T.; Mitou, G.; Shohat, G.; Sabanay, H.; Mizushima, N.; Yoshimori, T.; Kimchi, A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008, 15, 1875–1886. [Google Scholar] [CrossRef]
- Wickman, G.R.; Julian, L.; Mardilovich, K.; Schumacher, S.E.; Munro, J.; Rath, N.; Al Zander, S.; Mleczak, A.; Sumpton, D.; Morrice, N.; et al. Blebs produced by actin-myosin contraction during apoptosis release damage-associated molecular pattern proteins before secondary necrosis occurs. Cell Death Differ. 2013, 20, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Lieberthal, W.; Menza, S.A.; Levine, J.S. Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am. J. Physiol. 1998, 274, F315–F327. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Single, B.; Castoldi, A.F.; Kuhnle, S.; Nicotera, P. Intracellular adenosine triphosphate (ATP) concentration: A switch in the decision between apoptosis and necrosis. J. Exp. Med. 1997, 185, 1481–1486. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta 2015, 1852, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Chan, P.S. Oxidative stress and diabetic retinopathy. Exp. Diabetes Res. 2007, 2007, 43603. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Hombrebueno, J.R.; Cairns, L.; Dutton, L.R.; Lyons, T.J.; Brazil, D.P.; Moynagh, P.; Curtis, T.M.; Xu, H. Uncoupled turnover disrupts mitochondrial quality control in diabetic retinopathy. JCI Insightig. 2019, 4, e129760. [Google Scholar] [CrossRef]
- Al-Shabrawey, M.; Rojas, M.; Sanders, T.; Behzadian, A.; El-Remessy, A.; Bartoli, M.; Parpia, A.K.; Liou, G.; Caldwell, R.B. Role of NADPH oxidase in retinal vascular inflammation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3239–3244. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Szabo, C.; Kern, T.S. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes 2004, 53, 2960–2967. [Google Scholar] [CrossRef] [PubMed]
- Obrosova, I.G.; Li, F.; Abatan, O.I.; Forsell, M.A.; Komjati, K.; Pacher, P.; Szabo, C.; Stevens, M.J. Role of poly(ADP-ribose) polymerase activation in diabetic neuropathy. Diabetes 2004, 53, 711–720. [Google Scholar] [CrossRef]
- Burkart, V.; Wang, Z.Q.; Radons, J.; Heller, B.; Herceg, Z.; Stingl, L.; Wagner, E.F.; Kolb, H. Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic beta-cell destruction and diabetes development induced by streptozocin. Nat. Med. 1999, 5, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Gamez, J.A.; Rodriguez-Vargas, J.M.; Quiles-Perez, R.; Aguilar-Quesada, R.; Martin-Oliva, D.; de Murcia, G.; Menissier de Murcia, J.; Almendros, A.; Ruiz de Almodovar, M.; Oliver, F.J. PARP-1 is involved in autophagy induced by DNA damage. Autophagy 2009, 5, 61–74. [Google Scholar] [CrossRef]
- Del Nagro, C.; Xiao, Y.; Rangell, L.; Reichelt, M.; O’Brien, T. Depletion of the central metabolite NAD leads to oncosis-mediated cell death. J. Biol. Chem. 2014, 289, 35182–35192. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef]
- Li, W.; Liu, X.; Yanoff, M.; Cohen, S.; Ye, X. Cultured retinal capillary pericytes die by apoptosis after an abrupt fluctuation from high to low glucose levels: A comparative study with retinal capillary endothelial cells. Diabetologia 1996, 39, 537–547. [Google Scholar] [CrossRef]
- Mandarino, L.J.; Finlayson, J.; Hassell, J.R. High glucose downregulates glucose transport activity in retinal capillary pericytes but not endothelial cells. Investig. Ophthalmol. Vis. Sci. 1994, 35, 964–972. [Google Scholar]
- Mertens, S.; Noll, T.; Spahr, R.; Krutzfeldt, A.; Piper, H.M. Energetic response of coronary endothelial cells to hypoxia. Am. J. Physiol. 1990, 258, H689–H694. [Google Scholar] [CrossRef]
- Winkler, B.S.; Arnold, M.J.; Brassell, M.A.; Puro, D.G. Energy metabolism in human retinal Muller cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3183–3190. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gardiner, T.A.; Stitt, A.W. Pericyte and Vascular Smooth Muscle Death in Diabetic Retinopathy Involves Autophagy. Int. J. Transl. Med. 2022, 2, 26-40. https://doi.org/10.3390/ijtm2010003
Gardiner TA, Stitt AW. Pericyte and Vascular Smooth Muscle Death in Diabetic Retinopathy Involves Autophagy. International Journal of Translational Medicine. 2022; 2(1):26-40. https://doi.org/10.3390/ijtm2010003
Chicago/Turabian StyleGardiner, Tom A., and Alan W. Stitt. 2022. "Pericyte and Vascular Smooth Muscle Death in Diabetic Retinopathy Involves Autophagy" International Journal of Translational Medicine 2, no. 1: 26-40. https://doi.org/10.3390/ijtm2010003
APA StyleGardiner, T. A., & Stitt, A. W. (2022). Pericyte and Vascular Smooth Muscle Death in Diabetic Retinopathy Involves Autophagy. International Journal of Translational Medicine, 2(1), 26-40. https://doi.org/10.3390/ijtm2010003