
Exploring the Roles of Different DNA Repair Proteins in Short Inverted Repeat Mediated Genomic Instability: A Pilot Study



Abstract
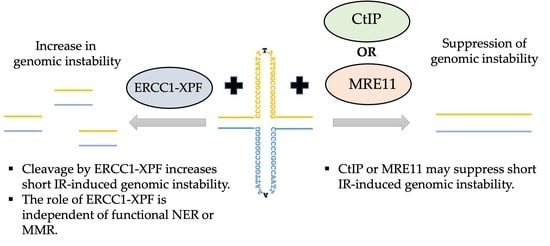
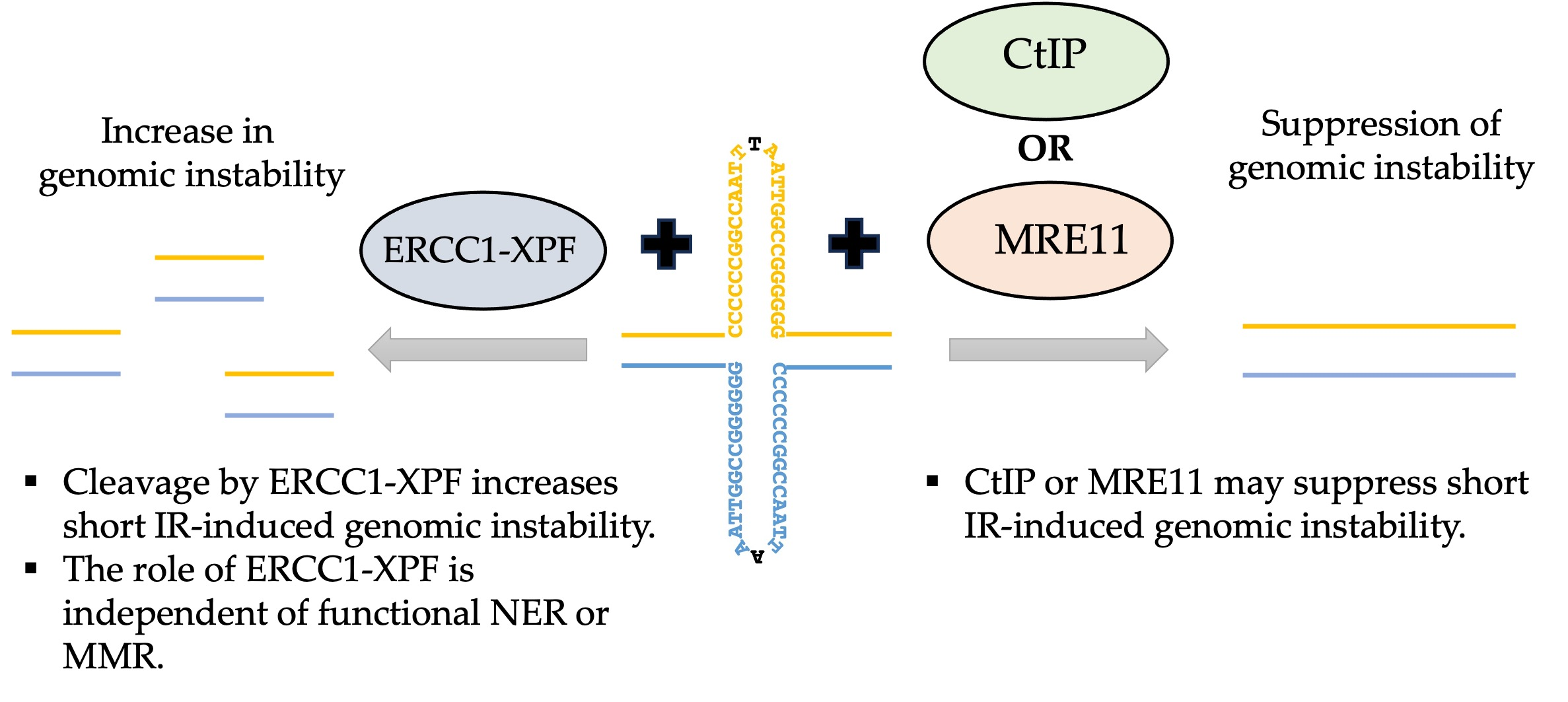
1. Introduction
2. Materials and Methods
2.1. Mutagenesis Assays in Mammalian Cell Lines
2.2. Cell Lines
2.3. siRNA-Mediated Knockdown of DNA Repair Proteins and Mutation Reporter Transfection
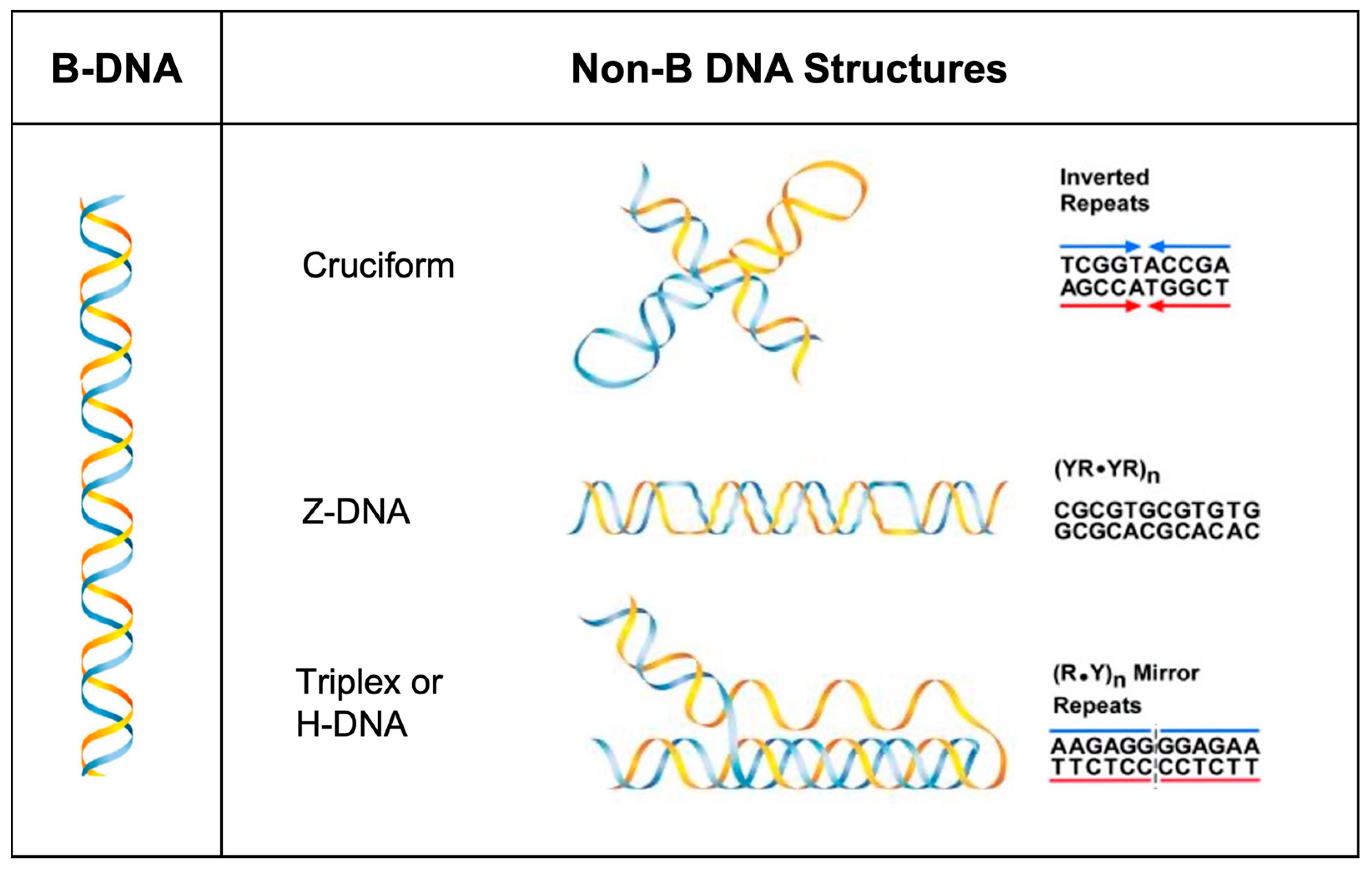
2.4. Mutation Frequency and Mutation Spectra Analyses
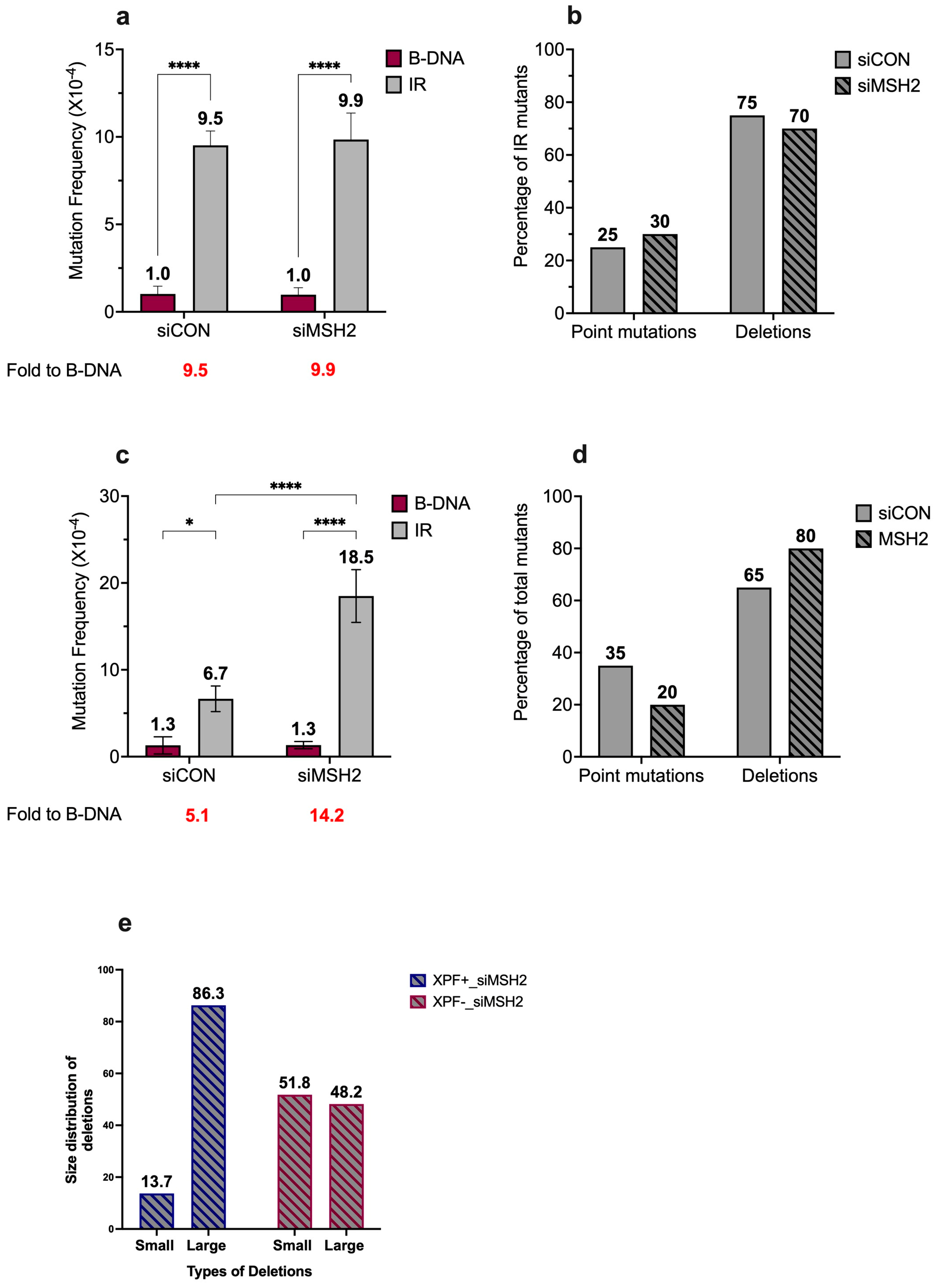
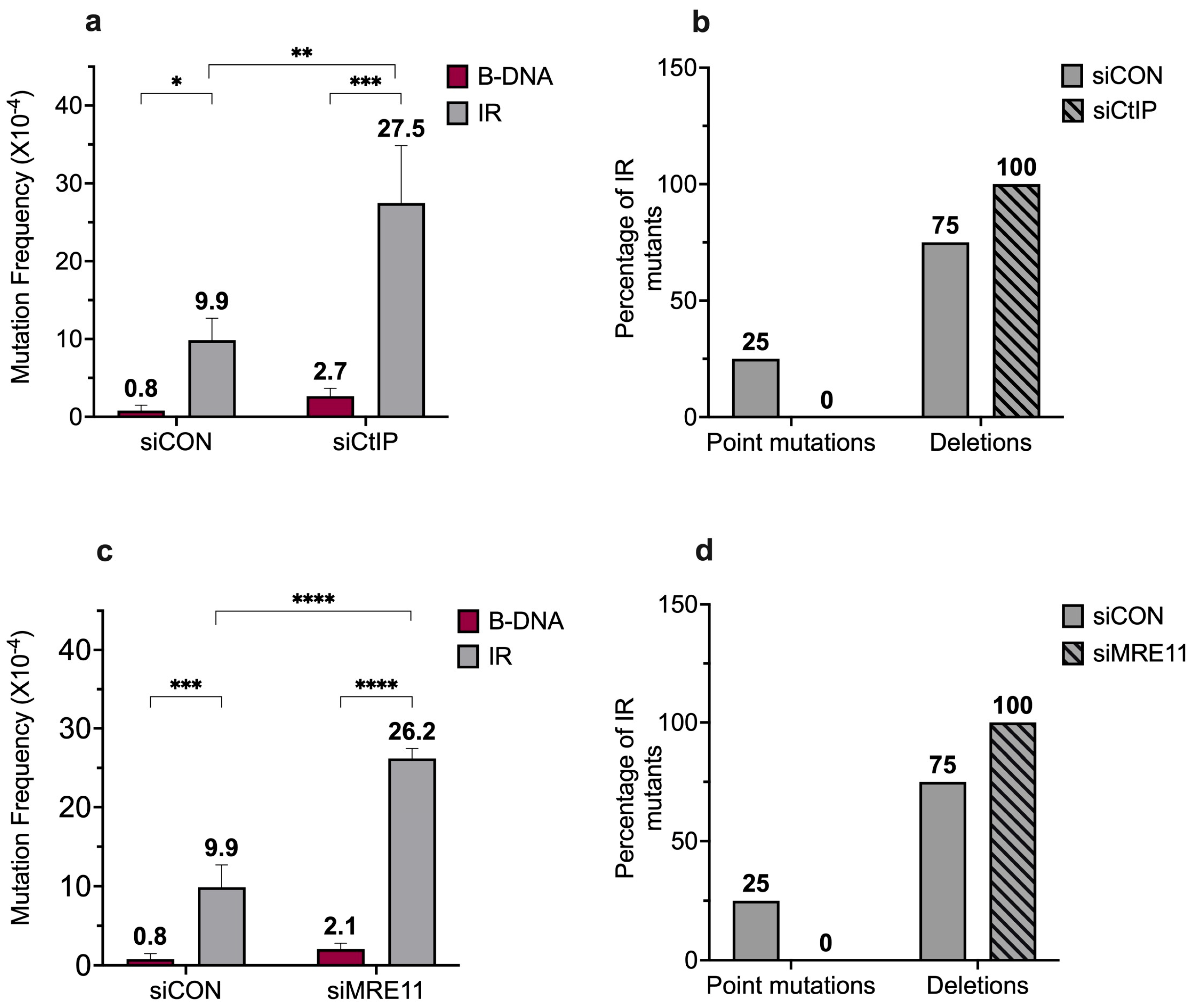
3. Results and Discussion
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sinden, R.R. DNA Structure and Function; Academic Press: San Diego, CA, USA, 1994. [Google Scholar]
- Wang, G.; Vasquez, K.M. Non-B DNA structure-induced genetic instability. Mutat. Res. 2006, 598, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, M.; Kaushik, S.; Roy, K.; Singh, A.; Mahendru, S.; Kumar, M.; Chaudhary, S.; Ahmed, S.; Kukreti, S. A bouquet of DNA structures: Emerging diversity. Biochem. Biophys. Rep. 2016, 5, 388–395. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, J.; Majima, T. Conformational changes of non-B DNA. Chem. Soc. Rev. 2011, 40, 5893–5909. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bacolla, A.; Wang, G.; Vasquez, K.M. Non-B DNA structure-induced genetic instability and evolution. Cell Mol. Life Sci. 2010, 67, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Vasquez, K.M. Effects of Replication and Transcription on DNA Structure-Related Genetic Instability. Genes 2017, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Vasquez, K.M. Impact of alternative DNA structures on DNA damage, DNA repair, and genetic instability. DNA Repair 2014, 19, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Vasquez, K.M. Dynamic alternative DNA structures in biology and disease. Nat. Rev. Genet. 2023, 24, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Wells, R.D. Non-B DNA conformations, genomic rearrangements, and human disease. J. Biol. Chem. 2004, 279, 47411–47414. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos-Soares, I.; Morganella, S.; Jain, N.; Hemberg, M.; Nik-Zainal, S. Noncanonical secondary structures arising from non-B DNA motifs are determinants of mutagenesis. Genome Res. 2018, 28, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos-Soares, I.; Chan, C.S.Y.; Ahituv, N.; Hemberg, M. High-throughput techniques enable advances in the roles of DNA and RNA secondary structures in transcriptional and post-transcriptional gene regulation. Genome Biol. 2022, 23, 159. [Google Scholar] [CrossRef]
- Bacolla, A.; Wojciechowska, M.; Kosmider, B.; Larson, J.E.; Wells, R.D. The involvement of non-B DNA structures in gross chromosomal rearrangements. DNA Repair 2006, 5, 1161–1170. [Google Scholar] [CrossRef]
- Wells, R.D. Non-B DNA conformations, mutagenesis and disease. Trends Biochem. Sci. 2007, 32, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Wells, R.D. Non-B DNA conformations as determinants of mutagenesis and human disease. Mol. Carcinog. 2009, 48, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Tainer, J.A.; Vasquez, K.M.; Cooper, D.N. Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic. Acids. Res. 2016, 44, 5673–5688. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M. DNA structures, repeat expansions and human hereditary disorders. Curr. Opin. Struct. Biol. 2006, 16, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, G.; Del Mundo, I.M.; McKinney, J.A.; Lu, X.; Bacolla, A.; Boulware, S.B.; Zhang, C.; Zhang, H.; Ren, P.; et al. Distinct Mechanisms of Nuclease-Directed DNA-Structure-Induced Genetic Instability in Cancer Genomes. Cell Rep. 2018, 22, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Carbajal, S.; Vijg, J.; DiGiovanni, J.; Vasquez, K.M. DNA structure-induced genomic instability in vivo. J. Natl. Cancer Inst. 2008, 100, 1815–1817. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Vasquez, K.M. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 13448–13453. [Google Scholar] [CrossRef] [PubMed]
- Weston-Hafer, K.; Berg, D.E. Limits to the role of palindromy in deletion formation. J. Bacteriol. 1991, 173, 315–318. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nasar, F.; Jankowski, C.; Nag, D.K. Long palindromic sequences induce double-strand breaks during meiosis in yeast. Mol. Cell Biol. 2000, 20, 3449–3458. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, G.; Bacolla, A.; Zhao, J.; Spitser, S.; Vasquez, K.M. Short Inverted Repeats Are Hotspots for Genetic Instability: Relevance to Cancer Genomes. Cell Rep. 2015, 10, 1674–1680. [Google Scholar] [CrossRef] [PubMed]
- McKinney, J.A.; Wang, G.; Mukherjee, A.; Christensen, L.; Subramanian, S.H.S.; Zhao, J.; Vasquez, K.M. Distinct DNA repair pathways cause genomic instability at alternative DNA structures. Nat. Commun. 2020, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Li, T.T.; Vasquez, K.M. Multi-Faceted Roles of ERCC1-XPF Nuclease in Processing Non-B DNA Structures. DNA 2022, 2, 231–247. [Google Scholar] [CrossRef]
- Parniewski, P.; Bacolla, A.; Jaworski, A.; Wells, R.D. Nucleotide excision repair affects the stability of long transcribed (CTG*CAG) tracts in an orientation-dependent manner in Escherichia coli. Nucleic. Acids. Res. 1999, 27, 616–623. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lin, Y.; Dion, V.; Wilson, J.H. Transcription promotes contraction of CAG repeat tracts in human cells. Nat. Struct. Mol. Biol. 2006, 13, 179–180. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wilson, J.H. Transcription-induced CAG repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol. Cell Biol. 2007, 27, 6209–6217. [Google Scholar] [CrossRef] [PubMed]
- Taghian, D.G.; Hough, H.; Nickoloff, J.A. Biased short tract repair of palindromic loop mismatches in mammalian cells. Genetics 1998, 148, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Owen, B.A.; Yang, Z.; Lai, M.; Gajec, M.; Badger, J.D., 2nd; Hayes, J.J.; Edelmann, W.; Kucherlapati, R.; Wilson, T.M.; McMurray, C.T. (CAG)(n)-hairpin DNA binds to Msh2-Msh3 and changes properties of mismatch recognition. Nat. Struct. Mol. Biol. 2005, 12, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Manley, K.; Shirley, T.L.; Flaherty, L.; Messer, A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat. Genet. 1999, 23, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.F. The Startling Role of Mismatch Repair in Trinucleotide Repeat Expansions. Cells 2021, 10, 1019. [Google Scholar] [CrossRef] [PubMed]
- Lahue, R.S. New developments in Huntington’s disease and other triplet repeat diseases: DNA repair turns to the dark side. Neuronal Signal. 2020, 4, NS20200010. [Google Scholar] [CrossRef] [PubMed]
- Mengoli, V.; Ceppi, I.; Sanchez, A.; Cannavo, E.; Halder, S.; Scaglione, S.; Gaillard, P.H.; McHugh, P.J.; Riesen, N.; Pettazzoni, P.; et al. WRN helicase and mismatch repair complexes independently and synergistically disrupt cruciform DNA structures. EMBO J. 2023, 42, e111998. [Google Scholar] [CrossRef] [PubMed]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Gellert, M. The 3’ to 5’ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, K.M.; Yuan, S.S.; Lee, E.Y.; Sung, P. Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J. Biol. Chem. 1998, 273, 21447–21450. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Gellert, M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999, 13, 1276–1288. [Google Scholar] [CrossRef] [PubMed]
- Lengsfeld, B.M.; Rattray, A.J.; Bhaskara, V.; Ghirlando, R.; Paull, T.T. Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol. Cell 2007, 28, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Andres, S.N.; Williams, R.S. CtIP/Ctp1/Sae2, molecular form fit for function. DNA Repair 2017, 56, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Y.; Truong, L.N.; Shi, L.Z.; Hwang, P.Y.; He, J.; Do, J.; Cho, M.J.; Li, H.; Negrete, A.; et al. CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol. Cell 2014, 54, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, J.; Vasquez, K.M. Methods to determine DNA structural alterations and genetic instability. Methods 2009, 48, 54–62. [Google Scholar] [CrossRef]
- Kraemer, K.H.; Seidman, M.M. Use of supF, an Escherichia coli tyrosine suppressor tRNA gene, as a mutagenic target in shuttle-vector plasmids. Mutat. Res. 1989, 220, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Ariza, R.R.; Roldan-Arjona, T.; Hera, C.; Pueyo, C. A method for selection of forward mutations in supF gene carried by shuttle-vector plasmids. Carcinogenesis 1993, 14, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Mandke, P. Elucidation of factors affecting short inverted repeat-induced genomic instability in eukaryotic systems. PhD Thesis, University of Texas at Austin, Austin, TX, USA, 2022. [Google Scholar]
- Levy, D.D.; Saijo, M.; Tanaka, K.; Kraemer, K.H. Expression of a transfected DNA repair gene (XPA) in xeroderma pigmentosum group A cells restores normal DNA repair and mutagenesis of UV-treated plasmids. Carcinogenesis 1995, 16, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Manandhar, M.; Lowery, M.G.; Boulware, K.S.; Lin, K.H.; Lu, Y.; Wood, R.D. Transcriptional consequences of XPA disruption in human cell lines. DNA Repair 2017, 57, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zacal, N.J.; Rainbow, A.J.; Zhu, X.D. XPF with mutations in its conserved nuclease domain is defective in DNA repair but functions in TRF2-mediated telomere shortening. DNA Repair 2007, 6, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, C.M.; Sancar, C. Mechanisms and Maps of Nucleotide Excision Repair. In DNA Damage, DNA Repair and Disease; Royal Society of Chemistry: London, UK, 2020; Volume 2, pp. 1–23. [Google Scholar] [CrossRef]
- Wood, R.D. Nucleotide excision repair in mammalian cells. J. Biol. Chem. 1997, 272, 23465–23468. [Google Scholar] [CrossRef]
- Zhao, J.; Jain, A.; Iyer, R.R.; Modrich, P.L.; Vasquez, K.M. Mismatch repair and nucleotide excision repair proteins cooperate in the recognition of DNA interstrand crosslinks. Nucleic. Acids. Res. 2009, 37, 4420–4429. [Google Scholar] [CrossRef] [PubMed]
- Young, S.J.; West, S.C. Coordinated roles of SLX4 and MutSbeta in DNA repair and the maintenance of genome stability. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 157–177. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.R.; Pluciennik, A.; Burdett, V.; Modrich, P.L. DNA mismatch repair: Functions and mechanisms. Chem. Rev. 2006, 106, 302–323. [Google Scholar] [CrossRef]
- Young, S.J.; Sebald, M.; Shah Punatar, R.; Larin, M.; Masino, L.; Rodrigo-Brenni, M.C.; Liang, C.C.; West, S.C. MutSbeta Stimulates Holliday Junction Resolution by the SMX Complex. Cell Rep. 2020, 33, 108289. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhang, R.; Wang, X.W.; Linke, S.P.; Sengupta, S.; Hickson, I.D.; Pedrazzi, G.; Perrera, C.; Stagljar, I.; Littman, S.J.; et al. The mismatch DNA repair heterodimer, hMSH2/6, regulates BLM helicase. Oncogene 2004, 23, 3749–3756. [Google Scholar] [CrossRef] [PubMed]
- Lobachev, K.S.; Rattray, A.; Narayanan, V. Hairpin- and cruciform-mediated chromosome breakage: Causes and consequences in eukaryotic cells. Front. Biosci. 2007, 12, 4208–4220. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, J.M.; Harper, J.W. GEN1/Yen1 and the SLX4 complex: Solutions to the problem of Holliday junction resolution. Genes Dev. 2010, 24, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Garner, E.; Kim, Y.; Lach, F.P.; Kottemann, M.C.; Smogorzewska, A. Human GEN1 and the SLX4-associated nucleases MUS81 and SLX1 are essential for the resolution of replication-induced Holliday junctions. Cell Rep. 2013, 5, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Brazda, V.; Laister, R.C.; Jagelska, E.B.; Arrowsmith, C. Cruciform structures are a common DNA feature important for regulating biological processes. BMC Mol. Biol. 2011, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, H.; Ohye, T.; Kogo, H.; Tsutsumi, M.; Kato, T.; Tong, M.; Emanuel, B.S.; Kurahashi, H. Two sequential cleavage reactions on cruciform DNA structures cause palindrome-mediated chromosomal translocations. Nat. Commun. 2013, 4, 1592. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, S.; Wollmuth, C.E.; Das, K.; Hile, S.E.; Regan, S.B.; Barnes, R.P.; Haouzi, A.; Lee, S.M.; House, N.C.M.; Guyumdzhyan, M.; et al. Sequence and Nuclease Requirements for Breakage and Healing of a Structure-Forming (AT)n Sequence within Fragile Site FRA16D. Cell Rep. 2019, 27, 1151–1164.e1155. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Kanagaraj, R.; Mihaljevic, B.; Schwendener, S.; Sartori, A.A.; Gerrits, B.; Shevelev, I.; Janscak, P. MRE11 complex links RECQ5 helicase to sites of DNA damage. Nucleic. Acids. Res. 2009, 37, 2645–2657. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Symington, L.S. DNA end resection: Many nucleases make light work. DNA Repair 2009, 8, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Caron, P.; Legube, G.; Paull, T.T. Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic. Acids. Res. 2014, 42, e19. [Google Scholar] [CrossRef] [PubMed]
- Moreau, S.; Morgan, E.A.; Symington, L.S. Overlapping functions of the Saccharomyces cerevisiae Mre11, Exo1 and Rad27 nucleases in DNA metabolism. Genetics 2001, 159, 1423–1433. [Google Scholar] [CrossRef]
- Lam, A.F.; Krogh, B.O.; Symington, L.S. Unique and overlapping functions of the Exo1, Mre11 and Pso2 nucleases in DNA repair. DNA Repair 2008, 7, 655–662. [Google Scholar] [CrossRef] [PubMed]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef] [PubMed]
- McKinney, J.A.; Wang, G.; Vasquez, K.M. Distinct mechanisms of mutagenic processing of alternative DNA structures by repair proteins. Mol. Cell Oncol. 2020, 7, 1743807. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Concentration | Company |
|---|---|---|
| MSH2 | 1:100 | Calbiochem (San Diego, CA, USA) (#PC57) |
| CtIP | 1:1000 | Cell Signaling Technology (Danvers, MA, USA) (#9201) |
| MRE11 | 1:1000 | Novus Biologicals (Centennial, CO, USA) (NB100-142) |
| Beta-actin | 1:5000 | Abcam (Cambridge, UK) (ab8227) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandke, P.; Vasquez, K.M. Exploring the Roles of Different DNA Repair Proteins in Short Inverted Repeat Mediated Genomic Instability: A Pilot Study. DNA 2024, 4, 141-153. https://doi.org/10.3390/dna4020008
Mandke P, Vasquez KM. Exploring the Roles of Different DNA Repair Proteins in Short Inverted Repeat Mediated Genomic Instability: A Pilot Study. DNA. 2024; 4(2):141-153. https://doi.org/10.3390/dna4020008
Chicago/Turabian StyleMandke, Pooja, and Karen M. Vasquez. 2024. "Exploring the Roles of Different DNA Repair Proteins in Short Inverted Repeat Mediated Genomic Instability: A Pilot Study" DNA 4, no. 2: 141-153. https://doi.org/10.3390/dna4020008
APA StyleMandke, P., & Vasquez, K. M. (2024). Exploring the Roles of Different DNA Repair Proteins in Short Inverted Repeat Mediated Genomic Instability: A Pilot Study. DNA, 4(2), 141-153. https://doi.org/10.3390/dna4020008