1. Introduction
To identify a biomarker by metabolomics sounds simple, and it may be in some cases, but then there are other cases where it may not be so simple. A lot depends on the metabolic pathway involved. In principle, it should just involve the appearance of a breakdown product of the substrate or a conjugated product of the substrate if that is what is involved in the particular pathways. In many cases one may not necessarily be looking for a biomarker but rather investigating the metabolic pathway and something appears which may be a biomarker. With advances in methods of detection of substances, it is becoming clear that well known metabolic pathways may be more complex than what may have long been thought. This is certainly true in the case of glycogen metabolism. In my particular case, I was not looking for a biomarker but rather investigating problems with enzyme replacement therapy (ERT) for a glycogen storage disease, Pompe disease.
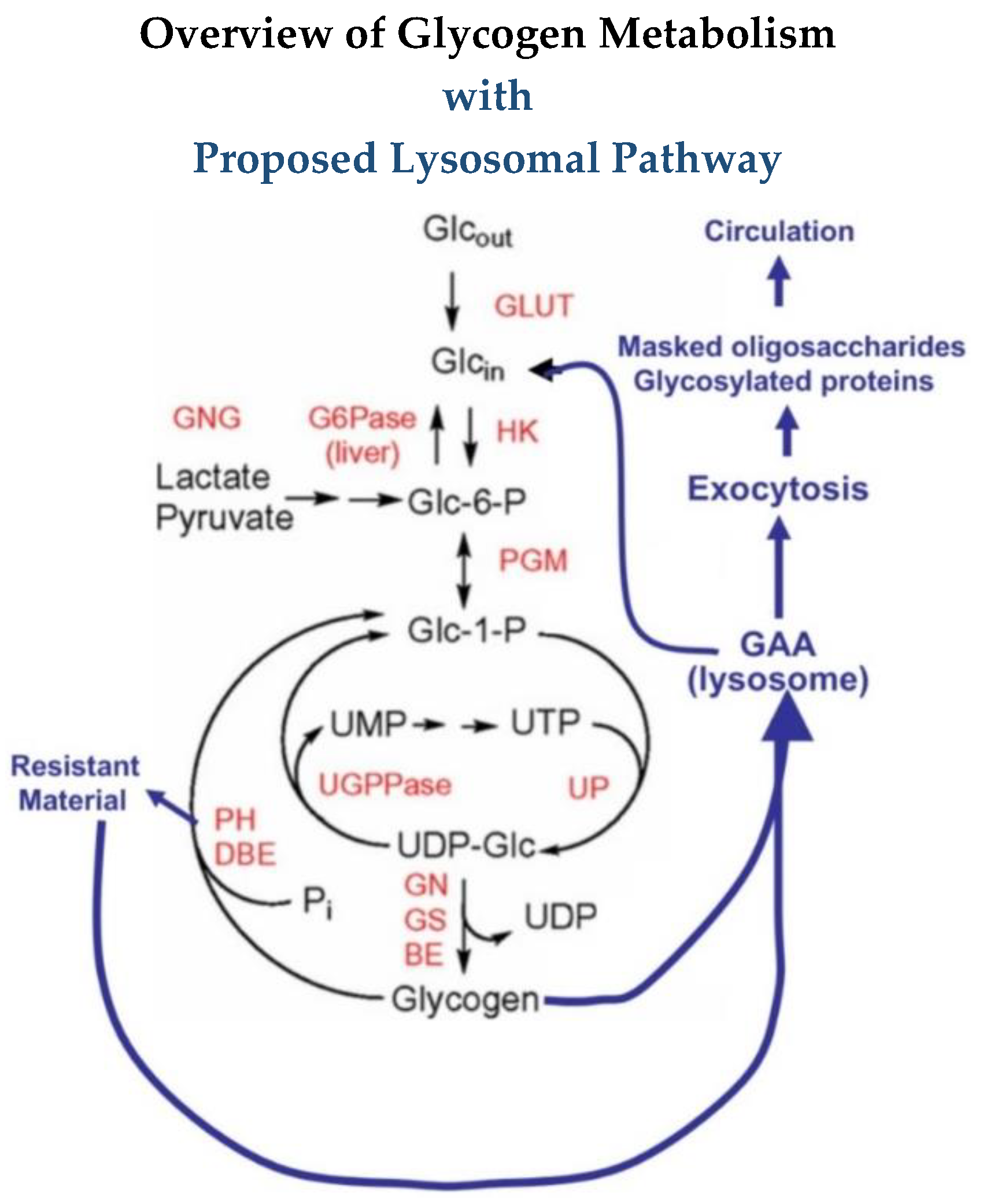
Glycogen, the storage form of glucose in animals, is a complex polymer of glucose consisting of chains of α-1,4 linked glucose residues with α-1,6 linked branch points about every 12 residues, and it consists of 12 layers with a molecular weight up to 10
7 kDa [
1]. Glycogen is organized into spherical particles of size similar to the calculated size of the spherical model [
2,
3]. At the reducing end is a protein, glycogenin, which functions as a self-glycosylating primer for the synthesis of the molecule. Glycogen is primarily degraded by phosphorylase for the linear chains and a debranching enzyme to cleave the 1,6 branches. Glycogen and these degradative enzymes are cytoplasmic in all cells but are most abundant in liver and muscle. In addition to the cytoplasmic components of glycogen metabolism, there is a lysosomal α-glucosidase which degrades glycogen in lysosomes. About 1–2% of the cell’s glycogen is localized in the lysosomes.
In 1963, H.G. Hers reported the deficiency of the lysosomal α-glucosidase (GAA) in Type II glycogenosis which became known as Pompe disease, as well as acid maltase deficiency [
4]. His initial report demonstrated the inability to degrade glycogen, but did not specifically report the deficiency of α-1,6-glucosidase activity. In 1964, the lysosomal α-glucosidase was shown to have α-1,6-glucosidase activity in dog liver [
5]. In 1970, Brown et al. reported the absence in α-1,6-glucosidase in human Pompe disease tissues [
6]. The enzyme was shown to be capable of transglucanase, transglucosylation, maltase and glucamylase activities in addition to α-1,4-glucosidase and α-1,6-glucosidase activities [
7,
8,
9,
10,
11]. The enzyme is inhibited by maltooligosaccharides above 5 mM concentration [
7,
8,
9,
10,
11]. The concentration of substrates in vitro or in vivo is unknown since the molecular weight of glycogen is variable and the molecular weight of the soluble fraction is unknown, and concentration only refers to things in solution. The enzyme has been reported to degrade glycogen by 91%, but the inability to completely degrade glycogen when it was 80% degraded by the lysosomal α-glucosidase followed by phosphorylase and debrancher [
7]. Another report stated glycogen was 95% converted to glucose by the rabbit liver lysosomal α-glucosidase; however, this was likely a comparison to the glucose released by acid as was a previous report by the same laboratory [
10]. It was not possible to detect very small amounts of other carbohydrates at that time. These two studies utilized commercially isolated glycogen which I have found to be partially degraded by chemically harsh procedures [
7,
10]. Many reports give the impression that only glucose is released from glycogen in vivo, but the data reported here indicate that oligosaccharides are released and then converted to glucose in vitro.
There are infantile, juvenile and adult forms of the disease with the infantile involving all tissues but primarily the heart and liver, as well as muscle and the later onset forms primarily involving muscle. The development of the knock-out mouse model for Pompe disease facilitated work which resulted in the development of enzyme replacement therapy [
12]. Since 2006, Pompe disease has been treated by enzyme replacement therapy with recombinant GAA (rhGAA). The rhGAA is modified by the addition of mannose-6-phosphate residues to take advantage of the Man-6-P receptor to facilitate its entry into tissues and lysosomes [
12,
13]. The rhGAA is a precursor form of the GAA which is then processed to the mature form by tissues along with the uptake into the cells and lysosomes. Most research over the last 30 years has focused on getting the rhGAA into tissues and the lysosomes.
The complexity of the structure of glycogen and the different activities of the enzyme are often not included in discussions about the deficiency in Pompe disease. As mentioned, GAA has α-1,4-glucosidase, α-1,6-glucosidase, endoglucanase and glucosyltransferase activities which facilitate its degradation of glycogen [
7,
8,
9,
10,
11]. This means it can remove a terminal α-1,4-linked glucose, a terminal α-1,6 linked glucose, it can transfer a glucose to another molecule, or it can cleave internally in a glucose chain and potentially attach it to another molecule. Since in diagrams of pathways, GAA is usually written with an arrow going to glucose, many readers may not be aware of the other activities. For the outer chains of glycogen, the cleavage of one glycosidic linkage may release a glucose residue but for the internal structure more bonds must be cleaved to release a glucose. This difference is reflected in the rate of glucose released over time. Some of the results presented here suggest that there may even be another level of organization of glycogen that has not been described.
The present work was begun based on the fact that there are some unresolved issues related to ERT. Following ERT in the Pompe mouse there is residual carbohydrate material, as determined by periodic acid-Schiff (PAS) staining, which accumulates in the cytoplasm [
14,
15]. This same observation has been made on biopsy tissue from patients following ERT [
16,
17]. This material has been referred to as glycogen in publications, but it has not been isolated and identified. This may be important since, from my prior work, I know that glycogen contains carbohydrates other than glucose, whereas most investigators are of the belief that glycogen only contains only glucose. It has also been noted that many patients have marked improvement for about 6 months on ERT after which the benefits are not as significant [
18]. Both of these observations date back over 15 years. In retrospect, it would seem plausible that initially rhGAA may clear a significant amount of storage material but after some time a portion of the stored material may not be degraded and begins to accumulate [
14,
18]. Therefore, my initial question at the beginning of this work was “Can rhGAA completely degrade Pompe glycogen?” Secondarily, can it completely degrade all glycogen? It is clear that the metabolomics of glycogen degradation by rhGAA is very complex and that we clearly do not understand the complete structure of glycogen.
As stated, GAA has α-1,4-glucosidase, α-1,6-glucosidase, transglucanase, transglucosylation, maltase and glucamylase activities which coupled with the variabilities in the structure of glycogen means that this is not a straight forward single step enzyme catalyzed reaction from substrate to product [
7,
8,
9,
10]. The α-1,6 glucosidase activity on glycogen cannot be studied kinetically since the actual substrate for the reaction is not known and there may be several possibilities involved. The assumption, conscious or not, has been that glycogen is a homopolymer of glucose. When dealing with a storage disease the term and scope of metabolomics takes on a totally different approach This is because we are no longer trying to determine where the substance goes and intermediates, but our goal is really to determine how every bit of that polymer can be degraded and where it goes. Therefore, this is very different than the traditional way we have learned biochemistry which was primarily related to biosynthesis and not degradation.
2. Materials and Methods
2.1. Source
Autopsy liver tissue was obtained from an 18-month-old female with Pompe disease (Type II glycogenosis) and from two adult male accident victims. The Pompe liver and the Control 1 liver tissues were obtained at autopsy several hours postmortem. In the case of Control 2, the patient was an organ donor on life support, thus the liver tissue was obtained immediately on termination of life support. All liver tissue was stored at −76 °C until the glycogen isolation. The case of the Pompe disease patient and an enzyme replacement trial with lysosomal α-glucosidase linked to low-density lipoprotein has been previously reported [
19]. Her death was 25 days after a second enzyme infusion.
2.2. Glycogen Substrates
The Sigma, Type IX bovine liver glycogen, Sigma-Aldrich, St. Louis, MO, USA, was extracted by the method of Bell and Young, [
20] which involves boiling water and TCA precipitation of proteins at elevated temperature. This method is quite harsh compared with the method used in this work for of isolation of the human glycogen.
2.3. Isolation of Human Glycogen
The human glycogen was isolated by the method of Mordoh et al. [
21] with the addition of five freeze thaw steps to ensure the rupture of lysosomes. This method was chosen because it was reported that the preparation appeared to be identical to native glycogen isolated from liver as judged by its rate of sedimentation and its appearance under the electron microscope [
21]. Glycogen isolated by this method is reported to be para crystalline [
22]. The glycogens were characterized for a number of parameters including average glucose chain length, protein content, amino acid composition, RNA content, phosphate content, β-amolysis, iodine absorbance, interior chain length and external chain length. The average chain lengths for the control glycogens were 19.9 and for the Pompe glycogen 12.6. The average interior chain lengths were 4.0 and 4.4 for Control 1 and Control 2, respectively. The average interior chain length for the Pompe glycogen was 3.7. The average exterior chain lengths were 15.9, 15.2 and 8.9 for Control 1, Control 2 and Pompe glycogens, respectively [
23,
24,
25,
26]. The protein content was less than one per cent for all but one sample. All glycogens were hydrated for at least 18 h before being used as substrates in assays. Glycogen solutions were never frozen. The tissues were obtained in late 1978 and early 1979 and stored at −76 °C until the glycogen was isolated. The isolations were carried out in parallel. The characterization of the glycogen was carried out in 1979. Dr. Metzenberg’s master’s thesis was submitted in 1980 [
23]. The glycogen samples have been stored in a vacuum desiccator over desiccant since that time. I was approached by Genzyme in 2008 as they were aware I had these samples.
2.4. Enzyme Assays
Recombinant human GAA was provided by Sanofi Genzyme, Cambridge, MA, USA. This is the 110 kDa precursor which is processed to the mature form by the tissue in ERT. In the case of administration to a patient this precursor was shown by Western blot to result in the 76 kDa active form [
27]. The incubation mixtures contained 500 μg of glycogen, 50 mM sodium acetate buffer at pH 4.6 and 10 μL or 25 μL rhGAA (5 μg/μL). The mixtures were incubated at 37 °C under toluene for inhibition of any possible microbial growth. At time points indicated in the figures, the reaction mixtures were mixed on a vortex mixer and centrifuged at 16,000×
g for 5 min to precipitate insoluble material. Aliquots of 100 μL or 200 μL were extracted and boiled for 5 min in screw cap vials. Samples were again centrifuged at 16,000×
g for 5 min to precipitate and insoluble material and the supernatants were analyzed by HPAEC-PAD using a PA-1 column. The incubation mixture was mixed on a vortex mixer before being replaced in the water bath.
2.5. Carbohydrate Analysis
HPAEC-PAD was performed on a Dionex DX-600 ion chromatograph, Thermo Fisher Scientific, Waltham, MA, USA, using a Dionex CarboPac PA-1 column. The eluent was 150 mM sodium hydroxide, isocratic from 0 to 5 min then a linear sodium acetate gradient from 5 to 25 min from 0 to 57% 500 mM NaOAc in 150 mM NaOH at a flow rate of 1 mL/min. Fractions were collected using a Gilson 202 fraction collector, Gilson Scientific, Middleton, WI, USA, dialyzed overnight against 18.3 megohm water in 1.0 mL chambers with a 500 MWCO membrane. Fractions were combined and taken to dryness in a Speed-Vac. Fractions were then hydrolyzed with 2 N TFA at 100 °C for two hours after which they were taken to dryness in a Speed-Vac. If it was determined that hydrolysis was incomplete, samples were hydrolyzed with 4 N TFA at 120 °C for 1 to 4 h. Monosaccharides and sugar alcohols were determined using a Dionex CarboPac MA-1 column with isocratic elution with 612 mM or 480 mM NaOH at a flow rate of 0.4 mL/min. The waveform for carbohydrate analysis had a potential of +0.1 V from 0 to 0.40 s, −2.0 V from 0.41 to 0.42 s, +0.6 V from 0.43 to 0.44 s and –0.1 V from 0.44 to 0.50 s with integration from 0.20 to 0.40 s. Data analysis was performed using Dionex Chromeleon software, Thermo Fisher Scientific, Waltham, MA.
2.6. Amyloglucosidase Assay
Assay mixtures consisted of 1 mL volume containing 500 µg of glycogen, 50 mM sodium acetate buffer, pH 4.6 and 100 µL amyloglucosidase, 1 mg/mL, A. niger, Sigma A-7420. The reactions were incubated at 55 °C. Incubations were for 2 or 5 h and boiled for 5 min to stop the reaction, after which they were centrifuged for 5 min at 16,000× g.
2.7. Maltooligosaccharides and Amyloglucosidase
Maltooligosaccharides and amyloglucosidase were purchased from Sigma Aldrich Chemical Company. Maltooligosaccharides and their determined glucose content were as follows: maltotriose, 67.2%; maltotetraose, 89.1%; maltopentaose, 97.7%; maltohexaose, 94.4%; maltoheptaose, 84.7%.
2.8. Acid Hydrolysis of Glycogen Residues
The insoluble precipitate, which appeared to be stuck to the bottom of the tube, was washed three times with water by agitation on a vortex mixer and centrifugation. Following the washing the material was hydrolyzed in 2 N TFA for 3 h at 100 °C, dried in a Speed-Vac, Savant, Thermo Fisher Scientific, Waltham, MA, USA, and taken up in water for chromatography.
2.9. Protein Determination
Protein determination on the TFA hydrolyzates was by a modification of the method of Lowry et al. [
28]. A control experiment of protein determination on bovine serum albumin (BSA) showed no significant difference between samples of before and after hydrolysis for comparison.
2.10. KOH Treatment of Glycogen
Control 2 glycogen, 30.8 mg, in 30% KOH was boiled for 60 min. It was precipitated in 90% EtOH, washed, re-precipitated and taken to dryness. The final weight was 30.2 mg. The starting glycogen was opaque in the solution. The final material was clear in a solution of 10 mg/mL indicative of a significant change. Reports in the literature indicate that this KOH treatment generally reduces the glycogen to about half of its original molecular weight. The protein content of Control 2 glycogen was 1.7%, and the protein content of the Control 2 glycogen, KOH-treated, was 0.16%.
3. Results
Metabolomics issues will be discussed first, then biomarkers later.
3.1. Enzymatic Hydrolysis of Glycogen
Glucose released from the Pompe liver glycogen and the control glycogens is shown in
Figure 1. A comparison of the glycogen samples revealed that more glucose was released from the Pompe glycogen in the initial 24 h than from the two control glycogens when 10 µL (5 µg/µL) of rhGAA was used in the 1ml reaction mixture containing 0.5 mg of glycogen. There was a noticeable lag period after which the glucose released from the controls approached the others. This lag period was not observed when the incubation was started with 25 µL of rhGAA (5 µg/µL) in the 1ml reaction mixture. It is interesting to note that each glycogen sample appears to be somewhat different by the rate of degradation and the time frame in which the intermediates appear.
A similar result was obtained for all glycogens analyzed although only one Pompe liver glycogen is shown. The centrifuging before sampling was carried out to prevent insoluble glycogen from being removed from the incubation tube. The replacement of buffer containing rhGAA at 120 h was carried out to replace volume lost due to sampling and to reduce the free glucose concentration which might inhibit the enzyme. The reactions were allowed to proceed until no more glucose was released. At that point, there was a visible residue which was stuck to the bottom of the tube.
The significance of these results of the glucose released is that whereas rhGAA incubation resulted in the release of increasing amounts of glucose with time, it did not release the minor constituents [
24]. In a time course of TFA hydrolysis, the minor constituents are not released until well after the glucans are hydrolyzed [
25]. This was taken to indicate that the monosaccharides other than glucose are not intercalated with the glucose in glycogen. However, since they are present in the hydrolyzate of the residual material after no more glucose is released, it appears that that glycogen is not homogeneous and that the glucose in the α-glucosidase resistant component cannot be accessed by the α-glucosidase. However, the enzyme is able to access almost all of the glucose. The monosaccharide composition of the α-glucosidase resistant material contains hexoses and alditols which are not released by the α-glucosidase action on glycogen as shown in Figure 5. This is also indicative of the heterogeneity of glycogen.
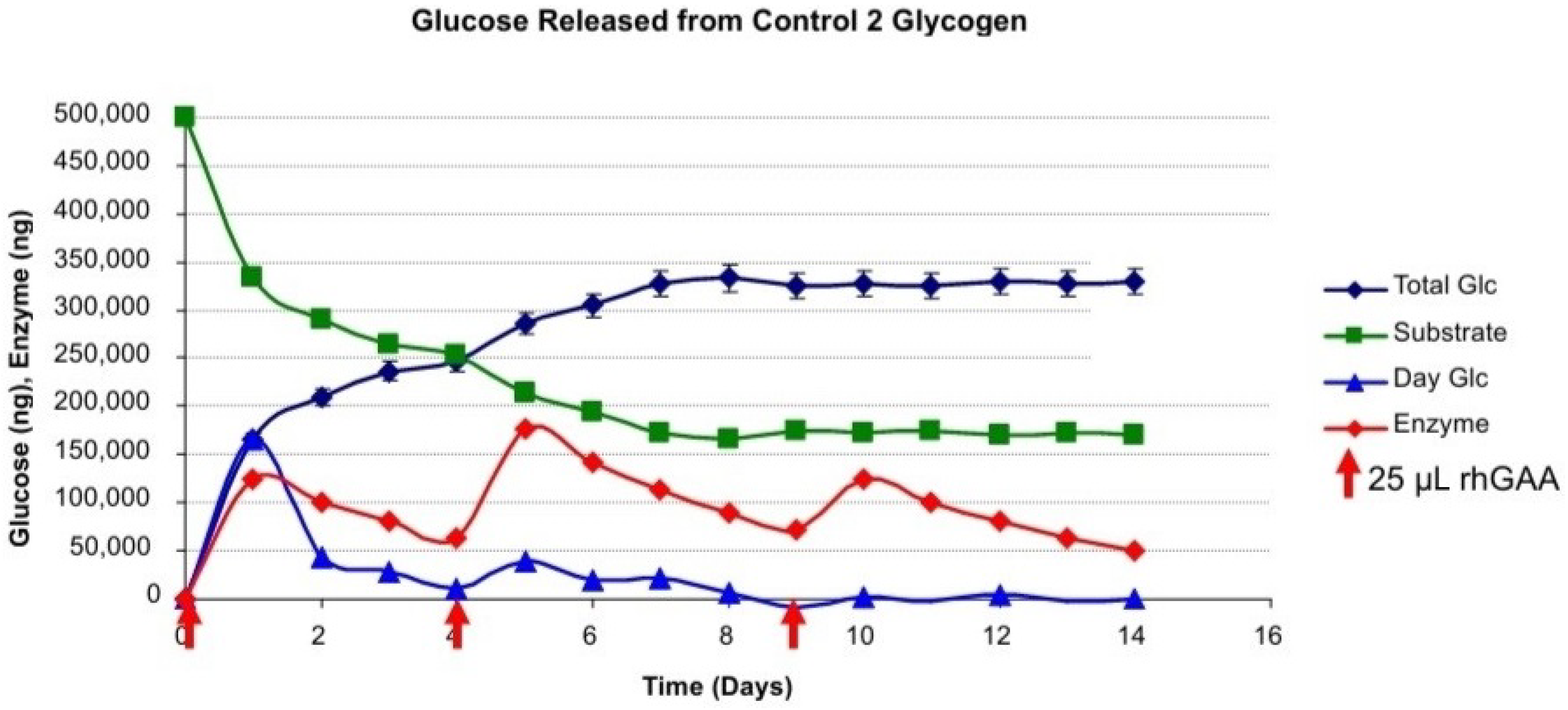
Figure 2 shows glucose released from Control 2 glycogen and the calculated daily remaining glycogen substrate, glucose released per day and the content of rhGAA remaining.
It may be that only glucose is released in vivo, but from work with glycogen as a substrate, it appears that oligosaccharides are also released in vitro and are then degraded to glucose likely within the lysosome. The oligosaccharides released into the medium during glycogen degradation by rhGAA typically are maltose, isomaltose, maltotriose and maltotetraose. Isomaltose is an α-1,6-linked glucan, whereas the others are α-1,4-linked glucans of 2,3 or 4 glucose residues, respectively.
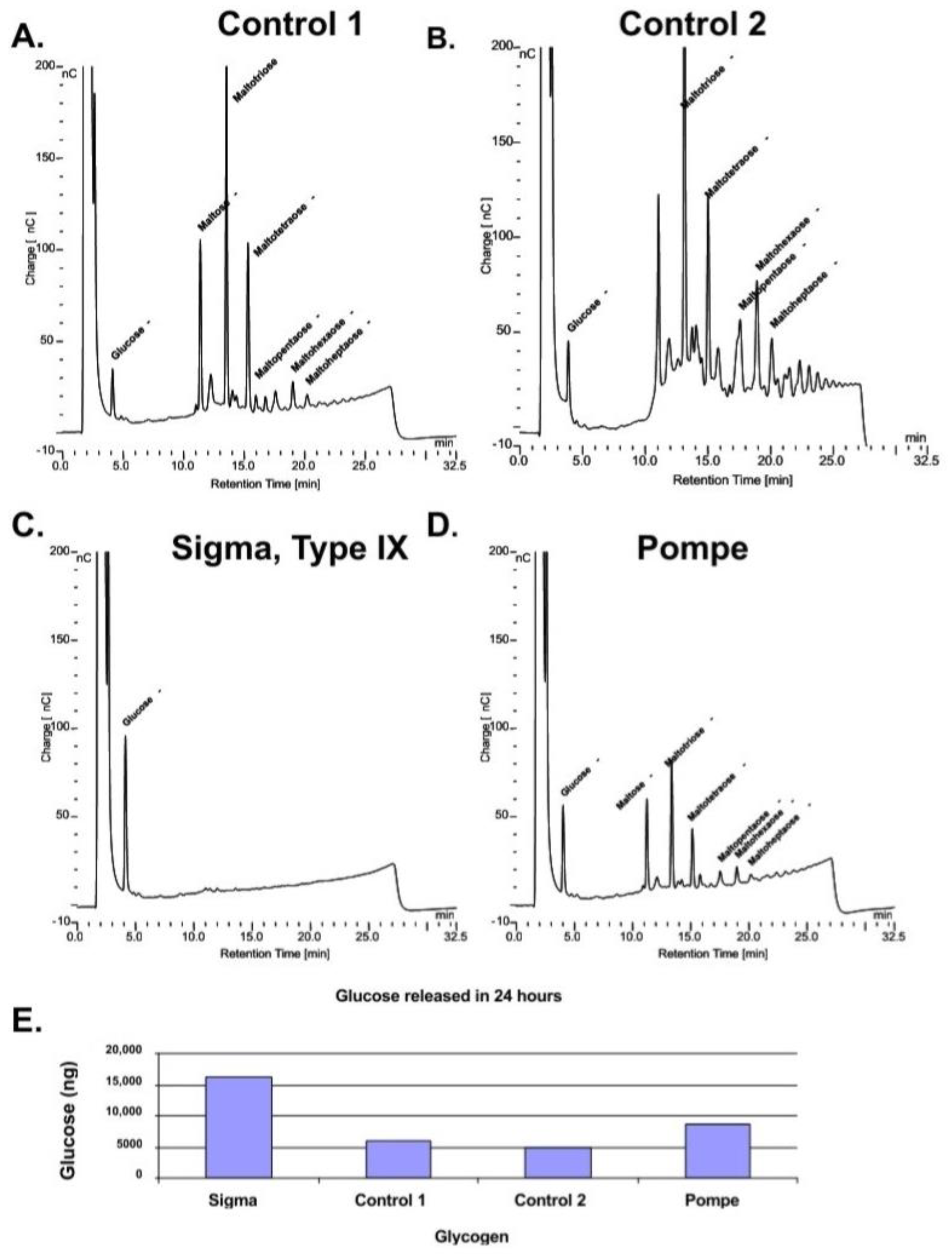
However, if the samples are incubated with rhGAA for 24 h and the incubation medium is replaced with 50 mM Tris-HCl, pH 6.7 for an additional 24 h a number of intermediates are released into the Tris-HCl solution as shown in the chromatograms in
Figure 3A–D. The the bar plot of glucose released after the first 24 h of degradation by rhGAA is shown in
Figure 3E. This shows that the rate of glucose released is reduced if the glycogen has a highly complex structure although oligosaccharides are released. The Sigma glycogen was completely degraded in the 24 h period. As mentioned, the Sigma glycogen is produced by an isolation procedure which is very harsh and as a result was completely degraded in 24 h. The human glycogens were isolated by a gentle procedure chosen for the reason that the glycogen was found to closely resemble glycogen in tissue [
22]. Another factor is the time period between death and freezing of the tissue. Control 1 and Pompe liver tissue was obtained at autopsy; however, Control 2 liver tissue was taken from an organ donor; it was diced and placed on ice immediately on removal and frozen on dry ice within minutes. The time period between death and when the tissue was frozen appears to have a significant effect on the degree of complexity of the glycogen subsequently isolated from the tissue. This rapid degradation of glycogen after death has been reported to occur within minutes for animal tissues [
29].
When the Tris-HCl buffer was washed out and the original incubation medium containing the rhGAA was replaced and the tubes returned to the water bath, no more glucose was released for 48 h. At that point the release of glucose resumed, and the rate of glucose release resembled that of incubations not interrupted by the Tris-HCl treatment. Obviously, oligosaccharides and higher structures were being degraded to a point where glucose was then released. In the case of the use of phosphate buffer to raise the pH no oligosaccharides were detected in the extract after 24 h. In addition, after removal of the phosphate buffer and replacement of the initial medium with rhGAA, no more glucose was released over several days. To determine if the rhGAA was still active, additional glycogen was added and it was degraded as usual with the release of glucose. If glycogen was incubated in the phosphate buffer for 24 h and the phosphate washed out before the incubation media with rhGAA added, the glycogen was degraded as usual. Therefore, if glycogen is incubated with rhGAA and then switched to phosphate buffer, the glycogen cannot be further degraded by switching back to the media with rhGAA. This is a very interesting observation, but I do not have an explanation for what is happening. This bears further investigation.
The differences in the rates of degradation of the different glycogens demonstrates that the rate of degradation of glycogen by rhGAA can only be compared by using glycogen from the same isolation batch. This means that the rate of glycogen degradation between any two experiments cannot be used as an indicator of enzyme activity unless both experiments were carried out using the same batch of glycogen.
The variable rates of degradation of different glycogen samples raises several questions: Does the glycogen from one individual vary the glycogen of another individual? Does the glycogen in an individual vary in structure, in addition to quantity, from time to time based on the metabolic state and perhaps other unknown factors? No two batches of isolated glycogen can be considered equivalent.
3.2. Specific Activity and Apparent Specific Activity
The apparent specific activity of the rhGAA on glycogen is lower than the specific activity on artificial or low molecular weight substrates such as 4-methylumbelliferyl α-
d-glucose or maltose. The problem with the 4-methylumbelliferyl or
p-nitrophenyl-α-
d-glucose substrates is that they are a very low molecular weight substrates compared with glycogen and not what the enzyme acts on in tissue. For this reason, specific activity for rhGAA was determined using maltose and the apparent specific activity against glycogen as substrates; therefore, the specific activity of the enzyme against them is much higher than against any natural substrate. The specific activity of 42.8 U/mg at pH 4.6 compares favorably with the specific activity of 32.7 U/mg at pH 4.5 and 10 mM maltose for the lysosomal α-glucosidase [
30]. It should also be kept in mind that a glycosyl hydrolase is a special case of a transferase which uses water as the acceptor alcohol.
Apparent Specific Activity: Glycogen Substrates
The true specific activity of GAA cannot be determined against glycogen due to a lack of knowledge of the glycogen structure that is the substrate the enzyme acts on [
8]. Experiments have demonstrated a difference in rhGAA activity with different glycogen substrates as shown in
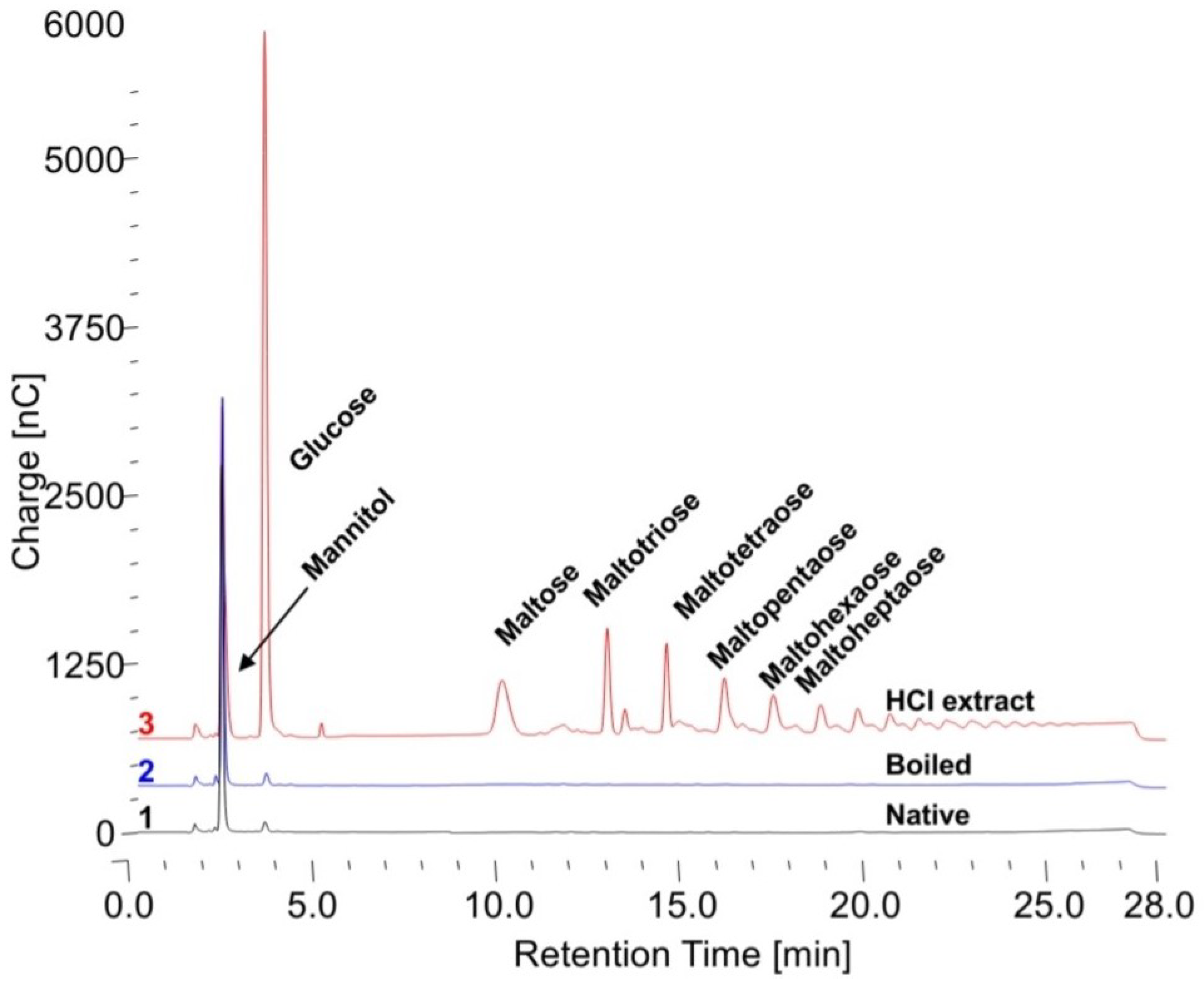
Figure 3. For this reason, both Sigma Type IX glycogen and Control 2 human glycogen were used at two different content levels in the reaction. It is not proper to refer to glycogen as a concentration since concentration refers to substances in solution and all of the glycogen is not in solution, thus content in the reaction mixture is used. To determine if configuration of the glycogen makes a difference in the apparent specific activity, in addition to native glycogen, glycogen was also boiled for 5 min before the assay and 0.1 N HCl extracts were also used. The free glucose in the HCl extracts was subtracted from that released by the enzyme.
The apparent specific activities against the native, boiled and 0.1 N HCl extracts of Sigma, Control 1, Control 2, Pompe and KOH-treated Control 2 glycogens are shown in
Table 1. These are the results from 24 h incubations at a wide range of substrate contents from 400 to 8000 mg/mL. In all cases, the human glycogens have the lowest apparent specific activity for the native glycogen, higher for the boiled glycogen and much higher for the HCl extract. The Sigma glycogen has very similar values for the native and boiled glycogen and somewhat higher for the HCl extract. The KOH Control 2-treated glycogen has values significantly higher than the native Control 2 glycogen and with less difference between them similar to the Sigma glycogen. These data demonstrate the difference between glycogens and the fact that the Sigma glycogen is significantly degraded which is consistent with the data in
Figure 3. These data again show that apparent specific activity of the enzyme is dependent on the glycogen substrate and that they are all different. Chromatograms of typical initial starting incubations for the native, boiled and HCl extracts are shown in
Figure 4. There is only some free glucose in the HCl extract; therefore, it was subtracted from the glucose liberated.
3.3. Residue from Enzymatic Degradation of Glycogen
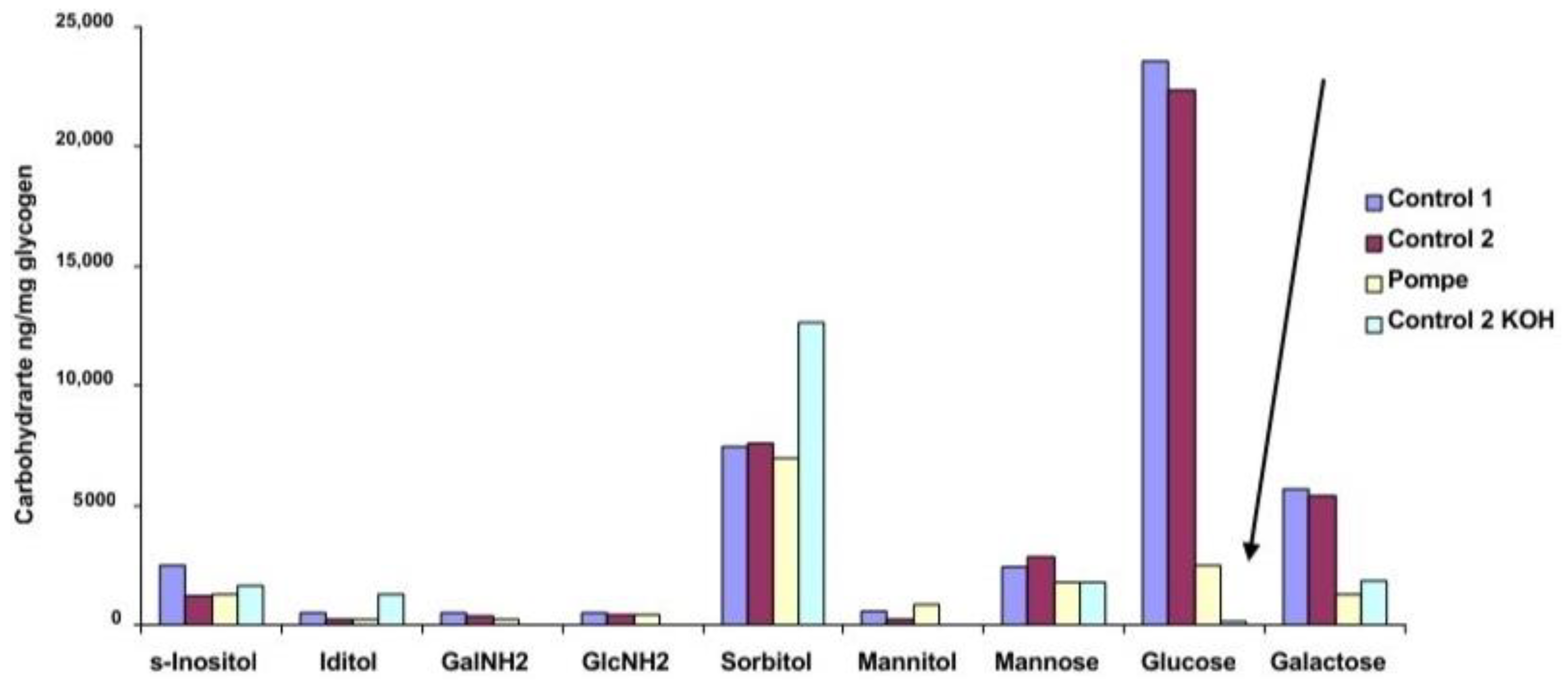
The carbohydrate composition of the residues from rhGAA degradation of glycogen is shown in
Figure 5 and the total carbohydrate and protein composition is shown in
Table 2. The difference in glucose content of Control 1 and Control 2 glycogen residues and the residues from Pompe glycogen and KOH-treated Control 2 glycogen is striking. The KOH treatment changes the structure enough that the rhGAA can access much more of the glucose which is consistent with the relative specific activity against the KOH-treated Control 2 indicated in
Table 1. The KOH-treated Control 2 glycogen has a less complex structure as a result of the KOH treatment. This difference is consistent with the results shown in
Figure 1 and
Figure 3 for the Control and Pompe glycogens. Clearly, rhGAA has greater access to the glucose containing structures in Pompe and KOH-treated Control 2 glycogens. The residue material is not a purified fraction but rather a mixture of the residues from all of the oligosaccharides of a glycogen which are shown in
Figure 5 and discussed further in Murray et al. [
24]. At this time, the nature of the association of the protein and carbohydrate components with the oligosaccharide components of glycogen is not known. However, if the chromatography of oligosaccharides, such as maltotriose, does not employ an acetate gradient the small peaks are not resolved [
24].
If the glycogens are degraded with amyloglucosidase, a similar residue with a similar carbohydrate composition to that following degradation with rhGAA. It is clearly due to the presence of saccharide residues other than α-glucans and the protein. A similar residue with a similar carbohydrate composition remains following amyloglucosidase degradation of corn starch.
The inability of amyloglucosidase to completely digest glycogen is interesting since there are a number of publications which claim that amyloglucosidase completely degrades glycogen [
31,
32,
33,
34,
35]. In some cases, investigators compared the amount of glucose released by the enzyme to the amount of glucose released by acid hydrolysis but in other cases it appears that they reached that conclusion based on the specificity of amyloglucosidase and the assumption that glycogen only contains glucose. It is clear that these earlier investigators were of the belief that glycogen is composed completely of glucose; therefore, they did not consider other monosaccharide components. Yet, insoluble residue is obvious from visual inspection of the incubation tubes after removal of the incubation medium. From the report of Jeffrey et al. that there is residual material present following degradation of glycogen by rhGAA and isolated lysosomal α-glucosidase [
7].
3.4. Treatment of Glycogen Residues with Amyloglucosidase and α-Mannosidase
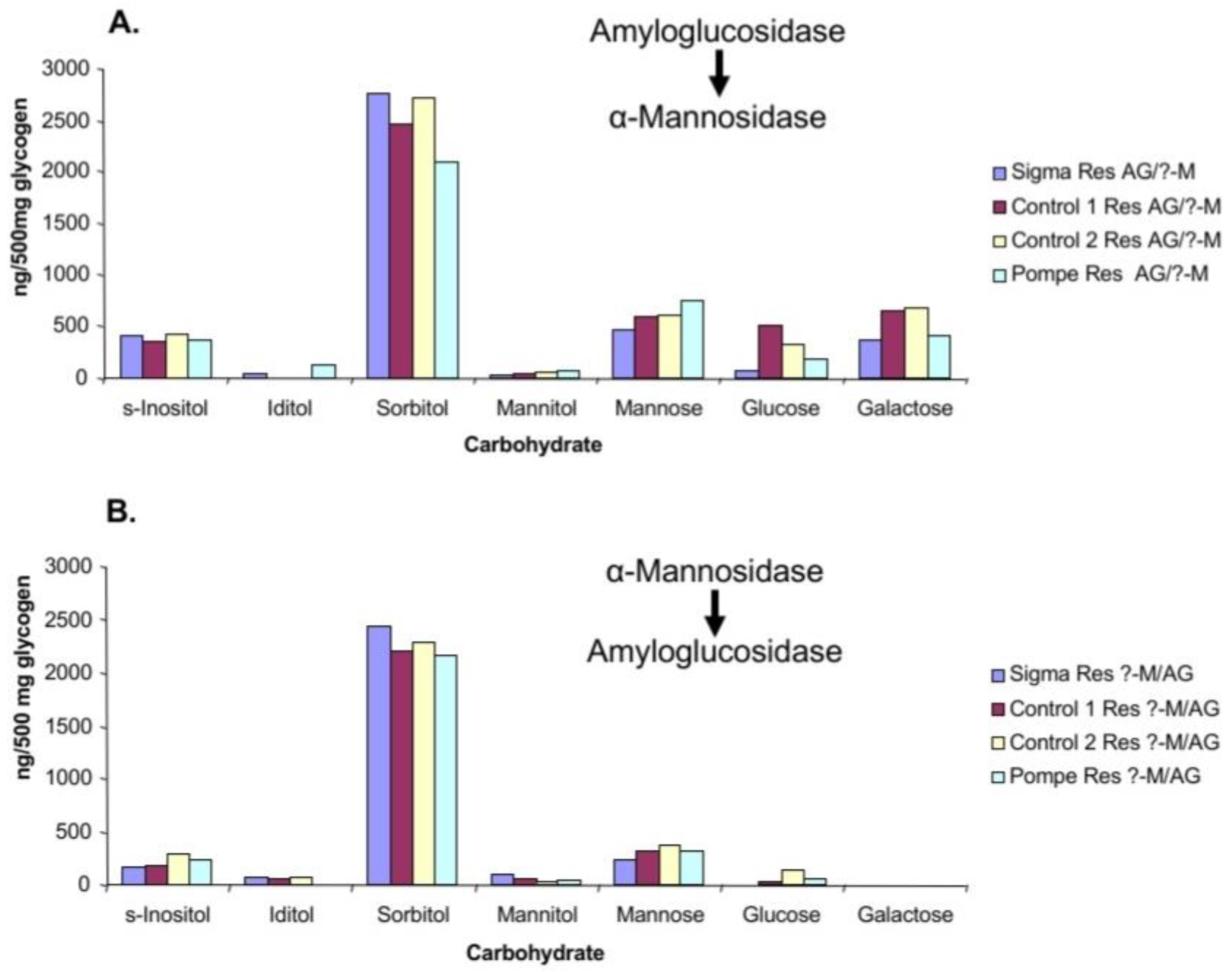
The residues were treated sequentially with α-mannosidase and amyloglucosidase as shown in
Figure 6A,B. Treatment with amyloglucosidase followed by α-mannosidase did not affect the glucose content of the residues. However, when the residues were treated first with α-mannosidase then amyloglucosidase there was a partial reduction in mannose and a significant reduction in both glucose and galactose. Therefore, it appears that part of the mannose impairs access to glucose and galactose by the amyloglucosidase.
On degradation of glycogen by rhGAA most of the glycogen is degraded to glucose.
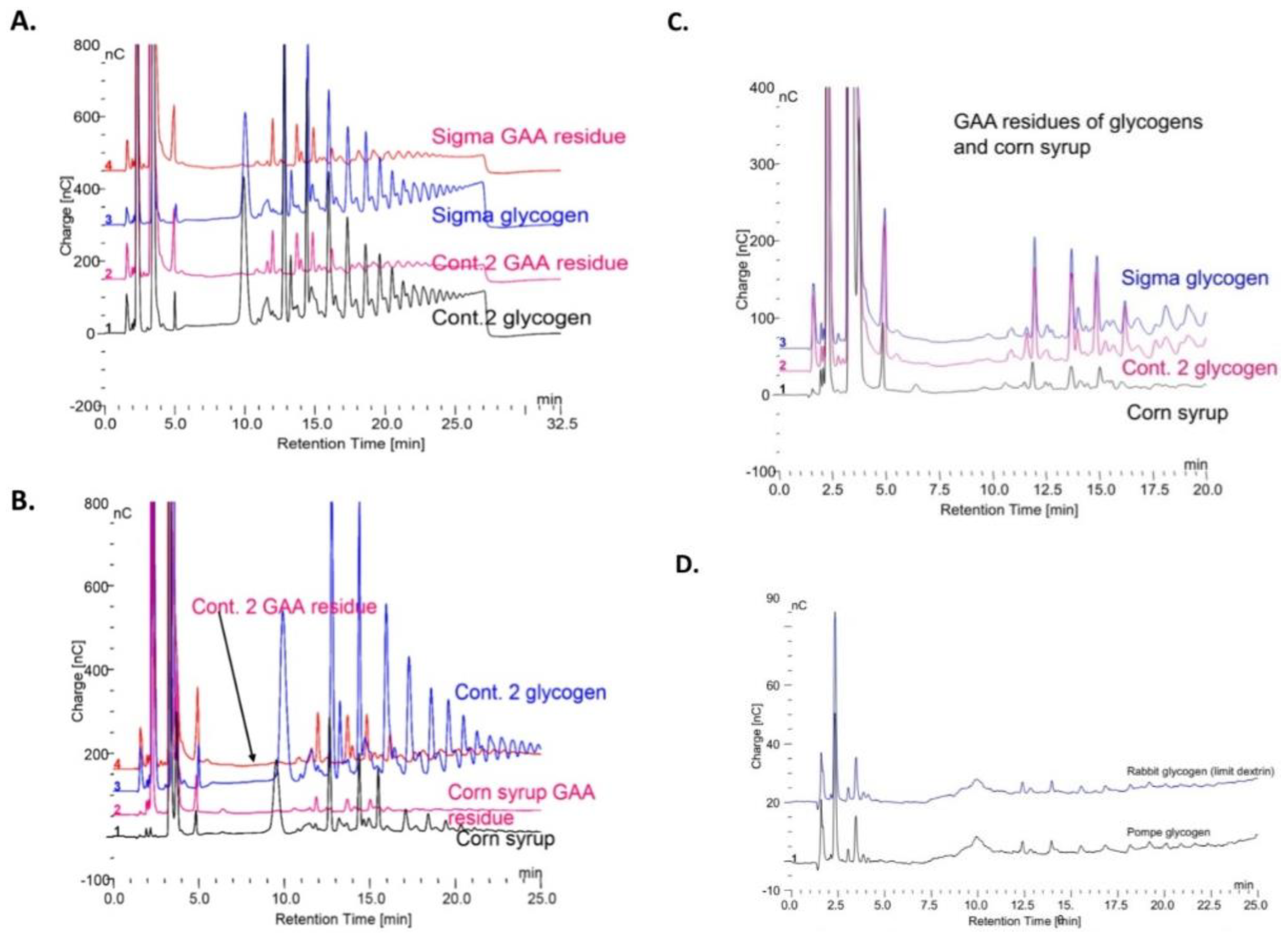
Figure 7A shows oligomers extracted from two different glycogens by boiling in 0.1 N HCl before and after the extracts were incubated with (rhGAA).
Figure 7B shows the HCl extract of Control 2 glycogen and corn syrup, a starch hydrolyzate, before and after incubation with rhGAA.
Figure 7C shows the oligomers from two glycogens and the maltooligosaccharides from corn syrup after incubation with rhGAA. The corn syrup was Karo syrup (1:100). The similarity of the rhGAA residues of the two glycogens and corn syrup is apparent. The similarity of these rhGAA residues to the rhGAA residues of rabbit limit dextrin and Pompe disease glycogen is shown in
Figure 7D. This residual material following glycogen degradation with rhGAA contains about 30 to 40% protein conjugated to inositols, iditol, sorbitol glucosamine galactosamine mannose glucose and galactose as shown earlier. The co-chromatography of the individual minor peaks for each major peak in the Sigma glycogen, Control 2 glycogen and corn starch hydrolyzates in these is taken as evidence that the glycogen residue is not a contaminant of glycogen.
3.5. Glycosylated Protein Biomarkers
The subjects of a previous paper were the glucan and a glycosylated protein which is glycosylated primarily with inositol and sorbitol [
36]. The enzyme releases about 70–75% of the glycogen as glucose by the action of rhGAA in vitro. After about four days of incubation of glycogen with rhGAA in vitro, the released glucose reaches a plateau, thus no more glucose is released. There is no detectable carbohydrate in the medium that elutes after glucose by HPAEC-PAD employing a CarboPac PA1 column. However, if the medium is boiled in 0.1 N HCl for 30 min, several oligosaccharides are detected. The oligosaccharides are also detected after incubation of the medium with trypsin. This appears to be a case of protein masking carbohydrate which is unusual but has been reported [
37,
38]. The soluble glycosylated protein in the medium is bound by Dowex 50W. This is taken as evidence of binding by a charged entity such as protein. The soluble glycosylated protein is not bound by Concanavalin A which binds mannose or glucose containing carbohydrates, including glycogen [
36]. Given these characteristics the possibility of lysosomal exocytosis, serum was analyzed, and the soluble glycosylated protein was found.
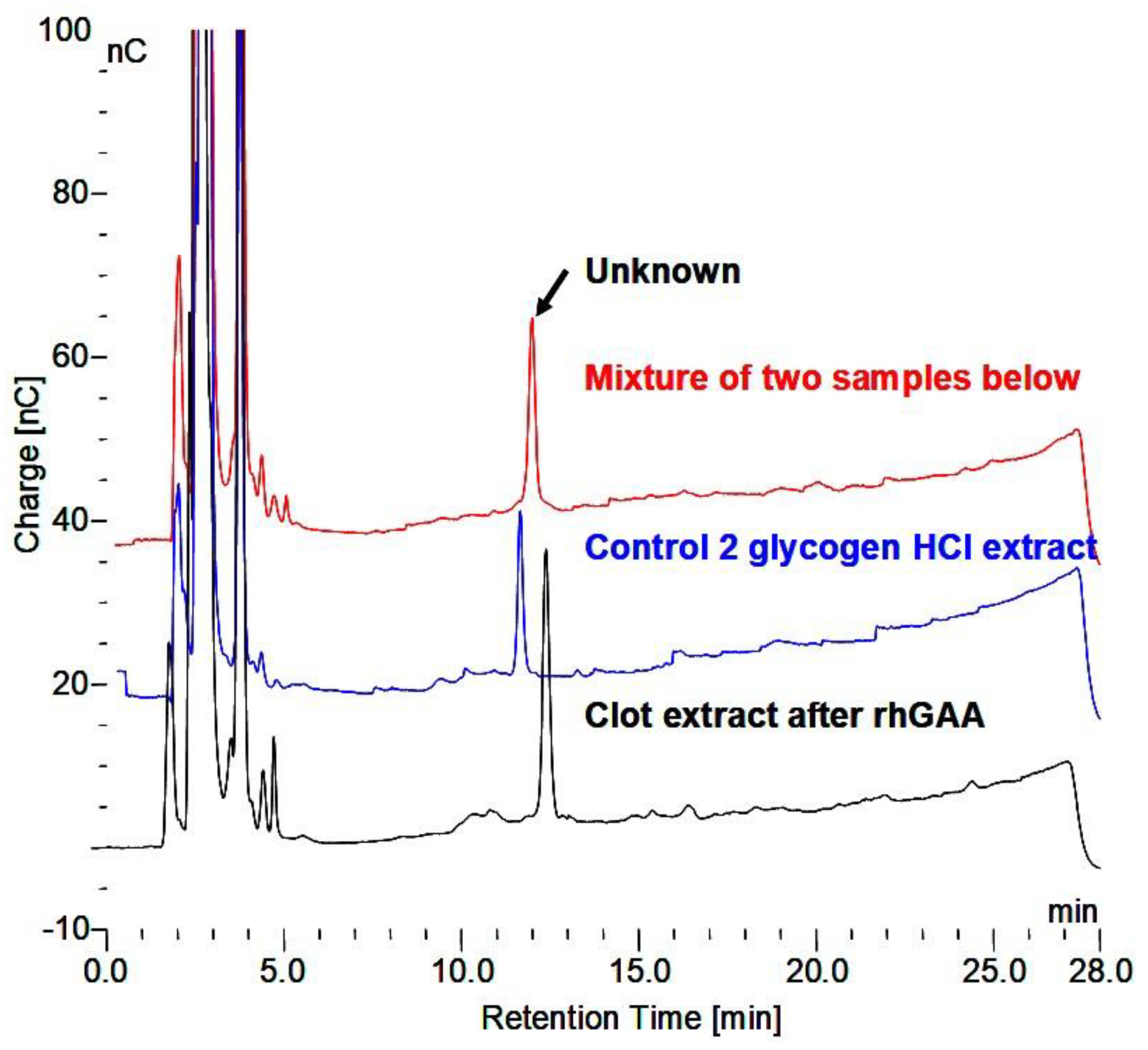
The comparison of their chromatographic identity of the glycosylated protein end product which cannot be further degraded by rhGAA and the glycosylated protein from serum are shown in
Figure 8 taken from Murray [
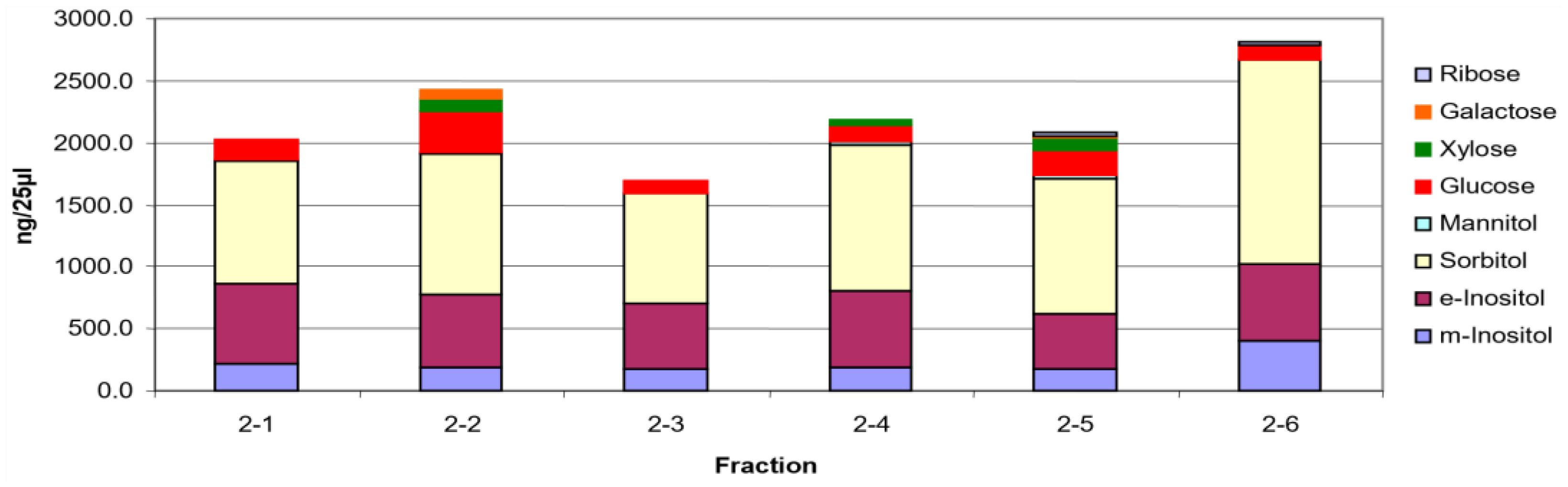
36]. In addition, the carbohydrate of this biomarker, which is labeled 2-2, is shown in
Figure 9 [
36]. The other glycosylated proteins for which the carbohydrate composition is shown are under investigation to increase our understanding of these masked glycosylated proteins.
Based on the carbohydrate composition of the fractions, the major components are the two inositols, sorbitol and mannitol are similar with variability in the hexose occurrence. It is important to be mindful these are the monosaccharide compositions of oligosaccharides which still contain peptide. Following incubation with rhGAA, all of them except 2-2 are converted increasing 2-2 and glucose. This suggests that glucose is a critical constituent even though it is quantitatively a minor constituent. At least some of the others appear to be converted to 2-2. However, they all contain peptide, but it is unknown if it is the same for all of them. Therefore, it is not possible to determine quantitative relationships. Based on more recent work, it is likely that there are multiple glycosylation sites with different monosaccharide composition involved.
The carbohydrate composition of these soluble glycosylated proteins is unique by consisting mainly of inositols and sorbitol with some iditol. Literature searches do not reveal any glycosylated proteins published with these as the major carbohydrate. In fact, a search does not reveal any publication of a glycosylated protein with sorbitol.
The biomarker glycosylated protein, found in normal individuals, is present in the serum of Pompe patients who are on ERT but it is not present in the serum of Pompe mice that are not on ERT as shown in
Figure 10. The Pompe mice not on ERT do have another glycosylated protein present in the serum which elutes at about 18 min retention time, which is not present in the serum of Pompe patients on ERT or normal individuals as shown in
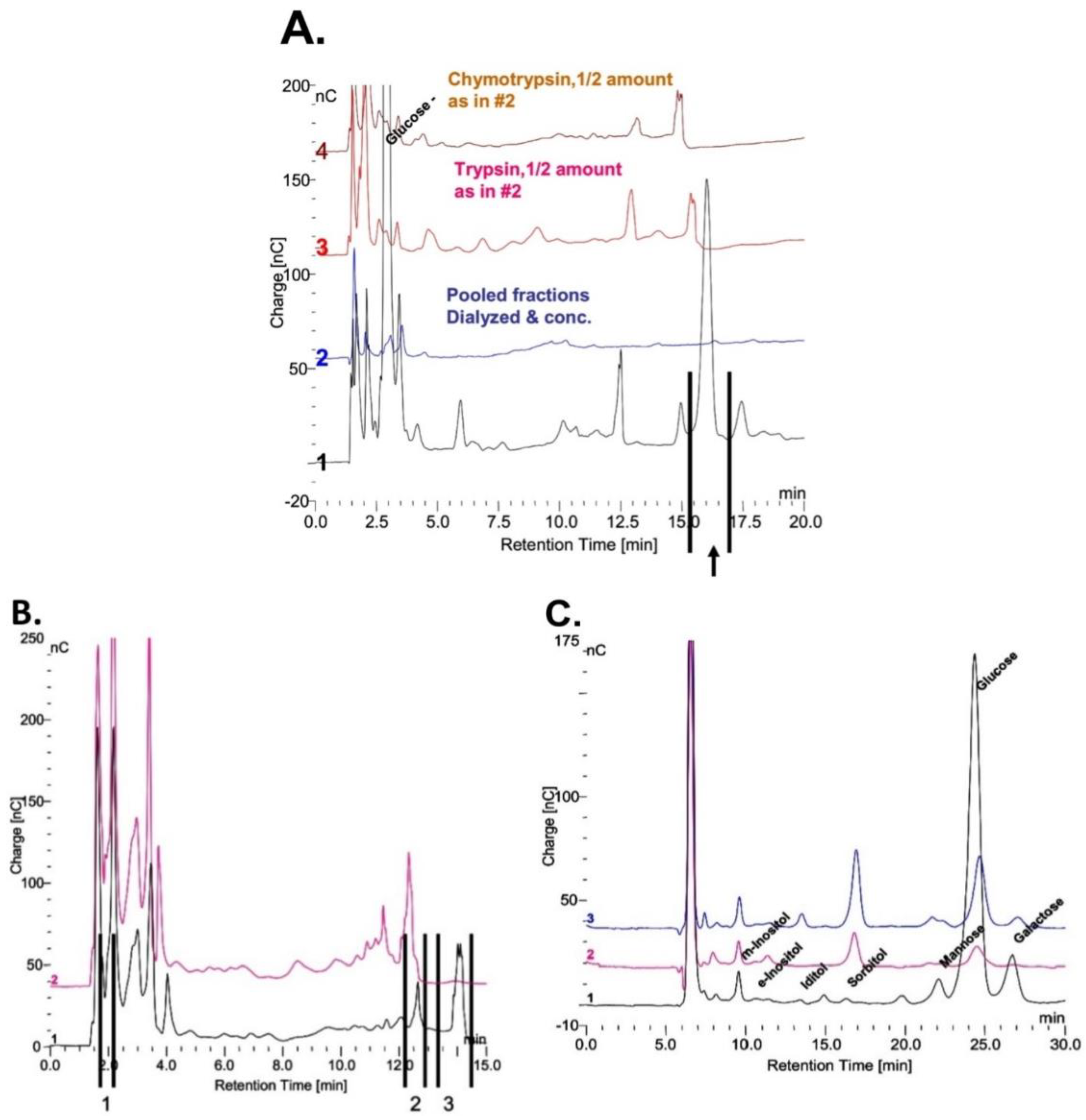
Figure 10. It is possible that his peak may be a biomarker for Pompe disease. This potential biomarker for Pompe disease was collected and incubated with trypsin and chymotrypsin as shown in
Figure 11A. The peaks were collected as shown in
Figure 11B, hydrolyzed in 4 N TFA and the monosaccharides are shown in the chromatograms in
Figure 11C. The result of this preliminary analysis is the demonstration that there are multiple glycosylation sites on the protein and that the monosaccharide composition of each glycosylation site is different. A similar result was obtained for the biomarker for GAA degradation. These results indicate that it will not be a simple task to determine the relationship between all of these glycosylated peptides.
The investigation of these glycosylated proteins is complicated by the fact that they the carbohydrate is masked and then unmasked by 0.1 N HCl at 100 °C then masked after dialysis against water. The biomarker at about 11 min and the other peak at 18 min in
Figure 10 are present after the HCl treatment. The five other oligosaccharides in
Figure 10 and the ones in
Figure 11 are all present after digestion with proteases. Therefore, it is not known just how many glycosylated proteins are involved. It could be as few as two and it is possible that they may be related since one is present when there is GAA activity in vivo and the other is present in the absence of GAA in vivo. The protein originates in glycogen, but it does not appear to be a known protein and the glycosylation is very unusual given the inositol, sorbitol and sometimes iditol.
The question of whether these in vivo fractions are intact components of glycogen or whether they have undergone some modification by GAA, or any other enzymes, is an open question since GAA does have glucanase, glucantransferase and glucosyltransferase activities under the same conditions in which it has glucosyl hydrolase activity [
7,
8,
9,
10]. As discussed earlier there is a commonly held belief that GAA only breaks glycogen down to glucose, but it breaks down glycogen to some oligosaccharides which then are later degraded to glucose as well as the glycosylated proteins.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}