Abstract
Thus far, in 2021, 219 countries with over 175 million people have been infected by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). SARS-CoV-2 is a positive sense, single-stranded RNA virus, and is the causal agent for coronavirus disease (COVID-19). Due to the urgency of the situation, virtual screening as a computational modeling method offers a fast and effective modality of identifying drugs that may be effective against SARS-CoV-2. There has been an overwhelming abundance of molecular docking against SARS-CoV-2 in the last year. Due to the massive volume of computational studies, this systematic review has been created to evaluate and summarize the findings of existing studies. Herein, we report on computational articles of drugs which target, (1) viral protease, (2) Spike protein-ACE 2 interaction, (3) RNA-dependent RNA polymerase, and (4) other proteins and nonstructural proteins of SARS-CoV-2. Based on the studies presented, there are 55 identified natural or drug compounds with potential anti-viral activity. The next step is to show anti-viral activity in vitro and translation to determine effectiveness into human clinical trials.
1. Introduction
Coronavirus disease (COVID-19), which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), was reported in Wuhan, Hubei Province, China on 17 November 2019, and was designated as a risk to public health on 30 January 2020 [1,2,3]. On 11 March 2020, the World Health Organization declared COVID-19 a pandemic [4]. COVID-19 follows an extremely heterogeneous route, where many patients are either asymptomatic or experience moderate signs of illness, while others experience an aggressive and rapid progression of the disease, in some cases leading to collapsed lungs and multi-organ failure [5]. At the time of writing, SARS-CoV-2 has been detected in almost all countries, with over 170 million confirmed cases and over 3.9 million deaths [6]. Thus far, some vaccines have been developed by different institutes, but there are controversies on the side effects, which result in hesitancy in vaccinations, in addition to the limitations in vaccinations between countries; thus, until the deterministic eradication, prolonged physical distancing, strict hygiene measures and expanded hospital capacity are necessary to halt the spread [7].
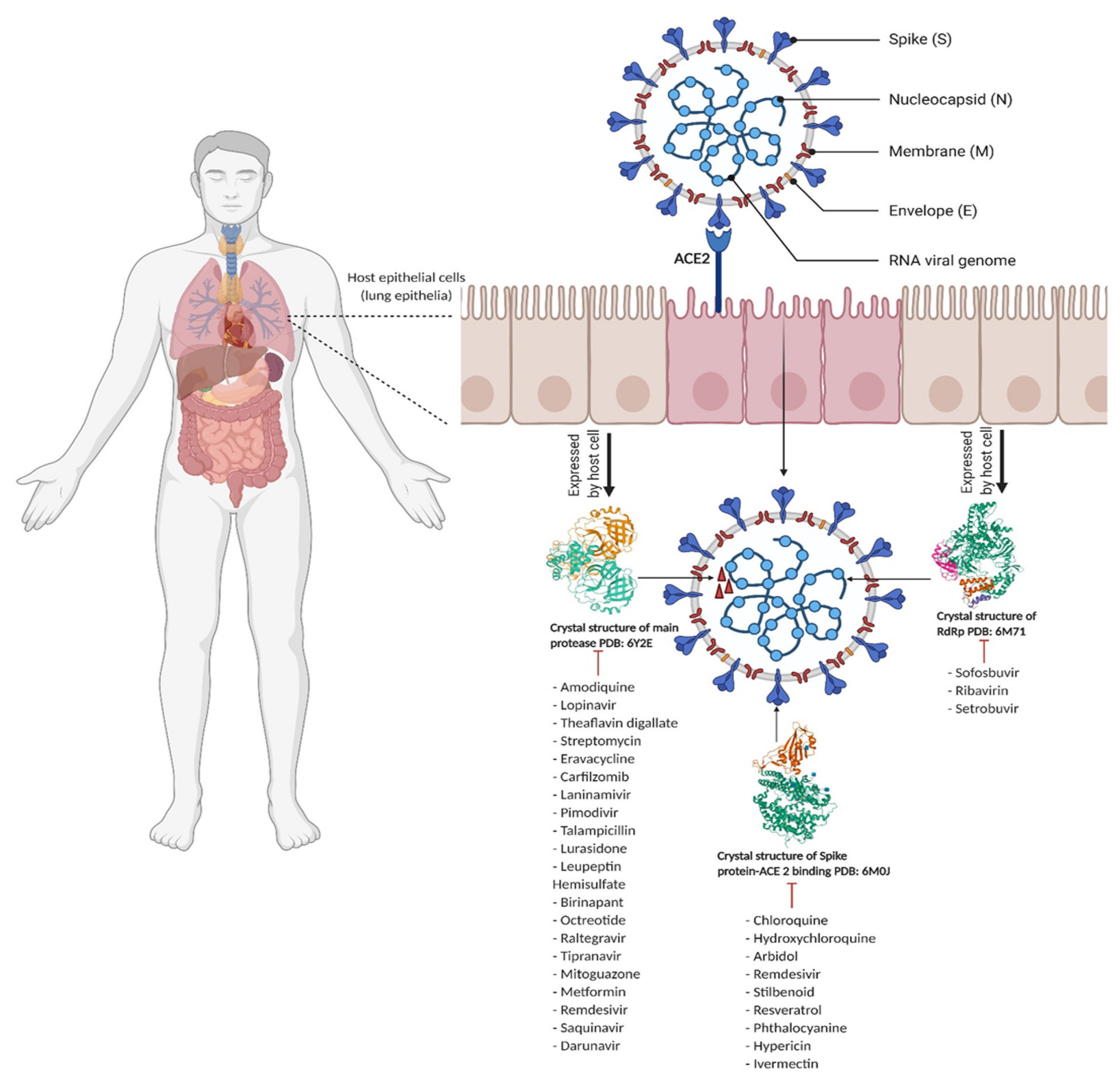
The Coronaviridae family comprises enveloped, single-stranded RNA and positive sense viruses. In the 1960s, human coronaviruses were reported in patients with the common cold [8]. Until now, α, β, γ and σ have been identified as Coronaviridae’s four genera. Both α coronavirus (NL63 and HCoV-229E) and the β coronavirus (SARS-CoV, HCoV-OC43, HCoV-HKU1 and MERS-CoV), have been detected in humans [9]. Virus genome sequencing results could detect β-CoV strain in all of five patients [10]. The SARS-CoV, MERS-CoV and SARS-CoV-2 phylogenetic tree is presented by Li et al. [11]. The SARS-CoV-2 virus contains four structural proteins: (1) Spike protein (S), (2) Envelope protein (E), (3) Nucleocapsid protein (N) and (4) Membrane protein (M) (Figure 1) [12]. In all types of emerged coronaviruses, the Spike protein appears as a “crown” [13]. Spike proteins act as mediators for the attachment of the virus to the host cell, and the fusion of host cell membranes with virion particles [14]. Both SARS-CoV and SARS-CoV-2 bind to angiotensin-converting enzyme 2 (ACE2) [15,16]. In ACE2–Spike protein fusion, the S protein is cleaved via proteolytic mechanisms into two subunits: S1 and S2 [17,18]. Each structural protein plays a significant role in the survival and life cycle of the virus, and among other proteins, the Spike protein serves major viral functions [19,20] and is the main target for vaccines for generation of neutralizing antibodies [21]. The Coronaviridae family Envelope protein (E) is composed of 76–109 amino acids, which vary from 8.4 to 12 kilodalton (kDa) in size [22]. The E protein is an integral membrane protein, and its structure is formed by hydrophilic terminals comprising 7–12 amino acids, a hydrophobic transmembrane domain of 25 amino acids, followed by a long hydrophilic carboxyl region [23]. The E protein is involved in protein pathogenesis and assembly, which can be migrated by ion channels [24]. One of the essential proteins in viral assembly and transcription is the ribonucleoprotein complex. In fact, in MERS-CoV, the genome is packed in N protein [25]. In SARS-CoV-2, 30,000 nucleotides build the virus genome [10]. During coronavirus infection in the host, of the four structural proteins, N protein is the most immunogenic [26], and so it is frequently targeted by vaccines and drugs. In fact IgM and IgG antibodies have been found against the N protein in recovered COVID-19 patients [27]. The N protein contains two domains: (1) the N-terminal and (2) the C-terminal domains which can bind to viral RNA genomes. In addition, the N protein is highly disordered and positively charged, which gives the opportunity to N protein for binding to nonspecific nucleic protein [26]. In addition to the structural proteins of SARS-CoV-2, there are a number of non-structural proteins such as the main protease (3CLpro, Mpro, Nsp5), Papin Like protease (PLpro, Nsp3) and RNA-dependent RNA polymerase (RdRp), which are necessary for viral replication and survival within the host [28], and as such, are targeted by drugs for anti-viral effects [29].
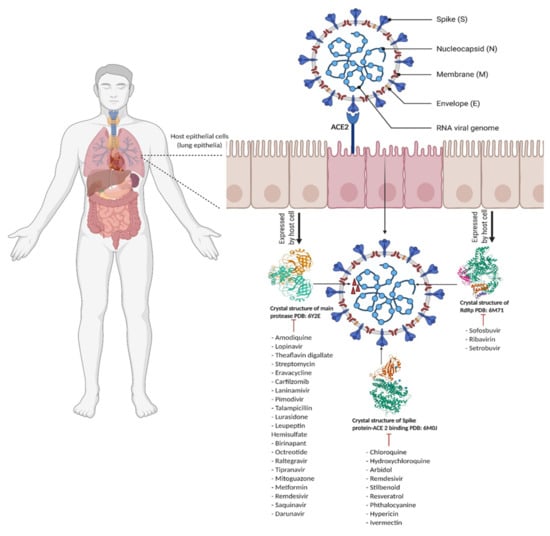
Figure 1.
Coronavirus entry process showing the Spike protein-ACE2 enzyme binding site and list of drugs that inhibit ACE2 mediated pathway (Created with Biorender.com) [45,46,47].
A report in 2019 demonstrated that the development of a human vaccine requires at least USD 200 million [30]. Given the current critical situation, drug repurposing can be a smart way to fast track drugs for human use whilst discovering new and novel effective therapeutic treatments [31]. In this way, various well-known drugs have already been used in clinical trials, including Azithromycin (an antibiotic) [32], Hydroxychloroquine (an anti-malarial drug) [33] and Remdesivir (an antiviral against Ebola) [34]. However, based on the clinical reports, most of them have disappointing results [35]. In order to alleviate the effects of the dire situation, as well finding a treatment, computational methods are being utilized as they are both cost-effective and fast [36]. Protein 3D structure is imperative for computational drug repurposing. The function of proteins is determined by their structural and chemical properties [37]. Three-dimensional structures of proteins can be predicted by either computational methods such as homology modeling, threading methods and ab initio, or can be evaluated via experimental methods such as protein x-ray crystallography, neutron diffraction, Cryogenic electron microscopy (Cryo-EM) and nuclear magnetic resonance (NMR) [38]. Computational, determination and prediction methods have been developed for protein structure determination, leading to accurate protein and protein–ligand complex structures. Permeation of computational strategies to various aspects of drug repurposing is possible by these advances [39]. As we can see, in comparison between virtual screening (VS), as a computational strategy, and experimental method high-throughput screening (HTS), VS is advantageous due to being a more direct, effective screening and cost effective [40,41]. VS can be divided into two methods: structure-based and ligand-based. In the case of unknown structural information for target, but known active ligand, the ligand-based method such as quantitative structure activity relationship (QSAR) method can be utilized. The up-side, when active site and structure of the target is available, molecular docking has been used since the early 1980s [42,43]. Molecular docking intends to predict the structure of a ligand within the constraint of a receptor active site, and, calculate the binding affinity or strength of ligand-target [44]. Molecular docking process passes through two interrelated steps: first sampling algorithm, then scoring function.
Due to numerous conformational degrees of freedom of protein and ligand, it would be an enormous number of binding modes between ligand and structure, and consequently too expensive to computationally afford. Thus, in order to relive the matter, various sampling algorithms with pros and cons have been developed such as: Matching Algorithms (MA) [48], Incremental Construction (IC) [49], Multiple Copy Simultaneous Search (MCSS) [50], Monte Carlo (MC) [51], Genetic Algorithm (GA) [52] and Molecular Dynamics (MD) [53]. At the next step, the correct poses are delineated from incorrect, as well binded molecules to inactive compounds are omitted by four scoring functions: force-field-based, empirical, knowledge-based and hybrid [54]. From a methodologies stand point, docking can be classified into three methods: rigid ligand and rigid receptor, which is the most simple method with markedly error; flexible ligand and rigid receptor, which is more accurate and widely used; and flexible ligand and flexible receptor docking, which needs high requirements because of a heavy computational burden [55].
Applying computational methods including docking and molecular dynamics, drugs can be repurposed for use in patients with COVID-19 [56]. Ever since the SARS-CoV-2 structure was characterized in February 2020 [57], various computational investigations have been conducted for drug repurposing, targeting both the structural and non-structural proteins of SARS-CoV-2. Due to the huge volume of computational studies published in the last months, we have identified and classified the papers into four subsets: (1) viral protease target, (2) Spike protein–ACE2 enzyme target, (3) RNA-dependent RNA polymerase (RdRp) target and (4) other proteins and non-structural proteins of SARS-CoV-2. In each subset, a summary of the computational studies is presented, identification of potential natural and drug compounds described and an overview of the need for further studies presented. Repurposing drugs already used safely for other diseases is a potential step towards the prevention or treatment of COVID-19 and a potential return to normality worldwide.
2. Methodology
2.1. Study Selection
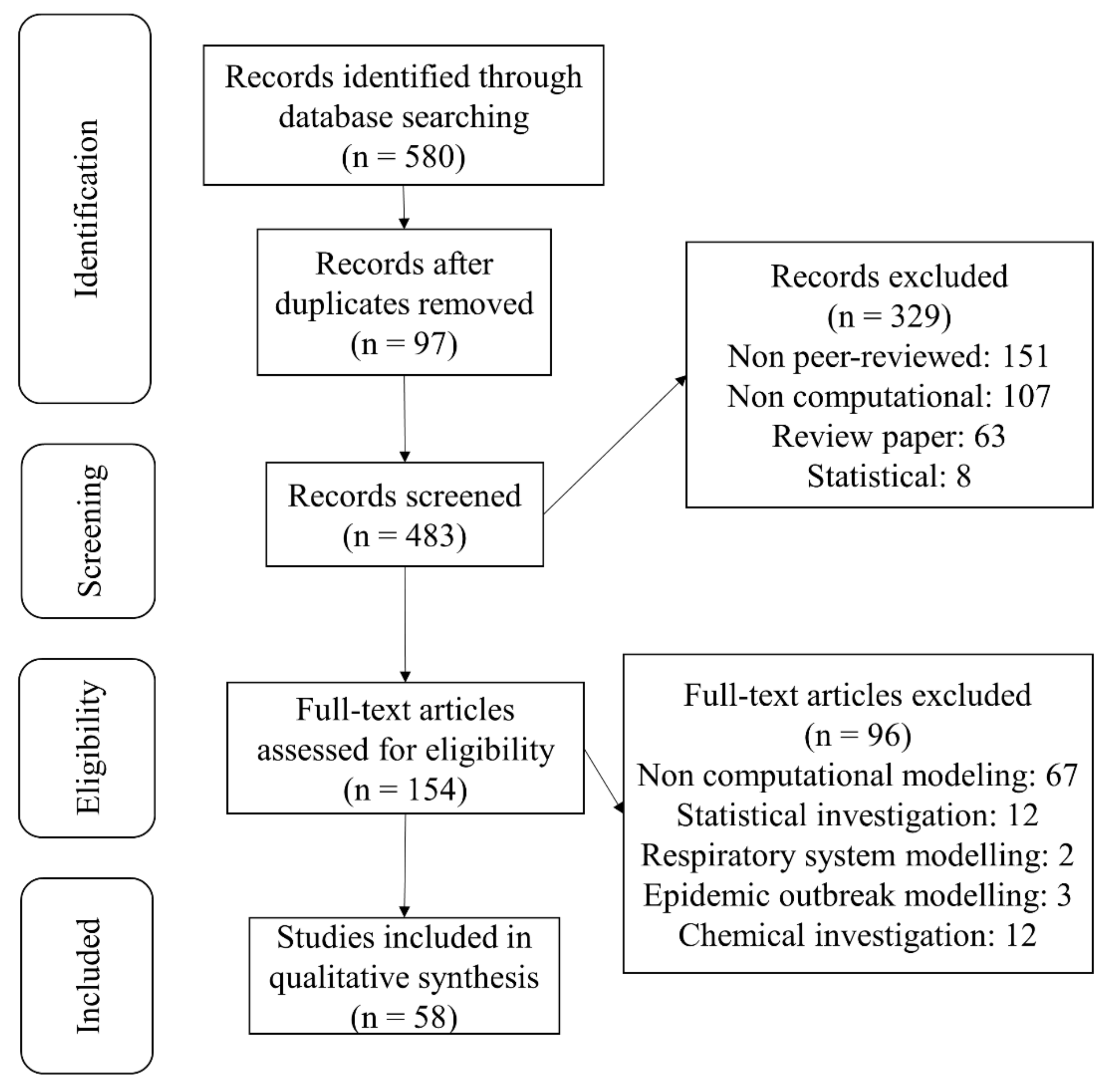
Herein, the principles of review preparation [58] regarding the role of computational modeling, specifically virtual screening in drug development against SARS-CoV-2 virus, were followed. This review classifies the various natural and drug compounds docked against different ingredients of SARS-CoV-2 to aid in confusion avoidance, also introducing potential conformers to researchers and allow effective methods and investigations. Research publication databases Scopus, PubMed, Medline, Google Scholar and Embase were searched, up to the 9th of May and including 2021, using appropriate terms related to SARS-CoV-2 computational modeling. All of the identified papers were in the English language and were published in peer-reviewed journals. The terms for screening of the considered field of study contain a term for coronavirus (“SARS-CoV-2” OR “corona” OR “coronavirus” OR “COVID-19” OR “anti-COVID-19” OR “anti-coronavirus”), a term for describing the drug development (“drug” OR “anti-viral” OR “drug development” OR “drug repurposing”) and a term for describing computational modeling (“computational modeling” OR “molecular dynamics” OR “molecular docking” OR “virtual screening”). The papers that qualified according to review standards were selected and reviewed. The PRISMA flow diagram and respective information, including the total number of identified papers and those excluded at different stages, is shown in Figure 2.
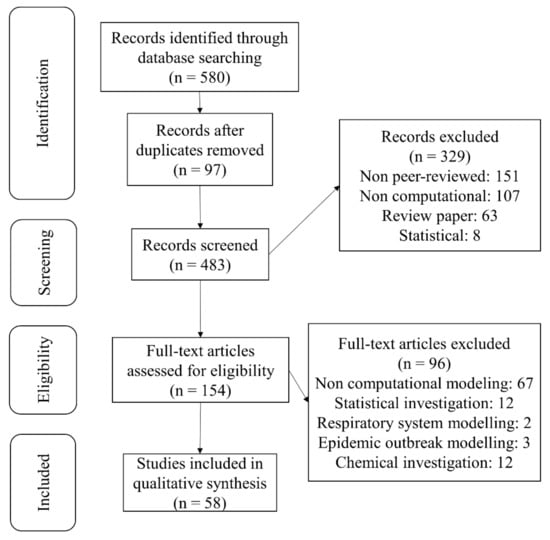
Figure 2.
PRISMA flow diagram showing the summary of search strategy and paper exclusion. A total of 580 studies were identified by searching databases. By going through the review steps, 58 original articles published in peer-reviewed journals are included regarding computational modeling of SARS-CoV-2.
2.2. Data Extraction
Based on the pathogenesis of SARS-CoV-2, there are 6 steps in the viral process: entry, primary replication, spread, secondary replication, secondary viremia and reached target organs [24,59]. The virus at the ‘reached target organs’ stage disrupts the organs and causes severe complications.
Inclusion criteria included papers that used computational studies with the following targets of SARS-CoV-2, (1) the main protease, (2) the spike protein-ACE2 interface, (3) RNA-dependent RNA polymerase (RdRp) and (4) other proteins and non-structural proteins of SARS-CoV-2. Exclusion criteria at the screening step included: non peer-reviewed, non-computational, review papers and statistical epidemiology. Following the research paper identification step, all abstracts were screened, and 329 papers were excluded at this screening step. After abstract screening at the identification stage, 483 papers were screened as full text, and 154 papers were found to be eligible for selection.
3. Computational Studies of Key SARS-CoV-2 Viral Proteins
3.1. Viral Proteases
The crystal structure of main protease and Papain Like protease can provide a basis for computational modeling and drug development [60]. The x-ray structure of main protease of SARS-CoV-2 and also the complex of main protease with an α-ketoamide inhibitor is reported [45]. As the first step, a peptidomimetic α-ketoamides of alphacoronavirus main protease were designed and synthesized [61]. At the next step, by incorporating the P3–P2 amide bond into a pyridine ring, the compound half-life in the plasma is enhanced [45]. The main function of the protease is to break up the polyproteins into non-structural proteins (Nsp) [62]. In the endoplasmic reticulum of the host cell, Nsps assemble and produce membranes as viral RNA synthesis sites [63]. Although the viral replication process is unquestionable [64], their defined role in viral replication is not clear [65,66]. Data on viral proteases can lead to a greater understanding of viral cleavage mechanisms [67], which is why they have been considered for computational modeling studies. Thus far, 33 molecular docking studies on the ligand-proteases docking by different sampling algorithms and scoring functions have been reported (Table 1); and for each study, promising candidate ligands, which are chosen based on the molecular docking and validated docking results by molecular dynamics simulation, are reported.

Table 1.
Summary of viral protease targets using virtual screening to identify potential repurposed FDA-approved drugs and natural compounds.
3.2. Spike Protein-ACE2 Enzyme Target
The Spike protein plays a key role in viral entry into host cells and in the pathogenesis of COVID-19. SARS-CoV-2 enters host cells by binding to ACE2 receptors [15,16,19,20]. At first, it was believed that ACE2 was an angiotensin conversion catalyzer in the endothelial cells of the kidney and heart [101,102]. Subsequently, however, the function of ACE2 in binding to SARS-CoV was discovered [103]. ACE2 can transport amino acids, whilst also serving as an important receptor for catalytic activities within the Coronaviridae family [15,104]. There are 380 amino acid differences between SARS-CoV and SARS-CoV-2, of which 5 amino acids are different between Spike protein-ACE2 binding interface [105]. As the Spike protein is regarded as a golden key at the viral entry stage, this protein has become a main target for drug repurposing. Both SARS-CoV-2 and SARS-CoV bind to ACE2 as a cell receptor and their binding mode is almost identical [106,107]. The crystal structure of Spike protein—ACE2 complex was crucial in identifying key amino acids in the interaction [46]. As a result, 13 computational studies have been published in the last three months (Table 2), screening for drugs and compounds, in order to block the ACE2 or the S protein–ACE2 complex.
Table 2.
Computational studies of spike protein or spike protein–ACE2 enzyme interface targeting of FDA re-purposed drugs and natural compounds.
3.3. RNA-Dependent RNA Polymerase Enzyme Target
RNA-dependent RNA polymerase (RdRp) is an enzyme that functions as a catalyzer in the RNA replication process within a virus life cycle [119]. In SARS-CoV-2, the non-structural protein 12 (Nsp12) is known as RdRp, and is associated with the non-structural protein 7 (Nsp7) and non-structural protein 8 (Nsp8), thus playing a key role in SARS-CoV-2 virus replication [120]. The structure of Nsp12 was demonstrated via cryo-electron microscopy [47]. It was noted that Nsp12 in SARS-CoV and SARS-CoV-2 possesses structural homology [121]. Remdesivir, Favipiravir and Ribavirin were shown to be potential drug candidates targeting RdRp protein [122]. Currently, there are five molecular docking against on RdRp and nucleocapsid protein as targets for drug screening (Table 3). Effects of anti-hepatitis C virus inhibitors on RdRp were investigated by Abdo Elifiky [123]. Through molecular dynamic and docking simulations, the binding energy of approved drugs and those against MERS and SARS-CoV were calculated and compared. As a result, Remdesivir, IDX-184, Sofosbuvir and Ribavitin are considered as potent drugs against SARS-CoV-2 [123]. Additionally, by molecular docking simulations, Tenofovir and Golidesivir were added to the list of anti-RdRp drug candidates [124].
Table 3.
Screening of FDA-repurposed drugs and natural compounds for RNA-dependent RNA polymerase enzyme target.
3.4. Other Proteins and Nonstructural Proteins of SARS-CoV-2
In relation to other proteins of SARS-CoV-2 including Nucleocapsid (N) protein, Envelope (E) protein and Membrane (M) protein and nonstructural proteins including Nsp7, Nsp8, Nsp10, Nsp13, Nsp14, Nsp15 and Nsp16, there have been nine computational studies published thus far that pass through our criteria. Among the 16 Nsps, Endoribonuclease (Nsp15) is considered the main viral interferon antagonist [128] and plays an important role in RNA processing [129]. The catalytic site of Nsp15 contains one lysine residue and two histidine residues and is structurally similar with RNase A [130,131]. By computational modelling on Nsp1, 2300 FDA approved drugs were repurposed, and Edoxudine and Remdesivir were identified as potential drugs. In addition, some natural small molecules including Glycyrrhizic acid, Gingeronone and Galangan were found to be promising candidates against Nsp1 [132]. With respect to virtual ligand screening and molecular docking studies of drugs against Nucleocapsid protein [133], 8987 drugs from PubChem and Asinex databases were assessed. According to calculated free binding energies, three drugs were noted to have stability during their interaction with SARS-CoV-2; Zidovudine from PubChem and 6799 and 5817 from Asinex databases. In other studies, 56,079 compounds from the Maybridge library and Asinex database and screened by virtual ligand binding followed by molecular dynamics, two binders, ZINC0000146942 and ZINC00003118440 were identified as inhibitors of the N-terminal domain of the N protein (Table 4) [134]. Additionally, two research studies investigated the Envelope (E) protein of SARS-CoV-2. Gupt et al. [24] studied the effects of phytochemicals on SARS-CoV-2 through the ion channels of E protein. It is concluded that Val25 and Phe26 amino acids can be effected by Vibsanol B and Macaflavanone E. In the Borkotohy study [135], the effects of 70 Indian plant compounds on membrane and envelope proteins were studied by GROMACS software, based on GROMOS 96-43a1 force field for 200 ns time period.
Table 4.
Nonstructural proteins of SARS-CoV-2. Summary of nine computational studies.
4. Discussion
The COVID-19 pandemic caused by SARS-CoV-2 virus first became apparent in November 2019 in China [1] and was subsequently declared a global pandemic [2]. Although Rappouli et al. [3] reported that the development of a human vaccine could take at least 15 years and at least USD 200 million, an alliance of the science society has now achieved this [4]. Besides the hesitation on the authenticity and reported side-effects in a small proportion of people of approved vaccines [141,142], the worldwide vaccination is time consuming, as well as public health being in danger from current virus mutations, which can lead to a prolonged pandemic leading towards a difficult situation [143]. In this rapidly evolving crisis, drug repurposing is considered the quickest way to identify potential drug treatments. Some of the repurposed drugs have already entered into human clinical trials and early data suggests that a number of drugs could be beneficial against SARS-CoV-2. For instance, based on the data analysis of National Institutes of Health, patients who received Remdesivir (a broad spectrum anti-viral), compared to those on placebo, showed significant differences in recovery time [144]. However, according to the report by Wang et al. [145], Remdesivir clinical trials were not found to be beneficial in a Chinese patient cohort. Accordingly, several other therapeutic agents have been developed and repurposed, although none have yet shown to significantly affect SARS-CoV-2 [146]. In addition, Hydroxychloroquine and Chloroquine, anti-malarial drugs, have shown to have anti-SARS-CoV-2 viral activity [147]. However, in a New York medical center clinical study of seventy COVID-19 patients treated with Hydroxychloroquine no beneficial effects were shown, in regard to patient survival [148]. Further, Dexamethasone (a corticosteroid) has also been identified as a potential drug for patients with COVID-19 showing reduced mortality rates in seriously ill patients [149]. However, due to the two properties of dexamethasone, which (1) limits the effect of cytokines [150] and (2) reduces the effects of B cells [151], further research is required.
An abundance of virtual screening models using various molecular docking methods and following that molecular dynamics have been produced. This large volume may overwhelm and confuse researchers. Thus, collection and classification of effective ligands on different ingredients of SARS-CoV-2 was aided by current review. At the first step, several reliable databases such as Google Scholar, Scopus, Embase, PubMed and Medline were investigated for drug repurposing through molecular docking against SARS-CoV-2, and almost all relevant papers published in peer-reviewed journals were collated and analyzed. All the studies, at the first step have identified numerous compounds to bind to SARS-CoV-2 proteins, and those with the lowest docking score (or highest binding affinity) are chosen for further investigation. The compounds are assessed by molecular dynamics to insure whether the conformation is stable, followed by RMSD calculations. In the case that the ligand could pass through the process, it is introduced as a potential compound, which could then be investigated clinically. By searching through search engines, many studies can be achieved. Molecular docking methodologies are various and consequently, diverse parameters are presented. In order to compare the results, we have chosen docking score as the benchmark. The results that had not reported docking scores were eliminated. Finally, 58 Studies are collected and classified in Table 1, Table 2, Table 3 and Table 4.
The viral main protease target section consisted of 33 articles. This subset contains the largest number of searches. Most of the studies focused on the identification of repurposed drugs and natural compounds for their ability to bind to the main protease of SARS-CoV-2 for potential anti-viral activity. For molecular docking, various software are utilized. Most studies have not presented the method of docking. Docking scores range from −4 to −16.35. Among 33 studies, Saquinavir is the only ligand presented with the highest binding affinity by four independent reportings [78,80,86,100]. In the first study [78], Saquinavir has interaction with His41 and Cys45 residues. It can form hydrogen bonds with Gly143, Ser144 and Cys145 with −36.3026 (kcal/mol) binding free energy. The second study [80] reports that Saquinavir forms 6 hydrogen binds in Cys145 and His41 as central dyad residues, as well with Gly143, Met49, Gln189 and Glu166, with −74.4061 (kcal/mol) free binding energy. Furthermore, in the third study [86], it was emphasized that Saqinavir interacts with Cys145 with −72.17 (kcal/mol) free binding energy; however, the number of hydrogen bonds was not mentioned. However, in the fourth study, no specific information was reported except docking score [100]. Besides Saquinavir, there are an additional two drugs with lower docking scores. Qamar et al. [76] reported a markedly lower docking score, -16.35 for Pubchem ID: 11,610,052 with His41, Cys145, Thr24, Thr25, Thr26, Cys44, Thr45, Ser46, Met49, Asn142, Gly143, His164, Glu166 and Gln189 as interacting residues. In addition, Simeprevier [68] with a docking score of −11.33 and free binding energy −252.54 (kcal/mol) also shows promise. The authors claimed that Simeprevier is more effective than Remdesivir, Chloroquine and Hydroxcychloroquine with −5.8, −7.5 and −6.7 docking scores, respectively. Simeprevier forms three hydrogen bonds with His163, Thr26 and Asn119 and interacts with central binding residues Cys145, His41 and Met49.
Regarding the Spike protein-ACE2 enzyme target, 13 original articles are identified and gathered in Table 2. As can be noted, there are a range of docking scores −101.17 for orientin compound with −70.6 (kcal/mol) binding free energy [111]. In this research, orientin is docked to ACE2 receptor and it has been observed that orientin forms five hydrogen bonds with three Lys26, Glu22 and Asn90 residues, as well as interactions with Lys94 and Glu22. The stability of orientin is checked via molecular dynamics simulation, and 4.6 Å is reported on average [111]. The second ligand in prioritization is phthalocyanine with −16.3 docking score and −66.6 (kcal/mol) [112]. It is reported that phthalocyanine establishes 11 interactions with the protein as well as hydrophobic interactions within itself, which increases its stability and complex strength. Moreover, coenzyme A with −11.555 docking score is introduced as a potential drug, but there is no information about the interactions or number of hydrogen binding [99]. Furthermore GR hydrochloride with −11.23 docking score is ranked fourth [115]. It is reported that it forms two hydrogen bonds with ACE2 receptor and interacts with Phe40, Ala348, Trp349, Gly352, Gly354, His378, Asp382, Tyr385, Ala386, Phe390, Arg393, Asn394, and His401 residues. Via computational modeling of RdRp enzyme target, five in silico studies were identified. The presented drugs identified as potential candidates with considerable docking score ranges about −9. In the first study [123], IDX-184 with −9 docking score is reported, which has 18 hydrogen bonds with RdRp and is more stable due to (i) the ability to establish a salt bridge with Asp514 and (ii) it forms two metal interactions with Glu702 and Asp635 residues. In addition, sofosbuvir with −9.3 docking score [124,125] forms eight hydrogen bonds with Asp343, Thr447, Lys442, Ser573, Asp524 and Cys513. Stability of these ligands are not evaluated by molecular dynamics, and thus are faced with strong hesitation. In the fourth study, it is reported that the compound CAS 833463-10-8 with −9.529 docking score can form three hydrogen bonds with Arg553, Tyr455 and Tyr619 [126]. The fifth study has introduced cryptomisirine with −9 docking score that forms 1 hydrogen bond with Thr556 residue [127].
In the other proteins and non-structural proteins of SARS-CoV-2 section, all the miscellaneous computational studies on N protein, M protein, E protein, Nsp7, Nsp8, Nsp10, Nsp13, Nsp14, Nsp15 and Nsp16 are collected and summarized, which can significantly help researchers in their future studies. Among these nine studies, docking score of Belachinal is the lowest with −11.46, which can effect E protein [24]. At the next stage, Nimbolin A with −11.2 docking score and binding free energy of −71.05 (kcal/mol), has been reported to have hydrophobic interactions with Leu18 (A), Leu19 (A), Ala22 (A), Leu19 (B), Leu18 (B), Ala22 (C), Phe23 (C), Phe26 (C), Asn15 (D), Leu18 (D), Leu19 (D), Val25 (D), Phe26 (D), Leu18 (E), Leu19 (E), and Ala22 (E) residues [135]. After collection and classification of various studies, only a limited number of natural or drug ligands have been translated to human clinical studies. At the time of writing the current article, no experimental or clinical studies have been reported on most of these identified natural and drug compounds, but their efficacy testing cannot be ignored. Thus far, Hydroxychloroquine and Chloroquine have been approved by the FDA, but because of disappointing outcomes and modest effects these have undergone controversial debates. A limited number of drugs have been investigated clinically or experimentally investigations such as Remdesivir, Lopinavir/Rotinavir, Sofosbuvir, Carvacol and Favipiravir. Gordon et al. has studied experimentally the effects of Remdesivir on nonstructural proteins of MERS-CoV and has introduced it as a high potency inhibitor against MERS-CoV, which have high structural similarity with SARS-CoV-2 [152]. Moreover, other research group after 14 days clinical trial of sofosbuvir has reported that duration of hospital stay is significantly reduced for the patients whom have received sofosbuvir [153]. Additionally, sofosbuvir is recommended by some pharmacologists to be considered in clinical trials [154]. Furthermore, it is reported that Lopinavir/ritonavir inhibitor, which is a well-known HIV medication drug, not only has undergone clinical trials [155], but also in some countries it can be used as an alternative drug. In a study on hemodialysis patients, lopinavir/ritonavir is identified as an effective drug through experimental investigation [156]. In addition, in a review paper by Javed et al. that is dedicated to experimental investigation as well clinical trials of carvacrol, it is claimed that due to wide range of experimental studies, carvacrol has protective effects against infective diseases within the scope of SARS-CoV-2 [157]. It should be mentioned that there are some articles about clinical trials of other compounds, but because they are preprints, we have ignored them. One of the important gaps of research is the lack of pharmacokinetic simulation for docked natural or drug compounds, which should be considered for future research; however, the value of current molecular docking and molecular dynamics is not diminished.
5. Conclusions
The year 2020 opened with an emerging threat to world health—the coronavirus outbreak. Genomic research suggested that SARS-CoV-2 was transmitted from bats, possibly via pangolins, to humans. Due to the complex nature of COVID-19 epidemiology and virology, there are still many unanswered questions. Discovering therapeutic anti-viral agents is currently the main focus due to the prevalence and rapid spread of the virus, as well as its relatively high death rate. In this critical situation, computational modeling, specifically virtual screening, offers the fastest, cheapest, and most effective method for discovering effective drugs. Based on the studies presented, there are almost 55 identified natural and drug compounds with potential anti-viral activity. However, much research is still required for their effectiveness in human clinical trials.
Author Contributions
Conceptualization, M.H. and A.H.A.; methodology, M.H.; investigation, N.R., M.K.H. and A.H.; writing—original draft preparation, M.H., A.H.A. and V.A.; writing—review and editing, V.A.; supervision, V.A.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
V.A. would like to thank the place-based planetary health grant PH098 for their support. V.A. would also like to thank The Pappas Family, the Greek Orthodox Archdiocese of Australia and the Victoria University vaccine appeal, whose generous philanthropic support made possible the preparation of this paper. The authors would also like to thank the Immunology and Translational Research Group for their significant contribution. The Mechanisms and Interventions in Health and Disease Program within the Institute for Health and Sport, Victoria University Australia are also appreciated for their support. M.K.H. and A.H. were supported by Victoria University Postgraduate Scholarships and M.K.H. was also supported by the Vice-Chancellors top-up Scholarship Award.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Atal, S.; Fatima, Z.; Balakrishnan, S. Approval of Itolizumab for COVID-19: A Premature Decision or Need of The Hour? BioDrugs 2020, 34, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Crisafulli, S.; Isgrò, V.; La Corte, L.; Atzeni, F.; Trifirò, G. Potential role of anti-interleukin (IL)-6 drugs in the treatment of COVID-19: Rationale, clinical evidence and risks. BioDrugs 2020, 34, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Ayres, J.S. A metabolic handbook for the COVID-19 pandemic. Nat. Metab. 2020, 2, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- COVID-19 CORONAVIRUS PANDEMIC. Available online: https://www.worldometers.info/coronavirus/ (accessed on 24 September 2020).
- Kissler, S.M.; Tedijanto, C.; Goldstein, E.; Grad, Y.H.; Lipsitch, M. Projecting the transmission dynamics of SARS-CoV-2 through the postpandemic period. Science 2020, 368, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108. [Google Scholar] [CrossRef]
- Wang, N.; Shang, J.; Jiang, S.; Du, L. Subunit vaccines against emerging pathogenic human coronaviruses. Front. Microbiol. 2020, 11, 298. [Google Scholar] [CrossRef]
- Ge, X.-Y.; Li, J.-L.; Yang, X.-L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020, 27, 763–767. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.R.; Patel, A.; Ramos, S.; Elwood, D.; Zhu, X.; Yan, J.; Gary, E.N.; Walker, S.N.; Schultheis, K.; Purwar, M.; et al. Immunogenicity of a DNA vaccine candidate for COVID-19. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Li, C.; Huang, A.; Xia, S.; Lu, S.; Shi, Z.; Lu, L.; Jiang, S.; Yang, Z.; Wu, Y. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microbes Infect. 2020, 9, 382–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Yang, Y.; Zhou, Y.; Lu, L.; Li, F.; Jiang, S. MERS-CoV spike protein: A key target for antivirals. Expert Opin. Ther. Targets 2017, 21, 131–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Wong, G.; Lu, G.; Yan, J.; Gao, G.F. MERS-CoV spike protein: Targets for vaccines and therapeutics. Antivir. Res. 2016, 133, 165–177. [Google Scholar] [CrossRef]
- Kuo, L.; Hurst, K.R.; Masters, P.S. Exceptional flexibility in the sequence requirements for coronavirus small envelope protein function. J. Virol. 2007, 81, 2249–2262. [Google Scholar] [CrossRef] [Green Version]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.K.; Vemula, S.; Donde, R.; Gouda, G.; Behera, L.; Vadde, R. In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. J. Biomol. Struct. Dyn. 2020, 39, 2617–2627. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-M.; Lin, S.-C.; Hsu, J.-N.; Chang, C.-K.; Chien, C.-M.; Wang, Y.-S.; Wu, H.-Y.; Jeng, U.-S.; Kehn-Hall, K.; Hou, M.-H. Structure-based stabilization of non-native protein–protein interactions of coronavirus nucleocapsid proteins in antiviral drug design. J. Med. Chem. 2020, 63, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Liu, G.; Ma, H.; Zhao, D.; Yang, Y.; Liu, M.; Mohammed, A.; Zhao, C.; Yang, Y.; Xie, J. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochem. Biophys. Res. Commun. 2020, 527, 618–623. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziebuhr, J.; Snijder, E.J.; Gorbalenya, A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Sun, J.; Mao, Z.; Wang, L.; Lu, Y.L.; Li, J. A guideline for homology modeling of the proteins from newly discovered betacoronavirus, 2019 novel coronavirus (2019-nCoV). J. Med. Virol. 2020, 92, 1542–1548. [Google Scholar] [CrossRef]
- Rappuoli, R.; Black, S.; Bloom, D.E. Vaccines and global health: In search of a sustainable model for vaccine development and delivery. Sci. Transl. Med. 2019, 11, eaaw2888. [Google Scholar] [CrossRef]
- Ciliberto, G.; Cardone, L. Boosting the arsenal against COVID-19 through computational drug repurposing. Drug Discov. Today 2020, 25, 946–948. [Google Scholar] [CrossRef]
- Rosenberg, E.S.; Dufort, E.M.; Udo, T.; Wilberschied, L.A.; Kumar, J.; Tesoriero, J.; Weinberg, P.; Kirkwood, J.; Muse, A.; DeHovitz, J.; et al. Association of treatment with hydroxychloroquine or azithromycin with in-hospital mortality in patients with COVID-19 in New York state. JAMA 2020, 323, 2493–2502. [Google Scholar] [CrossRef]
- Molina, J.M.; Delaugerre, C.; Le Goff, J.; Mela-Lima, B.; Ponscarme, D.; Goldwirt, L.; de Castro, N. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Med. Mal. Infect. 2020, 50, 30085–30088. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M. A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Bellera, C.L.; Llanos, M.; Gantner, M.E.; Rodriguez, S.; Gavernet, L.; Comini, M.; Talevi, A. Can drug repurposing strategies be the solution to the COVID-19 crisis? Expert Opin. Drug Discov. 2021, 16, 605–612. [Google Scholar] [CrossRef]
- Tabari, M.A.K.; Khoshhal, H.; Tafazoli, A.; Khandan, M.; Bagheri, A. Applying Computer Simulations in Battling with COVID-19, using pre-analyzed molecular and chemical data to face the pandemic. Inform. Med. Unlocked 2020, 21, 100458. [Google Scholar] [CrossRef] [PubMed]
- Hiranuma, N.; Park, H.; Baek, M.; Anishchenko, I.; Dauparas, J.; Baker, D. Improved protein structure refinement guided by deep learning based accuracy estimation. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, A.; Gopal, J.; Candavelou, M.; Gollapalli, S.; Karthikeyan, K. Computational approach for protein structure prediction. Healthc. Inform. Res. 2013, 19, 137. [Google Scholar] [CrossRef] [PubMed]
- Ou-Yang, S.-S.; Lu, J.-Y.; Kong, X.-Q.; Liang, Z.-J.; Luo, C.; Jiang, H. Computational drug discovery. Acta Pharmacol. Sin. 2012, 33, 1131–1140. [Google Scholar] [CrossRef] [Green Version]
- Moitessier, N.; Englebienne, P.; Lee, D.; Lawandi, J.; Corbeil, C.R. Towards the development of universal, fast and highly accurate docking/scoring methods: A long way to go. Br. J. Pharmacol. 2008, 153, S7–S26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Phatak, S.S. Homology modeling in drug discovery: Current trends and applications. Drug Discov. Today 2009, 14, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Moro, S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: An overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Fischer, D.; Norel, R.; Wolfson, H.; Nussinov, R. Surface motifs by a computer vision technique: Searches, detection, and implications for protein–ligand recognition. Proteins Struct. Funct. Bioinform. 1993, 16, 278–292. [Google Scholar] [CrossRef]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [Green Version]
- Miranker, A.; Karplus, M. Functionality maps of binding sites: A multiple copy simultaneous search method. Proteins Struct. Funct. Bioinform. 1991, 11, 29–34. [Google Scholar] [CrossRef]
- Hart, T.N.; Read, R.J. A multiple-start Monte Carlo docking method. Proteins Struct. Funct. Bioinform. 1992, 13, 206–222. [Google Scholar] [CrossRef]
- Oshiro, C.M.; Kuntz, I.D.; Dixon, J.S. Flexible ligand docking using a genetic algorithm. J. Comput. Aided Mol. Des. 1995, 9, 113–130. [Google Scholar] [CrossRef]
- Naqvi, A.A.; Mohammad, T.; Hasan, G.M.; Hassan, M. Advancements in docking and molecular dynamics simulations towards ligand-receptor interactions and structure-function relationships. Curr. Top. Med. Chem. 2018, 18, 1755–1768. [Google Scholar] [CrossRef]
- Tanchuk, V.Y.; Tanin, V.O.; Vovk, A.I.; Poda, G. A new, improved hybrid scoring function for molecular docking and scoring based on AutoDock and AutoDock Vina. Chem. Biol. Drug Des. 2016, 87, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Fu, A.; Zhang, L. Progress in molecular docking. Quant. Biol. 2019, 7, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Hodos, R.A.; Kidd, B.A.; Shameer, K.; Readhead, B.P.; Dudley, J.T. In silico methods for drug repurposing and pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 186–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, K.; Yazdanpanah, N.; Saghazadeh, A.; Rezaei, N. Computational drug discovery and repurposing for the treatment of Covid-19: A systematic review. Bioorganic Chem. 2021, 106, 104490. [Google Scholar] [CrossRef] [PubMed]
- Stewart, L.A.; Clarke, M.; Rovers, M.; Riley, R.D.; Simmonds, M.; Stewart, G.; Tierney, J.F. Preferred reporting items for a systematic review and meta-analysis of individual participant data: The PRISMA-IPD statement. JAMA 2015, 313, 1657–1665. [Google Scholar] [CrossRef]
- Schoggins, J.W. Interferon-stimulated genes: Roles in viral pathogenesis. Curr. Opin. Virol. 2014, 6, 40–46. [Google Scholar] [CrossRef]
- Mengist, H.M.; Fan, X.; Jin, T. Designing of improved drugs for COVID-19: Crystal structure of SARS-CoV-2 main protease M pro. Signal Transduct. Target. Ther. 2020, 5, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; Von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: Structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Hatada, R.; Okuwaki, K.; Mochizuki, Y.; Handa, Y.; Fukuzawa, K.; Komeiji, Y.; Okiyama, Y.; Tanaka, S. Fragment molecular orbital based interaction analyses on COVID-19 main protease-inhibitor N3 complex (PDB ID: 6LU7). J. Chem. Inf. Model. 2020, 60, 3593–3602. [Google Scholar] [CrossRef]
- Deng, X.; Hackbart, M.; Mettelman, R.C.; O’Brien, A.; Mielech, A.M.; Yi, G.; Kao, C.C.; Baker, S.C. Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc. Natl. Acad. Sci. USA 2017, 114, E4251–E4260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002, 76, 3697–3708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulferts, R.; Ziebuhr, J. Nidovirus ribonucleases: Structures and functions in viral replication. RNA Biol. 2011, 8, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Ciccozzi, M.; Benvenuto, D.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Angeletti, S. Response to Ribeiro da Silva et al, “Role of Nonstructural Proteins in the Pathogenesis of SARS-CoV-2”. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Wu, X.; Yang, Z.R. Mining viral protease data to extract cleavage knowledge. Bioinformatics 2002, 18, S5–S13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, F.S.; Amanlou, M. Anti-HCV and anti-malaria agent, potential candidates to repurpose for coronavirus infection: Virtual screening, molecular docking, and molecular dynamics simulation study. Life Sci. 2020, 258, 118205. [Google Scholar] [CrossRef]
- Kumar, Y.; Singh, H.; Patel, C.N. In silico prediction of potential inhibitors for the Main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health 2020, 13, 1210–1223. [Google Scholar] [CrossRef]
- Peele, K.A.; Chandrasai, P.; Srihansa, T.; Krupanidhi, S.; Sai, A.V.; Babu, D.J.; Indira, M.; Reddy, A.R.; Venkateswarulu, T. Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: A computational study. Inform. Med. Unlocked 2020, 19, 100345. [Google Scholar] [CrossRef]
- Wang, J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef]
- Havranek, B.; Islam, S.M. An in silico approach for identification of novel inhibitors as potential therapeutics targeting COVID-19 main protease. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Kumar, A.; Choudhir, G.; Shukla, S.K.; Sharma, M.; Tyagi, P.; Bhushan, A.; Rathore, M. Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. J. Biomol. Struct. Dyn. 2020, 39, 3760–3770. [Google Scholar] [CrossRef] [PubMed]
- Mittal, L.; Kumari, A.; Srivastava, M.; Singh, M.; Asthana, S. Identification of potential molecules against COVID-19 main protease through structure-guided virtual screening approach. J. Biomol. Struct. Dyn. 2020, 39, 3662–3680. [Google Scholar] [CrossRef] [PubMed]
- Joshi, T.; Sharma, P.; Joshi, T.; Pundir, H.; Mathpal, S.; Chandra, S. Structure-based screening of novel lichen compounds against SARS Coronavirus main protease (Mpro) as potentials inhibitors of COVID-19. Mol. Divers. 2020, 1–13. [Google Scholar] [CrossRef]
- Ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Naik, V.R.; Munikumar, M.; Ramakrishna, U.; Srujana, M.; Goudar, G.; Naresh, P.; Kumar, B.N.; Hemalatha, R. Remdesivir (GS-5734) as a therapeutic option of 2019-nCOV main protease–in silico approach. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Zia, K.; Ashraf, S.; Uddin, R.; Ul-Haq, Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn. 2020, 39, 2607–2616. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Singh, A.K.; Kushwaha, P.P.; Prajapati, K.S.; Shuaib, M.; Senapati, S.; Kumar, S. Identification of potential natural inhibitors of SARS-CoV2 main protease by molecular docking and simulation studies. J. Biomol. Struct. Dyn. 2020, 1–19. [Google Scholar] [CrossRef]
- Khan, A.; Ali, S.S.; Khan, M.T.; Saleem, S.; Ali, A.; Suleman, M.; Babar, Z.; Shafiq, A.; Khan, M.; Wei, D.-Q. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro). J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Evaluation of green tea polyphenols as novel corona virus (SARS CoV-2) main protease (Mpro) inhibitors–an in silico docking and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A.; ul Qamar, M.T.; Mirza, M.U.; Bhadane, R.; Alqahtani, S.M.; Muneer, I.; Froeyen, M.; Salo-Ahen, O.M. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CLpro. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef]
- Odhar, H.A.; Ahjel, S.W.; Albeer, A.A.M.A.; Hashim, A.F.; Rayshan, A.M.; Humadi, S.S. Molecular docking and dynamics simulation of FDA approved drugs with the main protease from 2019 novel coronavirus. Bioinformation 2020, 16, 236. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Sharma, P.P.; Shankar, U.; Kumar, D.; Joshi, S.K.; Pena, L.; Durvasula, R.; Kumar, A.; Kempaiah, P.; Poonam; et al. Discovery of New Hydroxyethylamine Analogs Against 3CLpro Protein Target of SARS-CoV-2: Molecular Docking, Molecular Dynamics Simulation and Structure-Activity Relationship Studies. J. Chem. Inf. Model. 2020, 60, 5754–5770. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Pitsillou, E.; Karagiannis, C.; Darmawan, K.K.; Ng, K.; Hung, A.; Karagiannis, T.C. Interaction of the prototypical α-ketoamide inhibitor with the SARS-CoV-2 main protease active site in silico: Molecular dynamic simulations highlight the stability of the ligand-protein complex. Comput. Biol. Chem. 2020, 87, 107292. [Google Scholar] [CrossRef] [PubMed]
- Al-Khafaji, K.; Al-Duhaidahawi, D.; Tok, T.T. Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pundir, H.; Joshi, T.; Joshi, T.; Sharma, P.; Mathpal, S.; Chandra, S.; Tamta, S. Using Chou’s 5-steps rule to study pharmacophore-based virtual screening of SARS-CoV-2 Mpro inhibitors. Mol. Divers. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Abdelrahman, A.H.; Hussien, T.A.; Badr, E.A.; Mohamed, T.A.; El, H.R.; Pare, P.W.; Efferth, T.; Hegazy, M.E.F. In silico Drug Discovery of Major Metabolites from Spices as SARS-CoV-2 Main Protease Inhibitors. Comput. Biol. Med. 2020, 126, 104046. [Google Scholar] [CrossRef] [PubMed]
- Baildya, N.; Khan, A.A.; Ghosh, N.N.; Dutta, T.; Chattopadhyay, A.P. Screening of potential drug from Azadirachta Indica (Neem) extracts for SARS-CoV-2: An insight from molecular docking and MD-simulation studies. J. Mol. Struct. 2021, 1227, 129390. [Google Scholar] [CrossRef]
- Kavitha, K.; Sivakumar, S.; Ramesh, B. 1, 2, 4 triazolo [1, 5-a] pyrimidin-7-ones as novel SARS-CoV-2 Main protease inhibitors: In silico screening and molecular dynamics simulation of potential COVID-19 drug candidates. Biophys. Chem. 2020, 267, 106478. [Google Scholar] [CrossRef]
- Tachoua, W.; Kabrine, M.; Mushtaq, M.; Ul-Haq, Z. An in-silico evaluation of COVID-19 main protease with clinically approved drugs. J. Mol. Graph. Model. 2020, 101, 107758. [Google Scholar] [CrossRef]
- Yepes-Pérez, A.F.; Herrera-Calderon, O.; Sánchez-Aparicio, J.-E.; Tiessler-Sala, L.; Maréchal, J.-D.; Cardona, G.-W. Investigating Potential Inhibitory Effect of Uncaria tomentosa (Cat’s Claw) against the Main Protease 3CLpro of SARS-CoV-2 by Molecular Modeling. Evid. Based Complementary Altern. Med. 2020, 2020, 4932572. [Google Scholar] [CrossRef]
- Hejazi, I.I.; Beg, M.A.; Imam, M.A.; Athar, F.; Islam, A. Glossary of phytoconstituents: Can these be repurposed against SARS CoV-2? A quick in silico screening of various phytoconstituents from plant Glycyrrhiza glabra with SARS CoV-2 main protease. Food Chem. Toxicol. 2021, 150, 112057. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, K.H.; Chowdhury, M.; Mahmud, S.; Tareq, A.M.; Hanif, N.B.; Banu, N.; Reza, A.; Emran, T.B.; Simal-Gandara, J. Drug Repurposing Approach against Novel Coronavirus Disease (COVID-19) through Virtual Screening Targeting SARS-CoV-2 Main Protease. Biology 2021, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, W.B.; Mendanha, S.A. Molecular dynamics simulation of docking structures of SARS-CoV-2 main protease and HIV protease inhibitors. J. Mol. Struct. 2021, 1225, 129143. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Abdelrahman, A.H.; Allemailem, K.S.; Almatroudi, A.; Moustafa, M.F.; Hegazy, M.-E.F. In silico evaluation of prospective anti-COVID-19 drug candidates as potential SARS-CoV-2 main protease inhibitors. Protein J. 2021, 40, 296–309. [Google Scholar] [CrossRef]
- Fakhar, Z.; Khan, S.; AlOmar, S.Y.; Alkhuriji, A.; Ahmad, A. ABBV-744 as a potential inhibitor of SARS-CoV-2 main protease enzyme against COVID-19. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Pushkaran, A.C.; Melge, A.R.; Puthiyedath, R.; Mohan, C.G. A phytochemical-based medication search for the SARS-CoV-2 infection by molecular docking models towards spike glycoproteins and main proteases. RSC Adv. 2021, 11, 12003–12014. [Google Scholar] [CrossRef]
- Hall, D.C., Jr.; Ji, H.-F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med. Infect. Dis. 2020, 35, 101646. [Google Scholar] [CrossRef]
- Qiao, Z.; Zhang, H.; Ji, H.-F.; Chen, Q. Computational view toward the inhibition of SARS-CoV-2 spike glycoprotein and the 3CL protease. Computation 2020, 8, 53. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Trask, A.J.; Jessup, J.A. Advances in biochemical and functional roles of angiotensin-converting enzyme 2 and angiotensin-(1–7) in regulation of cardiovascular function. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2281–H2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme–related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000, 87, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Hussain, M.; Jabeen, N.; Raza, F.; Shabbir, S.; Baig, A.A.; Amanullah, A.; Aziz, B. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J. Med Virol. 2020, 92, 1580–1586. [Google Scholar] [CrossRef] [Green Version]
- Abdelli, I.; Hassani, F.; Brikci, S.B.; Ghalem, S. In silico study the inhibition of Angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from western Algeria. J. Biomol. Struct. Dyn. 2020, 39, 3263–3276. [Google Scholar] [CrossRef]
- Ahmad, S.; Abbasi, H.W.; Shahid, S.; Gul, S.; Abbasi, S.W. Molecular Docking, Simulation and MM-PBSA Studies of Nigella Sativa Compounds: A Computational Quest to identify Potential Natural Antiviral for COVID-19 Treatment. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Wahedi, H.M.; Ahmad, S.; Abbasi, S.W. Stilbene-based natural compounds as promising drug candidates against COVID-19. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- Lakshmi, S.A.; Shafreen, R.M.B.; Priya, A.; Shunmugiah, K.P. Ethnomedicines of Indian origin for combating COVID-19 infection by hampering the viral replication: Using structure-based drug discovery approach. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Romeo, A.; Iacovelli, F.; Falconi, M. Targeting the SARS-CoV-2 spike glycoprotein prefusion conformation: Virtual screening and molecular dynamics simulations applied to the identification of potential fusion inhibitors. Virus Res. 2020, 286, 198068. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, O.V.; Rocha, G.B.; Paluch, A.S.; Costa, L.T. Repurposing approved drugs as inhibitors of SARS-CoV-2 S-protein from molecular modeling and virtual screening. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- Sinha, S.K.; Prasad, S.K.; Islam, M.A.; Gurav, S.S.; Patil, R.B.; AlFaris, N.A.; Aldayel, T.S.; AlKehayez, N.M.; Wabaidur, S.M.; Shakya, A. Identification of bioactive compounds from Glycyrrhiza glabra as possible inhibitor of SARS-CoV-2 spike glycoprotein and non-structural protein-15: A pharmacoinformatics study. J. Biomol. Struct. Dyn. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Malik, Y.S.; Tomar, S. Identification of SARS-CoV-2 cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. Front. Immunol. 2020, 11, 1664. [Google Scholar] [CrossRef]
- Trezza, A.; Iovinelli, D.; Santucci, A.; Prischi, F.; Spiga, O. An integrated drug repurposing strategy for the rapid identification of potential SARS-CoV-2 viral inhibitors. Sci. Rep. 2020, 10, 1–8. [Google Scholar]
- Unni, S.; Aouti, S.; Thiyagarajan, S.; Padmanabhan, B. Identification of a repurposed drug as an inhibitor of Spike protein of human coronavirus SARS-CoV-2 by computational methods. J. Biosci. 2020, 45, 1–20. [Google Scholar] [CrossRef]
- Khan, A.A.; Baildya, N.; Dutta, T.; Ghosh, N.N. Inhibitory efficiency of potential drugs against SARS-CoV-2 by blocking human angiotensin converting enzyme-2: Virtual screening and molecular dynamics study. Microb. Pathog. 2021, 152, 104762. [Google Scholar] [CrossRef]
- Hansen, J.L.; Long, A.M.; Schultz, S.C. Structure of the RNA-dependent RNA polymerase of poliovirus. Structure 1997, 5, 1109–1122. [Google Scholar] [CrossRef] [Green Version]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. USA 2014, 111, E3900–E3909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Song, W.; Huang, H.; Sun, Q. Pharmacological therapeutics targeting RNA-dependent RNA polymerase, proteinase and spike protein: From mechanistic studies to clinical trials for COVID-19. J. Clin. Med. 2020, 9, 1131. [Google Scholar] [CrossRef] [Green Version]
- Elfiky, A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020, 248, 117477. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci. 2020, 253, 117592. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A. SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. J. Biomol. Struct. Dyn. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Aouidate, A.; Ghaleb, A.; Chtita, S.; Aarjane, M.; Ousaa, A.; Maghat, H.; Sbai, A.; Choukrad, M.B.; Bouachrine, M.; Lakhlifi, T. Identification of a novel dual-target scaffold for 3CLpro and RdRp proteins of SARS-CoV-2 using 3D-similarity search, molecular docking, molecular dynamics and ADMET evaluation. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Borquaye, L.S.; Gasu, E.N.; Ampomah, G.B.; Kyei, L.K.; Amarh, M.A.; Mensah, C.N.; Nartey, D.; Commodore, M.; Adomako, A.K.; Acheampong, P. Alkaloids from Cryptolepis sanguinolenta as Potential Inhibitors of SARS-CoV-2 Viral Proteins: An In Silico Study. BioMed Res. Int. 2020, 2020, 5324560. [Google Scholar] [CrossRef]
- Yuen, C.-K.; Lam, J.-Y.; Wong, W.-M.; Mak, L.-F.; Wang, X.; Chu, H.; Cai, J.-P.; Jin, D.-Y.; To, K.K.-W.; Chan, J.F.-W.; et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microbes Infect. 2020, 1–29. [Google Scholar] [CrossRef]
- Kim, Y.; Jedrzejczak, R.; Maltseva, N.I.; Wilamowski, M.; Endres, M.; Godzik, A.; Michalska, K.; Joachimiak, A. Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 2020, 29, 1596–1605. [Google Scholar] [CrossRef]
- Bhardwaj, K.; Palaninathan, S.; Alcantara, J.M.O.; Yi, L.L.; Guarino, L.; Sacchettini, J.C.; Kao, C.C. Structural and functional analyses of the severe acute respiratory syndrome coronavirus endoribonuclease Nsp15. J. Biol. Chem. 2008, 283, 3655–3664. [Google Scholar] [CrossRef] [Green Version]
- Ricagno, S.; Egloff, M.-P.; Ulferts, R.; Coutard, B.; Nurizzo, D.; Campanacci, V.; Cambillau, C.; Ziebuhr, J.; Canard, B. Crystal structure and mechanistic determinants of SARS coronavirus nonstructural protein 15 define an endoribonuclease family. Proc. Natl. Acad. Sci. USA 2006, 103, 11892–11897. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Tiwari, V.; Sowdhamini, R. Computational search for potential COVID-19 drugs from FDA-approved drugs and small molecules of natural origin identifies several anti-virals and plant products. J. Biosci. 2020, 45, 100. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Imran, M.; Dhamija, P.; Suchal, K.; Handu, S. Virtual screening and dynamics of potential inhibitors targeting RNA binding domain of nucleocapsid phosphoprotein from SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Sarma, P.; Shekhar, N.; Prajapat, M.; Avti, P.; Kaur, H.; Kumar, S.; Singh, S.; Kumar, H.; Prakash, A.; Dhibar, D.P. In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). J. Biomol. Struct. Dyn. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borkotoky, S.; Banerjee, M. A computational prediction of SARS-CoV-2 structural protein inhibitors from Azadirachta indica (Neem). J. Biomol. Struct. Dyn. 2020, 1–17. [Google Scholar] [CrossRef]
- Krishnan, D.A.; Sangeetha, G.; Vajravijayan, S.; Nandhagopal, N.; Gunasekaran, K. Structure-based drug designing towards the identification of potential anti-viral for COVID-19 by targeting endoribonuclease NSP15. Inform. Med. Unlocked 2020, 20, 100392. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Gurjar, V.; Qamar, I.; Singh, N. Identification of Potential Inhibitors of SARS-COV-2 Endoribonuclease (EndoU) from FDA Approved Drugs: A Drug Repurposing Approach to find Therapeutics for COID19. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Chikhale, R.V.; Gurav, S.S.; Patil, R.B.; Sinha, S.K.; Prasad, S.K.; Shakya, A.; Shrivastava, S.K.; Gurav, N.S.; Prasad, R.S. Sars-cov-2 host entry and replication inhibitors from Indian ginseng: An in-silico approach. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Tazikeh-Lemeski, E.; Moradi, S.; Raoufi, R.; Shahlaei, M.; Janlou, M.A.M.; Zolghadri, S. Targeting SARS-COV-2 non-structural protein 16: A virtual drug repurposing study. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, L.; Manning, M.; Bonahoom, M.; Lotvola, A.; Yang, Z.; Yang, Z.-Q. Structural analysis, virtual screening and molecular simulation to identify potential inhibitors targeting 2′-O-ribose methyltransferase of SARS-CoV-2 coronavirus. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Hossain, M.K.; Hassanzadeganroudsari, M.; Feehan, J.; Apostolopoulos, V. COVID-19 vaccines in the pipeline, are antibodies adequate? Vaccines 2021, 9, 241. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.K.; Hassanzadeganroudsari, M.; Apostolopoulos, V. Why METH users are at high risk of fatality due to COVID-19 infection? Expert Rev. Vaccines 2020. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.K.; Hassanzadeganroudsari, M.; Apostolopoulos, V. The emergence of new strains of SARS-CoV-2. What does it mean for COVID-19 vaccines? Expert Rev. Vaccines 2021, 1–4. [Google Scholar] [CrossRef]
- Taccone, F.S.; Gorham, J.; Vincent, J.-L. Hydroxychloroquine in the management of critically ill patients with COVID-19: The need for an evidence base. Lancet Respir. Med. 2020, 8, 539–541. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic treatments for coronavirus disease 2019 (COVID-19): A review. JAMA 2020, 323, 1824–1836. [Google Scholar] [CrossRef]
- Ferner, R.E.; Aronson, J.K. Chloroquine and hydroxychloroquine in covid-19. BMJ 2020, 369, m1432. [Google Scholar] [CrossRef] [Green Version]
- Geleris, J.; Sun, Y.; Platt, J.; Zucker, J.; Baldwin, M.; Hripcsak, G.; Labella, A.; Manson, D.K.; Kubin, C.; Barr, R.G.; et al. Observational study of hydroxychloroquine in hospitalized patients with Covid-19. N. Engl. J. Med. 2020, 382, 2411–2418. [Google Scholar] [CrossRef]
- Theoharides, T.; Conti, P. Dexamethasone for COVID-19? Not so fast. J. Biol. Regul. Homeost Agents 2020, 34. [Google Scholar] [CrossRef]
- Chen, H.; Wang, F.; Zhang, P.; Zhang, Y.; Chen, Y.; Fan, X.; Cao, X.; Liu, J.; Yang, Y.; Wang, B.; et al. Management of cytokine release syndrome related to CAR-T cell therapy. Front. Med. 2019, 13, 610–617. [Google Scholar] [CrossRef]
- Giles, A.J.; Hutchinson, M.-K.N.; Sonnemann, H.M.; Jung, J.; Fecci, P.E.; Ratnam, N.M.; Zhang, W.; Song, H.; Bailey, R.; Davis, D. Dexamethasone-induced immunosuppression: Mechanisms and implications for immunotherapy. J. Immunother. Cancer 2018, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Götte, M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 2020, 295, 4773–4779. [Google Scholar] [CrossRef] [Green Version]
- Sadeghi, A.; Ali Asgari, A.; Norouzi, A.; Kheiri, Z.; Anushirvani, A.; Montazeri, M.; Hosamirudsai, H.; Afhami, S.; Akbarpour, E.; Aliannejad, R.; et al. Sofosbuvir and daclatasvir compared with standard of care in the treatment of patients admitted to hospital with moderate or severe coronavirus infection (COVID-19): A randomized controlled trial. J. Antimicrob. Chemother. 2020, 75, 3379–3385. [Google Scholar] [CrossRef]
- Nourian, A.; Khalili, H. Sofosbuvir as a potential option for the treatment of COVID-19. Acta Biomed. Atenei Parm. 2020, 91, 239. [Google Scholar]
- Sisay, M. 3CLpro inhibitors as a potential therapeutic option for COVID-19: Available evidence and ongoing clinical trials. Pharmacol. Res. 2020, 156, 104779. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Li, S.; Xiong, Y.; Tian, M.; Yu, J.; Xu, L.; Zhang, L.; Li, Z.; Ma, J.; Wen, F.; et al. COVID-19 pneumonia in a hemodialysis patient. Kidney Med. 2020, 2, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Javed, H.; Meeran, M.F.N.; Jha, N.K.; Ojha, S. Carvacrol, a Plant Metabolite Targeting Viral Protease (Mpro) and ACE2 in Host Cells Can Be a Possible Candidate for COVID-19. Front. Plant Sci. 2020, 11. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).