
Biological Analyses-Derived Translational Findings in the T Cell Receptor Alpha Chain Knockout Mouse as an Experimental Model for Ulcerative Colitis


Abstract
1. Introduction
2. Pathogenic Populations, Factors and Pathways in TCRα KO Mice during the Development of UC-like Colitis
2.1. Pathogenic T Cell Population
2.2. TCR Repertoire Analysis in IBD
2.3. Galectin-4
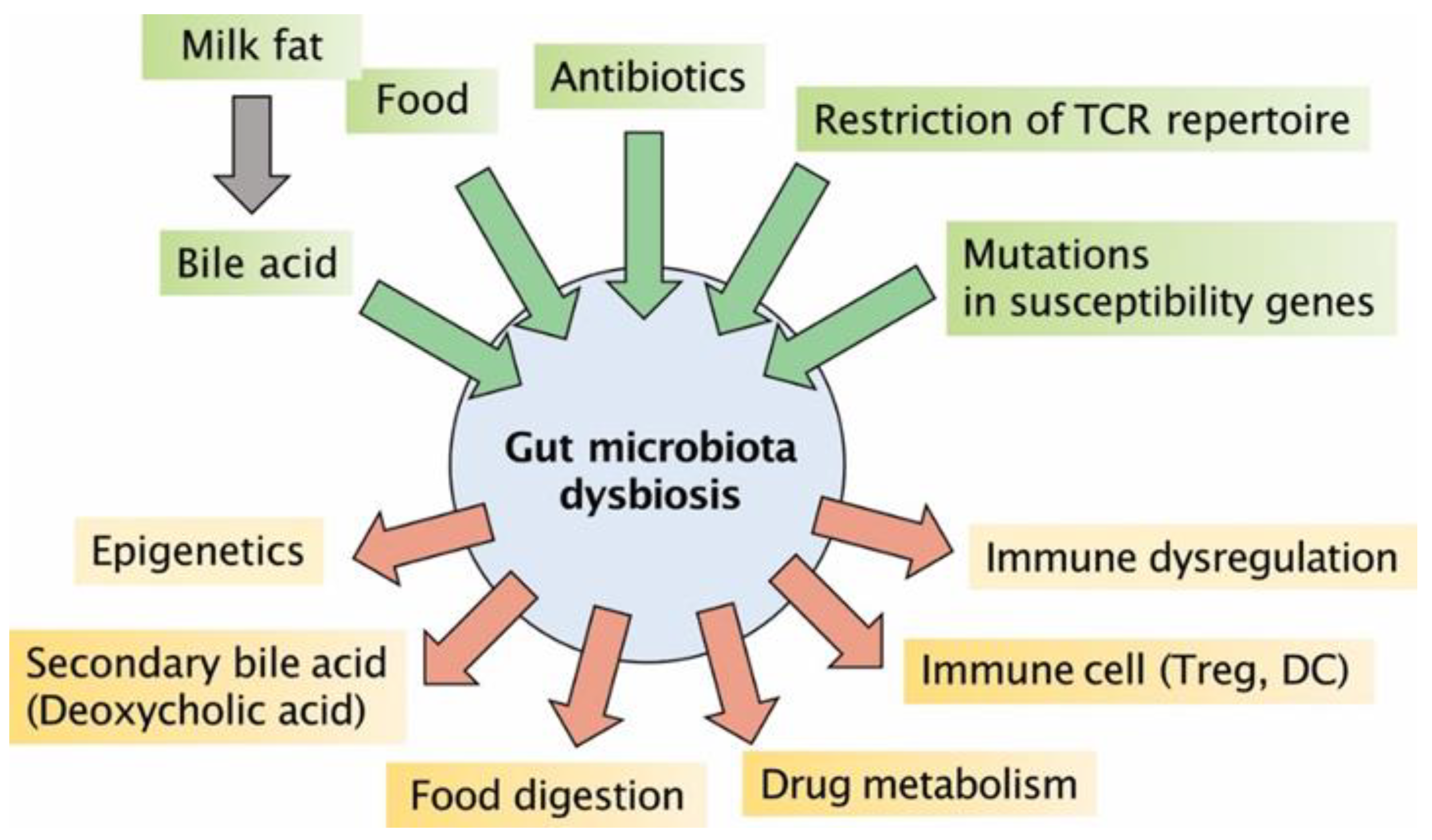
2.4. Gut Dysbiosis
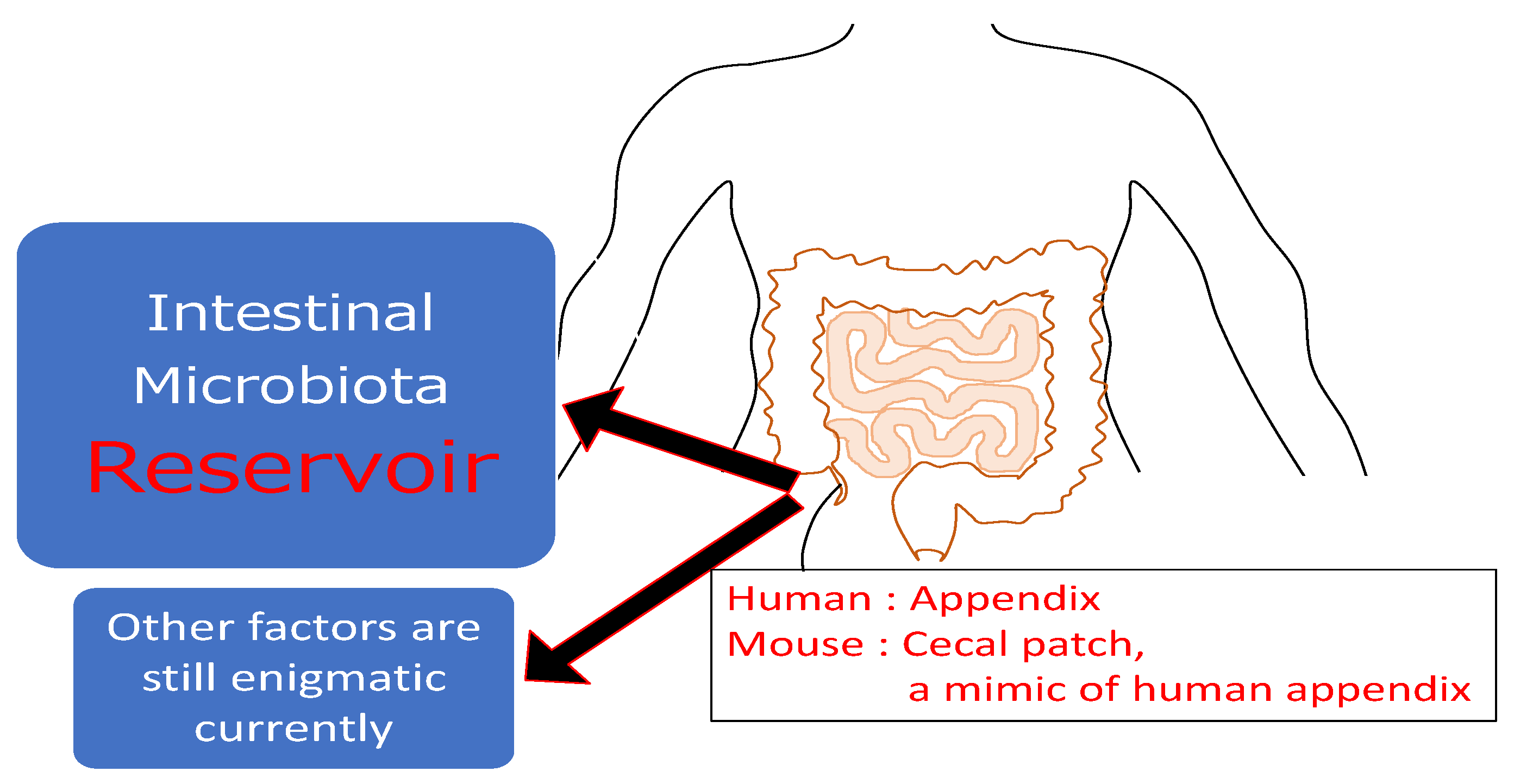
2.5. Lympoif Follicles in Cecal Patches (Appendix)
2.6. Chitinase 3-like 1
2.7. TNFR2 Signaling Pathway
2.8. PKCθ Signaling Pathway
2.9. NK Cells
2.10. Myeloid Dendritic-like Cells
3. Regulatory Populations, Factors, and Pathways in TCRα KO Mice during the Development of UC-like Colitis
3.1. Regulatory B Cells (Bregs)
3.2. IL-22 Signaling Pathway
3.3. Muc 1
3.4. Carbon Monoxide
3.5. Chitin-Microparticles
3.6. Regeneration/Detoxification-Associated Molecules
3.7. Elemental Diet
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sadlack, B.; Merz, H.; Schorle, H.; Schimpl, A.; Feller, A.C.; Horak, I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 1993, 75, 253–261. [Google Scholar] [CrossRef]
- Kuhn, R.; Lohler, J.; Rennick, D.; Rajewsky, K.; Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Mombaerts, P.; Mizoguchi, E.; Grusby, M.J.; Glimcher, L.H.; Bhan, A.K.; Tonegawa, S. Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell 1993, 75, 274–283. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Mizoguchi, E.; Chiba, C.; Spiekermann, G.M.; Tonegawa, S.; Nagler-Anderson, C.; Bhan, A.K. Cytokine imbalance and autoantibody production in T cell receptor-alpha mutant mice with inflammatory bowel disease. J. Exp. Med. 1996, 183, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Dianda, I.; Hanby, A.M.; Wright, N.A.; Sebesteny, A.; Hayday, A.C.; Owen, M.J. T cell receptor-alpha beta-deficient mice fail to develop colitis in the absence of a microbial environment. Am. J. Pathol. 1997, 150, 91–97. [Google Scholar] [PubMed]
- Gaskins, H.R.; Vondrak-Juergens, G.L.; McCracken, B.A.; Woolsey, J.H. Specific-pathogen-free conditions enhance inflammatory bowel disease in T-cell receptor knockout, but not C3H/HeJBir mice. Lab. Anim. Sci. 1997, 47, 650–655. [Google Scholar] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanisms of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar]
- Mizoguchi, A.; Takeuchi, T.; Himuro, H.; Okada, T.; Mizoguchi, E. Genetically engineered mouse models for studying inflammatory bowel disease. J. Pathol. 2016, 238, 205–219. [Google Scholar] [CrossRef]
- Mizoguchi, A. Animal models of inflammatory bowel disease. Prog. Mol. Biol. Trans. Sci. 2012, 105, 263–320. [Google Scholar]
- Mizoguchi, E.; Low, D.; Ezaki, Y.; Okada, T. Recent updates on the basic mechanisms and pathogenesis of inflammatory bowel diseases in experimental animal models. Intes. Res. 2020, 18, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Snapper, S.B.; Rosen, F.S.; Mizoguchi, E.; Cohen, P.; Khan, W.; Liu, C.H.; Hagemann, T.L.; Kwan, S.P.; Ferrini, R.; Davidson, L.; et al. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity 1998, 9, 81–91. [Google Scholar] [CrossRef]
- Beadling, C.; Johnson, K.W.; Smith, K.A. Isolation of interleukin 2-induced immediate-early genes. Proc. Natl. Acad. Sci. USA 1993, 90, 2719–2723. [Google Scholar] [CrossRef]
- Rothenberg, E.V.; Taghon, T. Molecular genetics of T cell development. Annu. Rev. Immunol. 2005, 23, 601–649. [Google Scholar] [CrossRef]
- Takahashi, I.; Iijima, H.; Katashima, R.; Itakura, M.; Kiyono, H. Clonal expansion of CD4+ TCRββ+ T cells in TCR α-chain-deficient mice by gut-derived antigens. J. Immunol. 1999, 162, 1843–1850. [Google Scholar]
- Barber, D.F.; Passoni, L.; Wen, L.; Geng, L.; Hayday, A.C. The expression in vivo of a second isoform of pT alpha: Implications for the mechanism of pT alpha action. J. Immunol. 1998, 161, 11–16. [Google Scholar] [PubMed]
- Morgan, N.V.; Goddard, S.; Cardno, T.S.; McDonald, D.; Rahman, F.; Barge, D.; Ciupek, A.; Straatman-Iwanowska, A.; Pasha, S.; Guckian, M.; et al. Mutation in the TCRα subunit constant gene (TRAC) leads to a human immunodeficiency disorder characterized by a lack of TCRαβ+ T cells. J. Clin. Investig. 2011, 121, 695–702. [Google Scholar] [CrossRef]
- Matsumura, K.; Nakase, H.; Kosugi, I.; Honzawa, Y.; Yoshino, T.; Matsuura, M.; Kawasaki, H.; Arai, Y.; Iwashita, T.; Nagasawa, T.; et al. Establishment of a novel mouse model of ulcerative colitis with concominanty cytomegalovirus infection: In vivo identification of cytomegalovirus persistent infected cells. Inflamm. Bowel Dis. 2013, 19, 1951–1963. [Google Scholar]
- Mizoguchi, A.; Mizoguchi, E.; Chiba, C.; Bhan, A.K. Role of appendix in the development of inflammatory bowel disease in TCR-α mutant mice. J. Exp. Med. 1996, 184, 707–715. [Google Scholar] [CrossRef]
- Nishio, J.; Baba, M.; Atarashi, K.; Tanoue, T.; Negishi, H.; Yanai, H.; Habu, S.; Hori, S.; Honda, K.; Taniguchi, T. Requirement of full TCR repertoire for regulatory T cells to maintain intestinal homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 12770–12775. [Google Scholar] [CrossRef]
- Curciarello, R.; Canziani, K.E.; Docena, G.H.; Muglia, C.I. Contribution of non-immune cells to activation and modulation of the intestinal inflammation. Front. Immunol. 2019, 10, 647. [Google Scholar] [CrossRef] [PubMed]
- Probert, C.S.J.; Saubermann, L.J.; Balk, S.; Blumberg, R.S. Repertoire of the αβ T-cell receptor in the intestine. Immunol. Rev. 2007, 215, 215–225. [Google Scholar] [CrossRef]
- Holtmeier, W.; Hennemann, A.; May, E.; Duchmann, R.; Caspary, W.F. T cell receptor δ repertoire in inflamed and noninflamed colon of patients with IBD analyzed by CDR3 spectratyping. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Werner, L.; Nunberg, M.Y.; Rechavi, E.; Lev, A.; Braun, T.; Haberman, Y.; Lahad, A.; Shteyer, E.; Schvimer, M.; Somech, R.; et al. Altered T cell receptor beta repertoire patterns in pediatric ulcerative colitis. Clin. Exp. Immunol. 2019, 196, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zvyagin, I.V.; Pogorelyy, M.V.; Ivanova, M.E.; Komech, E.A.; Shugay, M.; Bolotin, D.A.; Shelenkov, A.A.; Kurnosov, A.A.; Staroverov, D.B.; Chudakov, D.M.; et al. Distinctive properties of identical twins’ TCR repertoires revealed by high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 5980–5985. [Google Scholar] [CrossRef]
- Rosati, E.; Pogorelyy, M.V.; Marie Dowds, C.; Moller, F.T.; Sorensen, S.B.; Lebedev, Y.B.; Frey, N.; Schreiber, S.; Spehlmann, M.E.; Andersen, V.; et al. Identification of disease-associated traits and clonotypes in the T Cell receptor repertoire of monozygotic twins affected by inflammatory bowel diseases. J. Crohn Colitis 2020, 14, 778–790. [Google Scholar] [CrossRef]
- Kakuta, Y.; Nakano, T.; Naito, T.; Watanabe, K.; Izumiyama, Y.; Okamoto, D.; Ichikawa, R.; Moroi, R.; Kuroha, M.; Kanazawa, Y.; et al. Repertoire analysis of memory T-cell receptors in Japanese patients with inflammatory bowel disease. JGH Open 2020, 4, 624–631. [Google Scholar] [CrossRef]
- Günaltay, S.; Repsilber, D.; Helenius, G.; Nyhlin, N.; Bohr, J.; Hultgren, O.; Hultgren Hörnquist, E. Oligoclonal T-cell receptor 646 repertoire in colonic biopsies of patients with microscopic colitis and ulcerative colitis. Inflamm. Bowel Dis. 2017, 23, 932–945. [Google Scholar] [CrossRef]
- Hegazy, A.N.; West, N.R.; Stubbington, M.J.T.; Wendt, E.; Suijker, K.I.M.; Datsi, A.; This, S.; Danne, C.; Campion, S.; Duncan, S.H.; et al. Circulating and tissue-resident CD4+ T cells with reactivity to intestinal microbiota are abundant in healthy individuals and function is altered during inflammation. Gastroenterology 2017, 153, 1320–1337. [Google Scholar] [CrossRef]
- Zeissig, S.; Rosati, E.; Dowds, C.M.; Aden, K.; Bethge, J.; Schulte, B.; Pan, W.H.; Mishra, N.; Zuhayra, M.; Marx, M.; et al. Vedolizumab is associated with changes in innate rather than adaptive immunity in patients with inflammatory bowel disease. Gut 2019, 68, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Gamliel, A.; Werner, L.; Pinsker, M.; Salamon, N.; Weiss, B.; Shouval, D.S. Circulating α4β7+ memory T cells in pediatric ibd patients express a polyclonal T cell receptor repertoire. Clin. Exp. Gastroenterol. 2020, 13, 439–447. [Google Scholar] [CrossRef]
- Ideo, H.; Seko, A.; Ohkura, T.; Matta, K.L.; Yamashita, K. High-affinity binding of recombinant human galectin-4 to SO 3 → 3Gal β 1 → 3GalNAc pyranoside. Glycobiology 2002, 12, 199–208. [Google Scholar] [CrossRef]
- Hokama, A.; Mizoguchi, E.; Sugimoto, K.; Shimomura, Y.; Tanaka, Y.; Yoshida, M.; Rietdijk, S.T.; De Jong, Y.P.; Snapper, S.B.; Terhorst, C.; et al. Induced reactivity of intestinal CD4+ T cells with an epithelial cell lectin, galectin-4, contributes to exacerbation of intestinal inflammation. Immunity 2004, 20, 681–693. [Google Scholar] [CrossRef]
- Rechreche, H.; Mallo, G.V.; Montalto, G.; Dagorn, J.C.; Iovanna, J.L. Cloning and expression of the mRNA of human galectin-4, an S-type lectin down-regulated in colorectal cancer. Eur. J. Biochem. 1997, 248, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Gitt, M.A.; Colnot, C.; Poirier, F.; Nani, K.J.; Barondes, S.H.; Leffler, H. Galectin-4 and galectin-6 are two closely related lectins expressed in mouse gastrointestinal tract. J. Biol. Chem. 1998, 273, 2954–2960. [Google Scholar] [CrossRef]
- Hokama, A.; Mizoguchi, E.; Mizoguchi, A. Roles of galectins in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 5133–5137. [Google Scholar] [CrossRef]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Motta, J.P.; Flannigan, K.L.; Agbor, T.A.; Beatty, J.K.; Blackler, R.W.; Workentine, M.L.; Da Silva, G.J.; Wang, R.; Buret, A.G.; Wallace, J.L. Hydrogen sulfide protects from colitis and restores intestinal microbiota biofilm and mucus production. Inflamm. Bowel Dis. 2015, 21, 1006–1017. [Google Scholar] [CrossRef]
- Buret, A.; Ward, K.H.; Olson, M.E.; Costerton, J.W. An in vivo model to study the pathobiology of infectious biofilms on biomaterial surfaces. J. Biomed. Mater. Res. 1991, 25, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Palestrant, D.; Holzknecht, Z.E.; Collins, B.H.; Parker, W.; Miller, S.E.; Bollinger, R.R. Microbial biofilms in the gut: Visualization by electron microscopy and by acridine orange staining. Ultrastruct. Pathol. 2004, 28, 23–27. [Google Scholar] [CrossRef]
- Randal Bollinger, R.; Everett, M.L.; Palestrant, D.; Love, S.D.; Lin, S.S.; Parker, W. Human secretory immunoglobulin A may contribute to biofilm formation in the gut. Immunology 2003, 109, 580–587. [Google Scholar] [CrossRef]
- Banwell, J.G.; Howard, R.; Cooper, D.; Costerton, J.W. Intestinal microbial flora after feeding phytohemagglutinin lectins (Phaseolus vulgaris) to rats. Appl. Environ. Microbiol. 1985, 50, 68–80. [Google Scholar] [CrossRef]
- Yasuda, K.; Oh, K.; Ren, B.; Tickle, T.L.; Franzosa, E.A.; Wachtman, L.M.; Miller, A.D.; Westmoreland, S.V.; Mansfield, K.G.; Vallender, E.J.; et al. Biogeography of the intestinal mucosal and lumenal microbiome in the rhesus macaque. Cell Host Microbe 2015, 17, 385–391. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; Von Wright, A.; Vilpponen-Salmela, T.; Ben-Amor, K.; Akkermans, A.D.L.; De Vos, W.M. Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl. Environ. Microbiol. 2002, 68, 3401–3407. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Antonopoulos, D.A.; Zhu, X.; Harrell, L.; Hanan, I.; Alverdy, J.C.; Meyer, F.; Musch, M.W.; Young, V.B.; Chang, E.B. Laser capture microdissection and metagenomic analysis of intact mucosa-associated microbial communities of human colon. Appl. Microbiol. Biotechnol. 2010, 88, 1333–1342. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nava, G.M.; Friedrichsen, H.J.; Stappenbeck, T.S. Spatial organization of intestinal microbiota in the mouse ascending colon. ISME J. 2011, 5, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Swidsinski, A.; Loening-Baucke, V.; Lochs, H.; Hale, L.P. Spatial organization of bacterial flora in normal and inflamed intestine: A fluorescence in situ hybridization study in mice. World J. Gastroenterol. 2005, 11, 1131–1140. [Google Scholar] [CrossRef]
- Glasser, A.L.; Boudeau, J.; Barnich, N.; Perruchot, M.H.; Colombel, J.F.; Darfeuille-Michaud, A. Adherent invasive Escherichia coli strains from patients with Crohn′s disease survive and replicate within macrophages without inducing host cell death. Infect. Immun. 2001, 69, 5529–5537. [Google Scholar] [CrossRef]
- Wang, M.; Molin, G.; Ahrné, S.; Adawi, D.; Jeppsson, B. High proportions of proinflammatory bacteria on the colonic mucosa in a young patient with ulcerative colitis as revealed by cloning and sequencing of 16S rRNA genes. Dig. Dis. Sci. 2007, 52, 620–627. [Google Scholar] [CrossRef]
- Palmela, C.; Chevarin, C.; Xu, Z.; Torres, J.; Sevrin, G.; Hirten, R.; Barnich, N.; Ng, S.C.; Colombel, J.F. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 2018, 67, 574–587. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef]
- Swidsinski, A.; Loening-Baucke, V.; Vaneechoutte, M.; Doerffel, Y. Active Crohn’s disease and ulcerative colitis can be specifically diagnosed and monitored based on the biostructure of the fecal flora. Inflamm. Bowel Dis. 2008, 14, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Quévrain, E.; Maubert, M.A.; Michon, C.; Chain, F.; Marquant, R.; Tailhades, J.; Miquel, S.; Carlier, L.; Bermúdez-Humarán, L.G.; Pigneur, B.; et al. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut 2016, 65, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, Y.; Mizoguchi, E.; Sugimoto, K.; Kibe, R.; Benno, Y.; Mizoguchi, A.; Bhan, A.K. Regulatory role of B-1 B cells in chronic colitis. Int. Immunol. 2008, 20, 729–737. [Google Scholar] [CrossRef]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef]
- Wu, H.J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef]
- Romano, K.A.; Rey, F.E. Is maternal microbial metabolism an early-life determinant of health? Lab. Anim. 2018, 47, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Bjarnason, I.; Hayllar, J.; MacPherson, A.J.; Russell, A.S. Side effects of nonsteroidal anti-inflammatory drugs on the small and large intestine in humans. Gastroenterology 1993, 104, 1832–1847. [Google Scholar] [CrossRef]
- Kurahara, K.; Matsumoto, T.; Iida, M.; Honda, K.; Yao, T.; Fujishima, M. Clinical and endoscopic features of nonsteroidal anti-inflammatory drug-induced colonic ulcerations. Am. J. Gastroenterol. 2001, 96, 473–480. [Google Scholar] [CrossRef]
- Hale, L.P.; Gottfried, M.R.; Swidsinski, A. Piroxicam treatment of IL-10-deficient mice enhances colonic epithelial apoptosis and mucosal exposure to intestinal bacteria. Inflamm. Bowel Dis. 2005, 11, 1060–1069. [Google Scholar] [CrossRef]
- Nishiyori, A.; Nagakura, Y.; Ichikawa, K. Piroxicam accelerates development of colitis in T-cell receptor α chain-deficient mice. Eur. J. Pharmacol. 2009, 615, 241–245. [Google Scholar] [CrossRef]
- Feuerstein, J.D.; Isaacs, K.L.; Schneider, Y.; Siddique, S.M.; Falck-Ytter, Y.; Singh, S.; AGA Institute Clinical Guidelines Committee. AGA clinical practice guidelines on the management of moderate to severe ulcerative colitis. Gastroenterology. 2020, 158, 1450–1461. [Google Scholar] [CrossRef]
- Gajendran, M.; Loganathan, P.; Jimenez, G.; Catinella, A.P.; Ng, N.; Umapathy, C.; Ziade, N.; Hashah, J.G. A comprehensive review and update on ulcerative colitis. Dis. Mon. 2019, 65, 100851. [Google Scholar] [CrossRef] [PubMed]
- Andersson, P.; Söderholm, J.D. Surgery in ulcerative colitis: Indication and timing. Dig Dis. 2009, 27, 335–340. [Google Scholar] [CrossRef]
- Kobayashi, T.; Siegmund, B.; Le Berre, C.; Wei, S.C.; Ferrante, M.; Shen, B.; Bernstein, C.N.; Danese, S.; Peyrin-Biroulet, L.; Hibi, T. Ulcerative colitis. Nat. Rev. Dis. Primers 2020, 10, 74. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Mizoguchi, A.; Bhan, A.K. Insights from recent advances in animal models of inflammatory bowel disease. In Molecular Genetics of Inflammatory Bowel Disease; D’Amato, M., Rioux, J.D., Eds.; Springer: New York, NY, USA, 2013; pp. 45–83. [Google Scholar]
- Myrelid, P.; Landerholm, K.; Nordenvall, C.; Pinkney, T.D.; Andersson, R.E. Appendectomy and the risk of coloctomy in ulcerative colitis: A national cohort study. Am. J. Gastroenterol. 2017, 112, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Stellingwerf, M.E.; Sahami, S.; Winter, D.C.; Martin, S.T.; D′Haens, G.R.; Cullen, G.; Doherty, G.A.; Mulcahy, H.; Bemelman, W.A.; Buskens, C.J. Prospective cohort study of appendicectomy for treatment of therapy-refractory ulcerative colitis. Br. J. Surg. 2019, 106, 1697–1704. [Google Scholar] [CrossRef]
- Girard-Madoux, M.J.H.; Gomez de Agüero, M.; Ganal-Vonarburg, S.C.; Mooser, C.; Belz, G.T.; Macpherson, A.J.; Vivier, E. The immunological functions of the appendix: An example of redundancy? Semin. Immunol. 2018, 36, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Sahami, S.; Kooji, I.A.; Meijer, S.L.; Van den Brink, G.R.; Buskens, C.J.; Te Velde, A.A. The link between the appendix and ulcerative colitis: Clinical relevance and potential immunological mechanisms. Am. J. Gastroenterol. 2016, 111, 163–169. [Google Scholar] [CrossRef]
- Vitetta, L.; Chen, J.; Clarke, S. The vermiform appendix: An immunological organ sustaining a microbiome inoculum. Clin. Sci. 2019, 133, 1–8. [Google Scholar] [CrossRef]
- Masahata, K.; Umemoto, E.; Kayama, H.; Kotani, M.; Nakamura, S.; Kurakawa, T.; Kikuta, J.; Gotoh, K.; Motooka, D.; Sato, S.; et al. Generation of colonic IgA-secreting cells in the caecal patch. Nat. Commun. 2014, 5, 3704. [Google Scholar] [CrossRef]
- Palm, N.W.; De Zoete, M.R.; Cullen, T.W.; Barry, N.A.; Stefanowski, J.; Hao, L.; Degnan, P.H.; Hu, J.; Peter, I.; Zhang, W.; et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 2014, 158, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Bürgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Olszak, T.; An, D.; Zeissig, S.; Vera, M.P.; Richter, J.; Franke, A.; Glickman, J.N.; Siebert, R.; Baron, R.M.; Kasper, D.L.; et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 2012, 336, 489–493. [Google Scholar] [CrossRef]
- Mizoguchi, E. Chitinase 3-like 1 exacerbates intestinal inflammation by enhancing bacterial adhesion and invasion in colonic epithelial cells. Gastroenterology 2006, 130, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Low, D.; Tran, H.T.; Lee, I.A.; Dreux, N.; Reinecker, H.C.; Darfeuille-Michaud, A.; Barnich, N.; Mizoguchi, E. Chitin-bindingdomains of Escherichia coli chiA mediate interactions with intestinal epithelial cells in mice with colitis. Gastroenterology 2013, 145, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Low, D.; Subramaniam, R.; Lin, L.; Aomatsu, T.; Mizoguchi, A.; Ng, A.; DeGruttola, A.K.; Lee, C.G.; Elias, J.A.; Andoh, A.; et al. Chitinase 3-like 1 induces survival and proliferation of intestinal epithelial cells during chronic inflammation and colitis-associated cancer by regulating S100A9. Oncotarget 2015, 6, 36535–36550. [Google Scholar] [CrossRef]
- Chen, C.C.; Pekow, J.; Llado, V.; Kanneganti, M.; Lau, C.W.; Mizoguchi, A.; Mino-Kenudson, M.; Bissonnette, M.; Mizoguchi, E. Chitinase 3-like 1 (CHI3L1/YKL-40) expression in colonic epithelial cells as a potentially novel marker for colitis-associated neoplasia. Am. J. Pathol. 2011, 179, 1494–1503. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Subramaniam, R.; Okada, T.; Mizoguchi, A. A review of selected IBD biomarkers: From animal models to bedside. Diagnostics 2021, 11, 207. [Google Scholar] [CrossRef]
- Aomatsu, T.; Imaeda, H.; Matsumoto, K.; Kimura, E.; Yoden, A.; Tamai, H.; Fijiyama, Y.; Mizoguchi, E.; Andoh, A. Fecal chitinase 3-like 1 is a novel biological marker for disease activity in pediatric inflammatory bowel disease patients. Aliment. Pharmacol. Ther. 2011, 34, 941–948. [Google Scholar] [CrossRef]
- Low, D.; Poltrak, A.; DeGruttola, A.K.; Mizoguchi, A.; Mino-Kenudson, M.; Mizoguchi, E. High endogenous expression of Chitinase 3-like 1 and excessive epithelial proliferation with colonic tumor formation in MOLF/EiJ mice. PLoS ONE 2015, 10, e0139149. [Google Scholar] [CrossRef] [PubMed]
- Ober, C.; Tan, Z.; Sun, Y.; Possick, J.D.; Pan, L.; Nicolae, R.N.; Radford, S.; Parry, R.R.; Heinzmann, A.; Deichmann, K.A.; et al. Effect of variation in CHI3L1 on serum YKL-40 level, risk of asthma, and lung function. N. Engl. J. Med. 2008, 358, 1682–1691. [Google Scholar] [CrossRef]
- Kasaian, M.T.; Lee, J.; Brennan, A.; Danto, S.I.; Black, K.E.; Fitz, L.; Dixon, A.E. Proteomics analysis of serum and sputum analytes distinguishes controlled and poorly controlled asthmatics. Clin. Exp. Allergy 2018, 48, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Aardoom, M.A.; Veereman, G.; de Ridder, L. A review on the use of anti-TNF in children and adolescents with inflammatory bowel disease. Int. J. Mol. Sci. 2019, 20, 2529. [Google Scholar] [CrossRef] [PubMed]
- Wallach, D.; Varfotomeev, E.E.; Malinin, N.L.; Goltsev, Y.V.; Kovalenko, A.V.; Boldin, M.P. Tumor necrosis factor receptor and Fas signaling mechanisms. Ann. Rev. Immunol. 1999, 17, 331–367. [Google Scholar] [CrossRef] [PubMed]
- Schall, T.J.; Lewis, M.; Koller, K.J.; Lee, A.; Rice, G.C.; Wong, G.H.W.; Gatanaga, T.; Granger, G.A.; Lentz, R.; Raab, H.; et al. Molecular cloning and expression of a receptor for human tumor necrosis factor. Cell 1990, 61, 361–370. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Mizoguchi, A.; Takedatsu, H.; Caro, E.; de Jong, Y.P.; Ooi, C.J.; Xavier, R.J.; Terhorst, C.; Podolsky, D.K.; Bhan, A.K. Role of tumor necrosis factor receptor 2 (TNFR2) in colonic epithelial hyperplasia and chronic intestinal inflammation in mice. Gastroenterology 2002, 122, 134–144. [Google Scholar] [CrossRef]
- Li, X.; Lee, E.J.; Gawel, D.R.; Lilja, S.; Schafer, S.; Zhang, H.; Benson, M. Meta-analysis of expression profiling data indicates need for combinational biomarkers in pediatric ulcerative colitis. J. Immunol. Res. 2020, 2020, 8279619. [Google Scholar] [CrossRef]
- Spoettl, T.; Hausmann, M.; Klebl, F.; Dirmeier, A.; Klump, B.; Hoffmann, J.; Herfarth, H.; Timmer, A.; Rogler, G. Serum soluble TNF receptor I and II levels correlate with diseaseactivity in IBD patients. Inflamm. Bowel Dis. 2007, 13, 727–732. [Google Scholar] [CrossRef]
- Altman, A.; Villalba, M. Protein kinase C-θ (PKCθ): It′s all about location, location, location. Immunol. Rev. 2003, 192, 53–63. [Google Scholar] [CrossRef]
- Pfeifhofer, C.; Kofler, K.; Gruber, T.; Ghaffari Tabrizi, N.; Lutz, C.; Maly, K.; Leitges, M.; Baier, G. Protein kinase Cθ affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J. Exp. Med. 2003, 197, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, K.; Ogawa, A.; Shirane, K.; Shimomura, Y.; Sugimoto, K.; Mizoguchi, A. Protein kinase θ plays a fundamental role in different types of chronic colitis. Gastroenterology 2008, 134, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Zaiats Bittencourt, V.; Jones, F.; Tosetto, M.; Doherty, G.A.; Ryan, E.J. Dysregulation of metabolic pathways in circulating natural killer cells isolated from inflammatory bowel disease patients. J. Cron Colitis 2021, in press. [Google Scholar] [CrossRef]
- Poggi, A.; Benelli, R.; Vene, R.; Costa, D.; Ferrari, N.; Tosetti, F.; Zocchi, M.R. Human gut-associated natural killer cells in health and disease. Front. Immunol. 2019, 10, 961. [Google Scholar] [CrossRef]
- Egawa, S.; Hiwatashi, N. Natural killer cell activity in patients with inflammatory bowel disease. J. Clin. Lab. Immunol. 1986, 20, 187–192. [Google Scholar] [PubMed]
- Wu, Y.; Yao, J.; Xie, J.; Liu, Z.; Zhou, Y.; Pan, H.; Han, W. The role of autophagy in colitis-associated colorectal cancer. Signal Transduct. Target. Ther. 2018, 3, 31. [Google Scholar] [CrossRef]
- Kamada, N.; Hisamatsu, T.; Okamoto, S.; Chinen, H.; Kobayashi, T.; Sato, T.; Sakuraba, A.; Kitazume, M.T.; Sugita, A.; Koganei, K.; et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of crohn disease via IL-23/IFN-gamma axis. J. Clin. Investig. 2008, 118, 2269–2280. [Google Scholar]
- Mizoguchi, A.; Ogawa, A.; Takedatsu, H.; Sugimoto, K.; Shimomura, Y.; Shirane, K.; Nagahama, K.; Nagaishi, T.; Mizoguchi, E.; Blumberg, R.S.; et al. Dependence of intestinal granuloma formation on unique myeloid DC-like cells. J. Clin. Investig. 2007, 117, 605–615. [Google Scholar] [CrossRef]
- Barman, S.; Kayama, H.; Okuzaki, D.; Ogino, T.; Osawa, H.; Matsuno, H.; Mizushima, T.; Mori, M.; Nishimura, J.; Takeda, K. Identification of a human intestinal myeloid cell subset that regulates gut homeostasis. Int. Immunol. 2016, 28, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Honjo, T. Transgenic mouse models for B cell dominant autoimmune disease. Curr. Opin. Immunol. 1997, 9, 846–850. [Google Scholar] [CrossRef]
- Martin, F.; Chan, A.C. Pathogenic roles of B cells in human autoimmunity: Insights from the clinic. Immunity 2004, 20, 517–527. [Google Scholar] [CrossRef]
- Wolf, S.D.; Dittel, B.N.; Hardardottir, F.; Janeway, C.A., Jr. Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J. Exp. Med. 1996, 184, 2271–2278. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Mizoguchi, E.; Smith, R.N.; Preffer, F.I.; Bhan, A.K. Suppressive role of B cells in chronic colitis of T cell receptor α mutant mice. J. Exp. Med. 1997, 186, 1749–1756. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Bhan, A.K. A case for regulatory B cells. J. Immunol. 2006, 176, 705–710. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Mizoguchi, A.; Preffer, F.I.; Bhan, A.K. Regulatory role of mature B cells in a murine model of inflammatory bowel disease. Int. Immunol. 2000, 12, 597–605. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Mizoguchi, E.; Takedatsu, H.; Blumberg, R.S.; Bhan, A.K. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d-upregulation. Immunity 2002, 16, 219–230. [Google Scholar] [CrossRef]
- Mohd Jaya, F.N.; Garcia, S.G.; Borras, F.E.; Chan, G.C.F.; Franquesa, M. Paradoxical role of Breg-inducing cytokines in autoimmune diseases. J. Trans. Autoimm. 2019, 2, 100011. [Google Scholar] [CrossRef] [PubMed]
- Mauri, C.; Menon, M. Human regulatory B cells in health and disease; therapeutic potential. J. Clin. Investig. 2017, 127, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.; Blair, P.A.; Isenberg, D.A.; Mauri, C. A regulatory feedback between plasmacytoid dendritic cells and regulatory B cells is aberrant in systemic lupus erythematosus. Immunity 2016, 44, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Brand, D.; Zheng, S.G. Targeting IL-2: An unexpected effect in treating immunological diseases. Signal Transduct. Target. Ther. 2018, 3, 2. [Google Scholar] [CrossRef]
- Goetz, M.; Atreya, R.; Ghalibafian, M.; Galle, P.R.; Neurath, M.F. Exacerbation of ulcerative colitis after rituximab salvage therapy. Inflamm. Bowel Dis. 2007, 13, 1365–1368. [Google Scholar] [CrossRef]
- Zouali, M. B lymphocytes, the gastrointestinal tract and autoimmunity. Autoimm. Rev. 2021, 20, 102777. [Google Scholar] [CrossRef] [PubMed]
- Fagarasan, S.; Muramatsu, M.; Suzuki, R.; Nagaoka, H.; Hirai, H.; Honjo, T. Critical roles of activation induced cytidine deaminase in the homeostasis of gut flora. Science 2002, 298, 1424–1427. [Google Scholar] [CrossRef]
- Kim, M.; Qie, Y.; Park, J.; Kim, C.H. Gut microbial metabolites fuel host antibody responses. Cell Host Microbe 2016, 20, 202–214. [Google Scholar] [CrossRef]
- Wang, X.; Wong, K.; Ouyang, W.; Rutz, S. Targeting IL-10 family cytokines for the treatment of human diseases. Cold Spring Harb. Perspect. Biol. 2019, 11, a028548. [Google Scholar] [CrossRef]
- Ouyang, W.; O′Garra, A. IL-10 family cytokines IL-10 and IL-22: From basic science to clinical translation. Immunity 2019, 50, 871–879. [Google Scholar] [CrossRef]
- Dumoutier, L.; Lejeune, D.; Colau, D.; Renauld, J.C. Cloning and characterization of IL-22 binding protein, a natural antagonist of IL-10-related T cell-derived inducible factor/IL-22. J. Immunol. 2001, 166, 7090–7095. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Hansson, G.C. Immunological aspects of intestinal mucus and mucins. Nat. Rev. Immunol. 2016, 16, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mousemedel of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar]
- Lindemans, C.A.; Calafiore, M.; Mertelsmann, A.M.; O′Connor, M.H.; Dudakov, J.A.; Jenq, R.R.; Velardi, E.; Young, L.F.; Smith, O.M.; Lawrence, G.; et al. Interleukin-22 proteins intestinal-stem-cell-mediated epithelial regeneration. Nature 2015, 528, 560–564. [Google Scholar] [CrossRef]
- Andoh, A.; Zhang, Z.; Inatomi, O.; Fujino, S.; Deguchi, Y.; Araki, Y.; Tsujikawa, T.; Kitoh, K.; Kim–Mitsuyama, S.; Takayanagi, A.; et al. Interleukin-22, a member of the Il-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology 2005, 129, 969–984. [Google Scholar] [CrossRef]
- Huber, S.; Gagliani, N.; Zenewicz, L.A.; Huber, F.J.; Bosurgi, L.; Hu, B.; Hedl, M.; Zhang, W.; O′Connor, W.; Murphy, A.J.; et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012, 491, 259–263. [Google Scholar] [CrossRef]
- Pelczar, P.; Witkowski, M.; Perez, L.G.; Kempski, J.; Hammel, A.G.; Brockmann, L.; Kleinschmidt, D.; Wende, S.; Haueis, C.; Bedke, T.; et al. A pathogenic role for T cell-derived IL-22BP in inflammatory bowel disease. Science 2016, 354, 358–362. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Yano, A.; Himuro, H.; Ezaki, Y.; Sadanaga, T.; Mizoguchi, E. Clinical importance of IL-22 cascade in IBD. J. Gastroenterol. 2018, 53, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, G.F.; Fouser, L.A.; Artis, D. Border patrol: Regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 2011, 12, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Yi, Y.; Lu, T.; Ghilardi, N. The role of IL-22 in intestinal health and disease. J. Exp. Med. 2020, 217, e20192195. [Google Scholar] [CrossRef]
- Vancamelbeke, M.; Vanuytsel, T.; Farré, R.; Verstockt, S.; Ferrante, M.; Assche, G.V.; Rutgeerts, P.; Schuit, F.; Vermeire, S.; Arijs, I.; et al. Genetic and transcriptomic bases of intestinal epithelial barrier dysfunction in inflammatory bowel disease. Inflamm. Bowel Dis. 2017, 23, 1718–1729. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.; Lau, C.W.; Zhang, M.; Andoh, A.; Shi, H.N.; Mizoguchi, E.; Mizoguchi, A. The membrane-bound mucin Muc1 regulates T helper 17-cell responses and colitis in mice. Gastroenterology 2012, 142, 865–874. [Google Scholar] [CrossRef]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010, 464, 1371–1375. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Sheikh, S.Z.; Hegazi, R.A.; Kobayashi, T.; Onyiah, J.C.; Russo, S.M.; Matsuoka, K.; Sepulveda, A.R.; Li, F.; Otterbein, L.E.; Plevy, S.E. An anti-inflammatory role for carbon monoxide and heme oxygenase-1 in chronic Th2-mediated murine colitis. J. Immunol. 2011, 186, 5506–5513. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Debono, M.; Gordee, R.S. Antibiotics that inhibit fungal cell wall development. Ann. Rev. Microbiol. 1994, 48, 471–497. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.A.; Hartl, D.; Lu, W.; Lee, C.G.; Elias, J.A. TLR-2 and IL-17A in chitin-induced macrophage activation and acute inflammation. J. Immunol. 2008, 181, 4279–4286. [Google Scholar] [CrossRef]
- Lee, C.G.; Da Silva, C.A.; Lee, J.Y.; Hartl, D.; Elias, J.A. Chitin regulation of immune responses: An old molecule with new roles. Curr. Opin. Immunol. 2008, 20, 684–689. [Google Scholar] [CrossRef]
- Nagatani, K.; Wang, S.; Llado, V.; Lau, C.W.; Li, Z.; Mizoguchi, A.; Nagler, C.R.; Shibata, Y.; Reinecker, H.C.; Mora, R.J.; et al. Chitin microparticles for the control of intestinal inflammation. Inflamm. Bowel Dis. 2012, 18, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Mercer, B.; Cirone, A.M.; Brust, C.; Lee, Z.J.; Esiobu, N.; Li, Z.; Wei, J.; Dorey, C.K.; Shibata, Y.; et al. Dietary chitin particles called mimetic fungi ameliorate colitis in toll-like receptor 2/CD14-and sex-dependent manner. Infect. Immun. 2019, 87, e00006-19. [Google Scholar] [CrossRef] [PubMed]
- Sigsgaard, T.; Thorne, P.S.; Schlunssen, V.; Bonlokke, J.; Riddervold, I.S.; Hoppe, K.A.; Andersen, N.T.; Mackenzie, N.M. The change in nasal inflammatory markers after intranasal challenges with particulate chitin and lipopolysaccharide: A randomized, double-blind, placebo-controlled, crossover study with a positive control. Int. Forum Allergy Rhinol. 2015, 5, 16–23. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Xavier, R.J.; Reinecker, H.C.; Uchino, H.; Bhan, A.K.; Podolsky, D.K.; Mizoguchi, A. Colonic epithelial function phenotype varies with type and phase of experimental colitis. Gastroenterology 2003, 125, 148–161. [Google Scholar] [CrossRef]
- Xu, X.; Fukui, H.; Ran, Y.; Wang, X.; Inoue, Y.; Ebisudani, N.; Nishimura, H.; Tomita, T.; Oshima, T.; Watari, J.; et al. The link between type III Reg and STAT3-associated cytokines in inflamed colonic tissues. Mediat. Inflamm. 2019, 2019, 7859460. [Google Scholar] [CrossRef]
- Li, X.; Song, L.; Zhu, S.; Xiao, Y.; Huang, Y.; Hua, Y.; Chu, Q.; Ren, Z. Two strains of Lactobacilli effectively decrease the colonization of VRE in a mouse model. Front. Cell Infect. Microbiol. 2019, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Kishi, D.; Takahashi, I.; Kai, Y.; Tamagawa, H.; Iijima, H.; Obunai, S.; Nezu, R.; Ito, T.; Matsuda, H.; Kiyono, H. Alteration of V beta usage and cytokine production of CD4+ TCR beta beta homodimer T cells by elimination of Bacteroides vulgatus prevents colitis in TCR alpha-chain-deficient mice. J. Immunol. 2000, 165, 5891–5899. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Disease | Specimen | V Chain | J Chain | Authors | Year | Ref # | |||
|---|---|---|---|---|---|---|---|---|---|
| UC | Colon | TRBV4-1 | TRBJ2-2 | Günaltay S, et al. | 2017 | [29] | |||
| (Biopsy) | TRBV4-2 | TRBJ2-5 | TRBJ1-1 | ||||||
| TRBV4-3 | TRBJ2-2 | TRBJ2-5 | TRBJ1-2 | TRBJ2-6 | |||||
| TRBJ1-1 | TRBJ1-5 | ||||||||
| TRBV6-5 | TRBJ1-6 | ||||||||
| TRBV7-2 | TRBJ2-4 | ||||||||
| TRBV10-1 | TRBJ2-5 | ||||||||
| TRBV10-2 | TRBJ1-4 | ||||||||
| TRBV10-3 | TRBJ1-6 | TRBJ2-2 | TRBJ1-5 | TRBJ1-2 | |||||
| TRBJ2-6 | TRBJ1-4 | ||||||||
| TRBV24-1 | TRBJ1-2 | ||||||||
| TRBV28 | TRBJ1-6 | TRBJ1-4 | |||||||
| TRBV30 | TRBJ1-1 | ||||||||
| UC (Recurrence) | Colon | TRBV19 | TRBJ1-3 | TRBJ2-5 | |||||
| (Biopsy) | TRBV28 | TRBJ1-4 | TRBJ1-6 | ||||||
| TRBV30 | TRBJ2-1 | TRBJ1-1 | |||||||
| TRBV24-1 | TRBJ1-2 | ||||||||
| TRBV4-3 | TRBJ2-3 | ||||||||
| TRBV6-6 | TRBJ2-1 | ||||||||
| TRBV7-2 | TRBJ1-3 | TRBJ2-4 | |||||||
| TRBV10-2 | TRBJ1-4 | ||||||||
| TRBV10-3 | TRBJ1-1 | TRBJ2-6 | TRBJ1-2 | TRBJ1-5 | |||||
| TRBJ1-3 | TRBJ2-2 | TRBJ2-4 | TRBJ2-1 | ||||||
| TRBJ1-6 | TRBJ1-4 | TRBJ2-3 | |||||||
| pediatric UC | Blood, Colon | TRBV5 | TRBJ1-1 | TRBJ1-2 | TRBJ2-1 | TRBJ2-7 | Werner, L, et al. | 2019 | [24] |
| (Biopsy) | TRBV6 | ||||||||
| TRBV7 | |||||||||
| UC and CD | PBMC, LPMC | TRBV28 | TRBJ2-1 | Kakuta Y, et al. | 2020 | [31] | |||
| TRBV5-1 | TRBJ1-5 | ||||||||
| TRBV7-6 | TRBJ2-3 | ||||||||
| TRBV12-3 | TRBJ1-1 | ||||||||
| UC and CD | Blood | TRBV5-5 | TRBJ1-5 | Rosati, E et al. | 2020 | [26] | |||
| (Twins) | TRBV7-2 | TRBJ1-5 | TRBJ2-1 | TRBJ2-7 | |||||
| TRBV18 | TRBJ1-5 | ||||||||
| TRBV19 | TRBJ2-7 | ||||||||
| TRBV30 | TRBJ1-1 | ||||||||
| TRBV5-5 | TRBJ1-5 | ||||||||
| TRBV5-1 | TRBJ2-7 | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizoguchi, E.; Sadanaga, T.; Okada, T. Biological Analyses-Derived Translational Findings in the T Cell Receptor Alpha Chain Knockout Mouse as an Experimental Model for Ulcerative Colitis. Int. J. Transl. Med. 2021, 1, 187-204. https://doi.org/10.3390/ijtm1030014
Mizoguchi E, Sadanaga T, Okada T. Biological Analyses-Derived Translational Findings in the T Cell Receptor Alpha Chain Knockout Mouse as an Experimental Model for Ulcerative Colitis. International Journal of Translational Medicine. 2021; 1(3):187-204. https://doi.org/10.3390/ijtm1030014
Chicago/Turabian StyleMizoguchi, Emiko, Takayuki Sadanaga, and Toshiyuki Okada. 2021. "Biological Analyses-Derived Translational Findings in the T Cell Receptor Alpha Chain Knockout Mouse as an Experimental Model for Ulcerative Colitis" International Journal of Translational Medicine 1, no. 3: 187-204. https://doi.org/10.3390/ijtm1030014
APA StyleMizoguchi, E., Sadanaga, T., & Okada, T. (2021). Biological Analyses-Derived Translational Findings in the T Cell Receptor Alpha Chain Knockout Mouse as an Experimental Model for Ulcerative Colitis. International Journal of Translational Medicine, 1(3), 187-204. https://doi.org/10.3390/ijtm1030014