


Chemical Versus Enzymatic Nucleic Acid Modifications and Genomic Stability



Abstract
{kind=link}
{kind=link}
{kind=link}
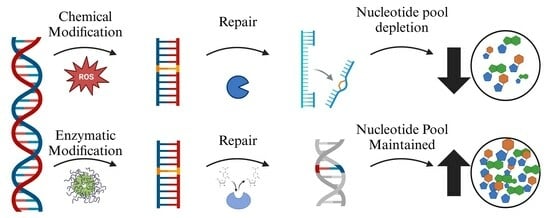
1. Introduction
1.1. The Fate of Chemically Modified Nucleic Acids and Repair Processes
1.2. Detection of Modified 2′Deoxy Nucleotides In Vivo
2. Chemical Modifications: Uncontrolled, Unregulated, and Devoid of Protein Inducers
2.1. Oxidation of Nucleotides
2.2. Halogenation of Nucleotides
2.3. Chemical Alkylation of Nucleobases Bases and DNA Crosslinks
2.4. Formation of Thymine Dimers
3. Enzyme-Driven Modification
3.1. Deaminated Ribonucleotides vs. Ribonucleotide Deamination
3.2. Signaling Alkylation: A Reversible Process Under Tight Regulation
3.3. The Aging Process and DNA Damage
4. Conclusions
Funding
Conflicts of Interest
References
- Broderick, K.; Moutaoufik, M.T.; Aly, K.A.; Babu, M. Sanitation enzymes: Exquisite surveillance of the noncanonical nucleotide pool to safeguard the genetic blueprint. Semin. Cancer Biol. 2023, 94, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Bebenek, K.; Roberts, J.D.; Kunkel, T.A. The effects of dNTP pool imbalances on frameshift fidelity during DNA replication. J. Biol. Chem. 1992, 267, 3589–3596. [Google Scholar] [CrossRef] [PubMed]
- Watt, D.L.; Buckland, R.J.; Lujan, S.A.; Kunkel, T.A.; Chabes, A. Genome-wide analysis of the specificity and mechanisms of replication infidelity driven by imbalanced dNTP pools. Nucleic Acids Res. 2016, 44, 1669–1680. [Google Scholar] [CrossRef] [PubMed]
- Mannava, S.; Moparthy, K.C.; Wheeler, L.J.; Natarajan, V.; Zucker, S.N.; Fink, E.E.; Im, M.; Flanagan, S.; Burhans, W.C.; Zeitouni, N.C.; et al. Depletion of Deoxyribonucleotide Pools Is an Endogenous Source of DNA Damage in Cells Undergoing Oncogene-Induced Senescence. Am. J. Pathol. 2013, 182, 142–151. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Moroz, O.V.; Wilson, K.S.; Murzin, A.G. House cleaning, a part of good housekeeping. Mol. Microbiol. 2006, 59, 5–19. [Google Scholar] [CrossRef]
- Nagy, G.N.; Leveles, I.; Vértessy, B.G. Preventive DNArepair by sanitizing the cellular (deoxy)nucleoside triphosphate pool. FEBS J. 2014, 281, 4207–4223. [Google Scholar] [CrossRef]
- Bilyard, M.K.; Becker, S.; Balasubramanian, S. Natural, modified DNA bases. Curr. Opin. Chem. Biol. 2020, 57, 1–7. [Google Scholar] [CrossRef]
- Gad, H.; Koolmeister, T.; Jemth, A.-S.; Eshtad, S.; Jacques, S.A.; Ström, C.E.; Svensson, L.M.; Schultz, N.; Lundbäck, T.; Einarsdottir, B.O.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar] [CrossRef]
- Gohil, D.; Sarker, A.H.; Roy, R. Base Excision Repair: Mechanisms and Impact in Biology, Disease, and Medicine. Int. J. Mol. Sci. 2023, 24, 14186. [Google Scholar] [CrossRef]
- Kemp, M.G.; Reardon, J.T.; Lindsey-Boltz, L.A.; Sancar, A. Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J. Biol. Chem. 2012, 287, 22889–22899. [Google Scholar] [CrossRef]
- Iyama, T.; Wilson, D.M. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjoras, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Krasikova, Y.; Rechkunova, N.; Lavrik, O. Nucleotide Excision Repair: From Molecular Defects to Neurological Abnormalities. Int. J. Mol. Sci. 2021, 22, 6220. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Raja, S.; Van Houten, B. The involvement of nucleotide excision repair proteins in the removal of oxidative DNA damage. Nucleic Acids Res. 2020, 48, 11227–11243. [Google Scholar] [CrossRef]
- Jacobs, A.L.; Schar, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef]
- Krokan, H.E.; Nilsen, H.; Skorpen, F.; Otterlei, M.; Slupphaug, G. Base excision repair of DNA in mammalian cells. FEBS Lett. 2000, 476, 73–77. [Google Scholar] [CrossRef]
- Fortini, P.; Pascucci, B.; Parlanti, E.; D’Errico, M.; Simonelli, V.; Dogliotti, E. The base excision repair: Mechanisms and its relevance for cancer susceptibility. Biochimie 2003, 85, 1053–1071. [Google Scholar] [CrossRef]
- Varma, S.J.; Calvani, E.; Gruning, N.M.; Messner, C.B.; Grayson, N.; Capuano, F.; Mulleder, M.; Ralser, M. Global analysis of cytosine and adenine DNA modifications across the tree of life. Elife 2022, 11, e81002. [Google Scholar] [CrossRef]
- Marchante-Gayon, J.M.; Nicolas Carcelen, J.; Potes Rodriguez, H.; Pineda-Cevallos, D.; Rodas Sanchez, L.; Gonzalez-Gago, A.; Rodriguez-Gonzalez, P.; Garcia Alonso, J.I. Quantification of modified nucleotides and nucleosides by isotope dilution mass spectrometry. Mass. Spectrom. Rev. 2024, 43, 998–1018. [Google Scholar] [CrossRef]
- Liu, Q.; Georgieva, D.C.; Egli, D.; Wang, K. NanoMod: A computational tool to detect DNA modifications using Nanopore long-read sequencing data. BMC Genom. 2019, 20, 78. [Google Scholar] [CrossRef]
- Liu, Q.; Fang, L.; Yu, G.; Wang, D.; Xiao, C.-L.; Wang, K. Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data. Nat. Commun. 2019, 10, 2449. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H. Understanding the Genetic Code. J. Bacteriol. 2019, 201, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Hayyan, M.; Hashim, M.A.; AlNashef, I.M. Superoxide Ion: Generation and Chemical Implications. Chem. Rev. 2016, 116, 3029–3085. [Google Scholar] [CrossRef] [PubMed]
- Jun, Y.W.; Kant, M.; Coskun, E.; Kato, T.A.; Jaruga, P.; Palafox, E.; Dizdaroglu, M.; Kool, E.T. Possible Genetic Risks from Heat-Damaged DNA in Food. ACS Cent. Sci. 2023, 9, 1170–1179. [Google Scholar] [CrossRef]
- Henderson, P.T.; Evans, M.D.; Cooke, M.S. Salvage of oxidized guanine derivatives in the (2’-deoxy)ribonucleotide pool as source of mutations in DNA. Mutat. Res. 2010, 703, 11–17. [Google Scholar] [CrossRef]
- Kamiya, H. Mutagenic potentials of damaged nucleic acids produced by reactive oxygen/nitrogen species: Approaches using synthetic oligonucleotides and nucleotides: Survey and Summary. Nucleic Acids Res. 2003, 31, 517–531. [Google Scholar] [CrossRef]
- Rudd, S.G.; Valerie, N.C.K.; Helleday, T. Pathways controlling dNTP pools to maintain genome stability. DNA Repair 2016, 44, 193–204. [Google Scholar] [CrossRef]
- Pai, C.C.; Kearsey, S.E. A Critical Balance: dNTPs and the Maintenance of Genome Stability. Genes 2017, 8, 57. [Google Scholar] [CrossRef]
- Kent, T.; Rusanov, T.D.; Hoang, T.M.; Velema, W.A.; Krueger, A.T.; Copeland, W.C.; Kool, E.T.; Pomerantz, R.T. DNA polymerase theta specializes in incorporating synthetic expanded-size (xDNA) nucleotides. Nucleic Acids Res. 2016, 44, 9381–9392. [Google Scholar] [CrossRef]
- Longley, M.J.; Nguyen, D.; Kunkel, T.A.; Copeland, W.C. The Fidelity of Human DNA Polymerase γ with and without Exonucleolytic Proofreading and the p55 Accessory Subunit. J. Biol. Chem. 2001, 276, 38555–38562. [Google Scholar] [CrossRef]
- Gedik, C.M.; Collins, A. Establishing the background level of base oxidation in human lymphocyte DNA: Results of an interlaboratory validation study. FASEB J. 2005, 19, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Miura, T.; Furuichi, M.; Tominaga, Y.; Tsuchimoto, D.; Sakumi, K.; Nakabeppu, Y. A genome-wide distribution of 8-oxoguanine correlates with the preferred regions for recombination and single nucleotide polymorphism in the human genome. Genome Res. 2006, 16, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, H. 2-Hydroxy-dATP is incorporated opposite G by Escherichia coli DNA polymerase III resulting in high mutagenicity. Nucleic Acids Res. 2000, 28, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, H.; Kasai, H. Formation of 2-hydroxydeoxyadenosine triphosphate, an oxidatively damaged nucleotide, and its incorporation by DNA polymerases. Steady-state kinetics of the incorporation. J. Biol. Chem. 1995, 270, 19446–19450. [Google Scholar] [CrossRef]
- Satou, K. Mutagenic effects of 2-hydroxy-dATP on replication in a HeLa extract: Induction of substitution and deletion mutations. Nucleic Acids Res. 2003, 31, 2570–2575. [Google Scholar] [CrossRef]
- Juan, C.A.; Perez de la Lastra, J.M.; Plou, F.J.; Perez-Lebena, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Ramana, K.V.; Srivastava, S.; Singhal, S.S. Lipid Peroxidation Products in Human Health and Disease. Oxidative Med. Cell. Longev. 2013, 2013, 583438. [Google Scholar] [CrossRef]
- Maddu, N. Diseases Related to Types of Free Radicals; IntechOpen: London, UK, 2019. [Google Scholar]
- Kanner, J. Dietary advanced lipid oxidation endproducts are risk factors to human health. Mol. Nutr. Food Res. 2007, 51, 1094–1101. [Google Scholar] [CrossRef]
- Davies, M.J. Protein oxidation and peroxidation. Biochem. J. 2016, 473, 805–825. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. Hypochlorite-induced damage to DNA, RNA, and polynucleotides: Formation of chloramines and nitrogen-centered radicals. Chem. Res. Toxicol. 2002, 15, 83–92. [Google Scholar] [CrossRef]
- Lao, V.V.; Herring, J.L.; Kim, C.H.; Darwanto, A.; Soto, U.; Sowers, L.C. Incorporation of 5-chlorocytosine into mammalian DNA results in heritable gene silencing and altered cytosine methylation patterns. Carcinogenesis 2009, 30, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Fedeles, B.I.; Freudenthal, B.D.; Yau, E.; Singh, V.; Chang, S.C.; Li, D.; Delaney, J.C.; Wilson, S.H.; Essigmann, J.M. Intrinsic mutagenic properties of 5-chlorocytosine: A mechanistic connection between chronic inflammation and cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4571–E4580. [Google Scholar] [CrossRef] [PubMed]
- Chancharoen, M.; Yang, Z.; Dalvie, E.D.; Gubina, N.; Ruchirawat, M.; Croy, R.G.; Fedeles, B.I.; Essigmann, J.M. 5-Chloro-2′-deoxycytidine Induces a Distinctive High-Resolution Mutational Spectrum of Transition Mutations In Vivo. Chem. Res. Toxicol. 2024, 37, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Loechler, E.L. A violation of the Swain-Scott principle, and not SN1 versus SN2 reaction mechanisms, explains why carcinogenic alkylating agents can form different proportions of adducts at oxygen versus nitrogen in DNA. Chem. Res. Toxicol. 1994, 7, 277–280. [Google Scholar] [CrossRef]
- Drablos, F.; Feyzi, E.; Aas, P.A.; Vaagbo, C.B.; Kavli, B.; Bratlie, M.S.; Pena-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Alkylation damage in DNA and RNA--repair mechanisms and medical significance. DNA Repair 2004, 3, 1389–1407. [Google Scholar] [CrossRef]
- Frischmann, M.; Bidmon, C.; Angerer, J.; Pischetsrieder, M. Identification of DNA adducts of methylglyoxal. Chem. Res. Toxicol. 2005, 18, 1586–1592. [Google Scholar] [CrossRef]
- Donald, W.K.; Pollock, R.E.; Weichselbaum, R.R.; Bast, R.C.; Gansler, T.S.; Holland, J.F.; Frei, E. Alkylating Agents. In Holland-Frei Cancer Medicine, 6th ed.; Holland-Frei, Ed.; BC Decker: Hamilton, ON, USA, 2003. [Google Scholar]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef]
- Klapacz, J.; Pottenger, L.H.; Engelward, B.P.; Heinen, C.D.; Johnson, G.E.; Clewell, R.A.; Carmichael, P.L.; Adeleye, Y.; Andersen, M.E. Contributions of DNA repair and damage response pathways to the non-linear genotoxic responses of alkylating agents. Mutat. Res. Rev. Mutat. Res. 2016, 767, 77–91. [Google Scholar] [CrossRef]
- Hamilton, J.T.; McRoberts, W.C.; Keppler, F.; Kalin, R.M.; Harper, D.B. Chloride methylation by plant pectin: An efficient environmentally significant process. Science 2003, 301, 206–209. [Google Scholar] [CrossRef]
- Soll, J.M.; Sobol, R.W.; Mosammaparast, N. Regulation of DNA Alkylation Damage Repair: Lessons and Therapeutic Opportunities. Trends Biochem. Sci. 2017, 42, 206–218. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.P.; Cho, Y.J.; Huang, H.; Kim, H.Y.; Kozekov, I.D.; Kozekova, A.; Wang, H.; Minko, I.G.; Lloyd, R.S.; Harris, T.M.; et al. Interstrand DNA cross-links induced by α,β-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc. Chem. Res. 2008, 41, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Kozekov, I.D.; Nechev, L.V.; Moseley, M.S.; Harris, C.M.; Rizzo, C.J.; Stone, M.P.; Harris, T.M. DNA interchain cross-links formed by acrolein and crotonaldehyde. J. Am. Chem. Soc. 2003, 125, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Housh, K.; Jha, J.S.; Haldar, T.; Amin, S.B.M.; Islam, T.; Wallace, A.; Gomina, A.; Guo, X.; Nel, C.; Wyatt, J.W.; et al. Formation and repair of unavoidable, endogenous interstrand cross-links in cellular DNA. DNA Repair 2021, 98, 103029. [Google Scholar] [CrossRef]
- Price, N.E.; Gates, K.S. Novel Processes Associated with the Repair of Interstrand Cross-Links Derived from Abasic Sites in Duplex DNA: Roles for the Base Excision Repair Glycosylase NEIL3 and the SRAP Protein HMCES. Chem. Res. Toxicol. 2024, 37, 199–207. [Google Scholar] [CrossRef]
- Donnellan, L.; Simpson, B.; Dhillon, V.S.; Costabile, M.; Fenech, M.; Deo, P. Methylglyoxal induces chromosomal instability and mitotic dysfunction in lymphocytes. Mutagenesis 2021, 36, 339–348. [Google Scholar] [CrossRef]
- Boiteux, S.; Guillet, M. Abasic sites in DNA: Repair and biological consequences in Saccharomyces cerevisiae. DNA Repair 2004, 3, 1–12. [Google Scholar] [CrossRef]
- Puyo, S.; Montaudon, D.; Pourquier, P. From old alkylating agents to new minor groove binders. Crit. Rev. Oncol. Hematol. 2014, 89, 43–61. [Google Scholar] [CrossRef]
- Arecco, A.; Mun, B.J.; Mathews, C.K. Deoxyribonucleotide pools as targets for mutagenesis by N-methyl-N-nitrosourea. Mutat. Res. Rev. Mutat. Res. 1988, 200, 165–175. [Google Scholar]
- Ikehata, H.; Ono, T. The Mechanisms of UV Mutagenesis. J. Radiat. Res. 2011, 52, 115–125. [Google Scholar] [CrossRef]
- Mitchell, D.L.; Jen, J.; Cleaver, J.E. Sequence specificity of cyclobutane pyrimidine dimers in DNA treated with solar (ultraviolet B) radiation. Nucleic Acids Res. 1992, 20, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Premi, S.; Han, L.; Mehta, S.; Knight, J.; Zhao, D.; Palmatier, M.A.; Kornacker, K.; Brash, D.E. Genomic sites hypersensitive to ultraviolet radiation. Proc. Natl. Acad. Sci. USA 2019, 116, 24196–24205. [Google Scholar] [CrossRef] [PubMed]
- Rauer, C.; Nogueira, J.J.; Marquetand, P.; Gonzalez, L. Cyclobutane Thymine Photodimerization Mechanism Revealed by Nonadiabatic Molecular Dynamics. J. Am. Chem. Soc. 2016, 138, 15911–15916. [Google Scholar] [CrossRef] [PubMed]
- Pathak, M.A.; Kramer, D.M.; Gungerich, U. Formation of thymine dimers in mammalian skin by ultraviolet radiation in vivo. Photochem. Photobiol. 1972, 15, 177–185. [Google Scholar] [CrossRef]
- Cannistraro, V.J.; Taylor, J.-S. Acceleration of 5-Methylcytosine Deamination in Cyclobutane Dimers by G and Its Implications for UV-Induced C-to-T Mutation Hotspots. J. Mol. Biol. 2009, 392, 1145–1157. [Google Scholar] [CrossRef]
- Tommasi, S.; Denissenko, M.F.; Pfeifer, G.P. Sunlight induces pyrimidine dimers preferentially at 5-methylcytosine bases. Cancer Res. 1997, 57, 4727–4730. [Google Scholar]
- Moirangthem, R.; Gamage, M.N.; Rokita, S.E. Dynamic accumulation of cyclobutane pyrimidine dimers and its response to changes in DNA conformation. Nucleic Acids Res. 2023, 51, 5341–5350. [Google Scholar] [CrossRef]
- Beukers, R.; Eker, A.P.; Lohman, P.H. 50 years thymine dimer. DNA Repair 2008, 7, 530–543. [Google Scholar] [CrossRef]
- Gessner, P.; Lum, J.; Frenguelli, B.G. The mammalian purine salvage pathway as an exploitable route for cerebral bioenergetic support after brain injury. Neuropharmacology 2023, 224, 109370. [Google Scholar] [CrossRef]
- Zhang, Q.; Tretyakova, N. Incorporation of inosine into DNA by human polymerase eta (Polη): Kinetics of nucleotide misincorporation and structural basis for the mutagenicity. Biochem. J. 2023, 480, 1479–1483. [Google Scholar] [CrossRef]
- Shapiro, R.; Pohl, S.H. The reaction of ribonucleotides with nitrous acid. Side products and kinetics. Biochemistry 1968, 7, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Kow, Y.W. Repair of deaminated bases in DNA. Free Radic. Biol. Med. 2002, 33, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Ventura, I.; Russo, M.T.; De Luca, G.; Bignami, M. Oxidized purine nucleotides, genome instability and neurodegeneration. Mutat. Res. 2010, 703, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Alseth, I.; Dalhus, B.; Bjoras, M. Inosine in DNA and RNA. Curr. Opin. Genet. Dev. 2014, 26, 116–123. [Google Scholar] [CrossRef]
- Sakumi, K.; Abolhassani, N.; Behmanesh, M.; Iyama, T.; Tsuchimoto, D.; Nakabeppu, Y. ITPA protein, an enzyme that eliminates deaminated purine nucleoside triphosphates in cells. Mutat. Res. 2010, 703, 43–50. [Google Scholar] [CrossRef]
- Cader, M.Z.; De Almeida Rodrigues, R.P.; West, J.A.; Sewell, G.W.; Md-Ibrahim, M.N.; Reikine, S.; Sirago, G.; Unger, L.W.; Iglesias-Romero, A.B.; Ramshorn, K.; et al. FAMIN Is a Multifunctional Purine Enzyme Enabling the Purine Nucleotide Cycle. Cell 2020, 180, 278–295.e223. [Google Scholar] [CrossRef]
- Iyer, L.M.; Zhang, D.; Rogozin, I.B.; Aravind, L. Evolution of the deaminase fold and multiple origins of eukaryotic editing and mutagenic nucleic acid deaminases from bacterial toxin systems. Nucleic Acids Res. 2011, 39, 9473–9497. [Google Scholar] [CrossRef]
- Gaded, V.; Anand, R. Nucleobase deaminases: A potential enzyme system for new therapies. RSC Adv. 2018, 8, 23567–23577. [Google Scholar] [CrossRef]
- Bass, B.L.; Nishikura, K.; Keller, W.; Seeburg, P.H.; Emeson, R.B.; O’Connell, M.A.; Samuel, C.E.; Herbert, A. A standardized nomenclature for adenosine deaminases that act on RNA. RNA 1997, 3, 947–949. [Google Scholar]
- Gao, Z.W.; Wang, X.; Zhang, H.Z.; Lin, F.; Liu, C.; Dong, K. The roles of adenosine deaminase in autoimmune diseases. Autoimmun. Rev. 2021, 20, 102709. [Google Scholar] [CrossRef]
- Parkman, R.; Weinberg, K.; Crooks, G.; Nolta, J.; Kapoor, N.; Kohn, D. Gene therapy for adenosine deaminase deficiency. Annu. Rev. Med. 2000, 51, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Cader, M.Z.; Boroviak, K.; Zhang, Q.; Assadi, G.; Kempster, S.L.; Sewell, G.W.; Saveljeva, S.; Ashcroft, J.W.; Clare, S.; Mukhopadhyay, S.; et al. C13orf31 (FAMIN) is a central regulator of immunometabolic function. Nat. Immunol. 2016, 17, 1046–1056. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Skon-Hegg, C.; Zhang, J.; Wu, X.; Sagolla, M.; Ota, N.; Wuster, A.; Tom, J.; Doran, E.; Ramamoorthi, N.; Caplazi, P.; et al. LACC1 Regulates TNF and IL-17 in Mouse Models of Arthritis and Inflammation. J. Immunol. 2019, 202, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Yla-Herttuala, S. ADA-SCID Gene Therapy Endorsed By European Medicines Agency For Marketing Authorization. Mol. Ther. 2016, 24, 1013–1014. [Google Scholar] [CrossRef]
- Cui, D.; Xu, X. DNA Methyltransferases, DNA Methylation, and Age-Associated Cognitive Function. Int. J. Mol. Sci. 2018, 19, 1315. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef]
- Jin, B.; Robertson, K.D. DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 2013, 754, 3–29. [Google Scholar] [CrossRef]
- Bird, J.G.; Basu, U.; Kuster, D.; Ramachandran, A.; Grudzien-Nogalska, E.; Towheed, A.; Wallace, D.C.; Kiledjian, M.; Temiakov, D.; Patel, S.S.; et al. Highly efficient 5′ capping of mitochondrial RNA with NAD+ and NADH by yeast and human mitochondrial RNA polymerase. eLife 2018, 7, e42179. [Google Scholar] [CrossRef]
- Kareta, M.S.; Botello, Z.M.; Ennis, J.J.; Chou, C.; Chedin, F. Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L. J. Biol. Chem. 2006, 281, 25893–25902. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Wang, Z.; Schramm, V.L. Human DNMT1 transition state structure. Proc. Natl. Acad. Sci. USA 2016, 113, 2916–2921. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Ehrenhofer-Murray, A.; Jurkowski, T.P.; Lyko, F.; Reuter, G.; Ankri, S.; Nellen, W.; Schaefer, M.; Helm, M. Mechanism and biological role of Dnmt2 in Nucleic Acid Methylation. RNA Biol. 2017, 14, 1108–1123. [Google Scholar] [CrossRef]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schubeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Peng, B.; Hurt, E.M.; Hodge, D.R.; Thomas, S.B.; Farrar, W.L. DNA hypermethylation and partial gene silencing of human thymine-DNA glycosylase in multiple myeloma cell lines. Epigenetics 2006, 1, 138–145. [Google Scholar] [CrossRef][Green Version]
- Riggs, A.D.; Xiong, Z. Methylation and epigenetic fidelity. Proc. Natl. Acad. Sci. USA 2004, 101, 4–5. [Google Scholar] [CrossRef]
- Howard, J.H.; Frolov, A.; Tzeng, C.W.; Stewart, A.; Midzak, A.; Majmundar, A.; Godwin, A.; Heslin, M.; Bellacosa, A.; Arnoletti, J.P. Epigenetic downregulation of the DNA repair gene MED1/MBD4 in colorectal and ovarian cancer. Cancer Biol. Ther. 2009, 8, 94–100. [Google Scholar] [CrossRef]
- Bochtler, M.; Kolano, A.; Xu, G.L. DNA demethylation pathways: Additional players and regulators. BioEssays 2017, 39, 1–13. [Google Scholar] [CrossRef]
- Carey, N.; Marques, C.J.; Reik, W. DNA demethylases: A new epigenetic frontier in drug discovery. Drug Discov. Today 2011, 16, 683–690. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Iwan, K.; Rahimoff, R.; Kirchner, A.; Spada, F.; Schroder, A.S.; Kosmatchev, O.; Ferizaj, S.; Steinbacher, J.; Parsa, E.; Muller, M.; et al. 5-Formylcytosine to cytosine conversion by C-C bond cleavage in vivo. Nat. Chem. Biol. 2018, 14, 72–78. [Google Scholar] [CrossRef]
- Schiesser, S.; Pfaffeneder, T.; Sadeghian, K.; Hackner, B.; Steigenberger, B.; Schroder, A.S.; Steinbacher, J.; Kashiwazaki, G.; Hofner, G.; Wanner, K.T.; et al. Deamination, oxidation, and C-C bond cleavage reactivity of 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxycytosine. J. Am. Chem. Soc. 2013, 135, 14593–14599. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.A.; Shen, L.; Yang, W.; Wolfenden, R. Three Pyrimidine Decarboxylations in the Absence of a Catalyst. Biochemistry 2017, 56, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-M. Detecting DNA hydroxymethylation: Exploring its role in genome regulation. BMB Rep. 2024, 57, 135–142. [Google Scholar] [CrossRef]
- Harman, D. The aging process. Proc. Natl. Acad. Sci. USA 1981, 78, 7124–7128. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.-G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef]
- Rai, P. Oxidation in the nucleotide pool, the DNA damage response and cellular senescence: Defective bricks build a defective house. Mutat. Res. 2010, 703, 71–81. [Google Scholar] [CrossRef]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortez, J.R.; Migaud, M.E. Chemical Versus Enzymatic Nucleic Acid Modifications and Genomic Stability. DNA 2025, 5, 19. https://doi.org/10.3390/dna5020019
Cortez JR, Migaud ME. Chemical Versus Enzymatic Nucleic Acid Modifications and Genomic Stability. DNA. 2025; 5(2):19. https://doi.org/10.3390/dna5020019
Chicago/Turabian StyleCortez, Jonathan R., and Marie E. Migaud. 2025. "Chemical Versus Enzymatic Nucleic Acid Modifications and Genomic Stability" DNA 5, no. 2: 19. https://doi.org/10.3390/dna5020019
APA StyleCortez, J. R., & Migaud, M. E. (2025). Chemical Versus Enzymatic Nucleic Acid Modifications and Genomic Stability. DNA, 5(2), 19. https://doi.org/10.3390/dna5020019