
Oxidation of Antipsychotics




1
Institute of Personalized Psychiatry and Neurology, Shared Core Facilities, V. M. Bekhterev National Medical Research Centre for Psychiatry and Neurology, 192019 Saint Petersburg, Russia
2
Shared Core Facilities “Molecular and Cell Technology”, V. F. Voino-Yasenetsky Krasnoyarsk State Medical University, 660022 Krasnoyarsk, Russia
3
International Centre for Education and Research in Neuropsychiatry, Samara State Medical University, 443099 Samara, Russia
*
Authors to whom correspondence should be addressed.
Encyclopedia 2022, 2(2), 974-989; https://doi.org/10.3390/encyclopedia2020064
Submission received: 21 February 2022
/
Revised: 25 April 2022
/
Accepted: 5 May 2022
/
Published: 19 May 2022
(This article belongs to the Section Medicine & Pharmacology)
Definition
:Antipsychotics (APs) are psychotropic drugs that generally have a psycholeptic effect, capable of reducing psychotic symptoms and psychomotor agitation. This class of drugs is widely used in psychiatric practice, especially for the treatment of psychosis in schizophrenia and other psychotic disorders. Most APs pass through a biotransformation process, or metabolism, after they enter the body before being eliminated. There are three phases of AP metabolism. Cytochrome P450 (CYP) monooxygenase (mixed-function oxidase) plays a central role in most AP biotransformation. CYP’s functional activity depends on gene–drug and drug–drug interaction and influences on the occurrence of adverse drug reactions (ADRs). So, it is extremely important for a practicing psychiatrist to know the oxidation pathway of APs, since most of them are metabolized in the liver. This is important both to prevent ADRs and to avoid unwanted drug–drug interactions, which will undoubtedly increase the effectiveness and safety of AP therapy.
1. Introduction
Antipsychotics (APs) are a class of psychotrophic medication primarily used to manage psychosis (including delusions, hallucinations, paranoia, or disordered thought), especially for the treatment of psychosis in schizophrenia and other psychotic disorders [1,2]. They, along with mood stabilizers, are also the first line of treatment for bipolar affective disorder [3]. First-generation APs (FGAs), conventional or typical antipsychotics, have significant potential to cause extrapyramidal syndrome (akathisia, acute distonic reactions, tardive dyskinesia, pseudo-Parkinson’s, and others) [4]. The main difference between FGAs and second generation APs (SGA) is the predisposition to cause these type of adverse drug reactions (ADRs) [5]. In other respects, such as other ADRs and their mechanism of action, the two classes have substantial overlap and comparable efficacy [6].
Most APs pass through a biotransformation process, or metabolism, after they enter the body before being eliminated [7]. In the course of biotransformation, APs are converted into more water-soluble suspensions, and, therefore, are subsequently more easily excreted from the body.
In the process of AP metabolism, most initial APs lose their pharmacological action and are removed from the body through excretion. During biotransformation, produced metabolites usually are more polar or charged than the parent APs, which increases the rate of clearance; this modification can also decrease reabsorption in the tubules [8].
In the process of biotransformation, APs usually become less pharmacologically active or completely inactive compounds, but also newly formed metabolites can be equally pharmacologically active and even more pharmacologically active compounds if the original APs was a prodrug. As a result of AP biotransformation new metabolites are formed: With changed and new pharmacological actions, these new metabolites may have both lower and higher potencies in comparison with initial APs; new metabolites may also have a toxic effect, or new metabolites may be active, if the parent APs were prodrugs [9].
Biotransformation reactions of drugs and endogenous substances often develop over sequential stages, such reactions occur with the participation of enzymes and enzyme systems. Most APs undergo biotransformation in the liver, also some APs are metabolized in other organs and tissues [10].
Biotransformation reactions occur with the participation of specific enzymes or enzyme systems. These enzymes can catalyze both xenobiotic metabolism, in this case APs, and substances with endogenic origin, such as hormones. Most often, AP biotransformation reactions occur in the liver; however, individual APs undergo these reactions to a greater or lesser extent in other organs and tissues of the human body.
The process of AP biotransformation is quite changeable, and this variability depends on many factors, for example:
- -
- Nutritional status;
- -
- Hormonal status;
- -
- Genetic factors;
- -
- Previous therapy with Aps or other classes of drugs;
- -
- Concomitant somatic, neurological, or mental status (for example, diseases of the cardiovascular and respiratory systems may decrease biotransformation, etc.);
- -
- I age of the patient (for example, very old patients or children often have a greater sensitivity to APs, due in part to the involutional or immature state of the enzyme systems by which APs are metabolized);
- -
- Functional state of the liver [11].
2. Phases of Antipsychotic Metabolism
2.1. Phase I of Antipsychotic Metabolism
As a result of phase I reactions, the initial APs usually become less active. These reactions are non-synthetic or happen in the absence of conjugation processes. Important to note is that when the formed metabolites after reaction I become sufficiently polar, they may be immediately excreted from the human body. Otherwise, the following reaction occurs, which combines the formed metabolites with new functional groups to form greatly polar, and therefore more water-soluble, active metabolites by unmasking or inserting a polar functional group (–OH, –SH, –NH2) that enable the following stages of biotransformation [13]. APs metabolized via phase I reactions have longer half-lives. Geriatric patients have decreased phase I metabolism; thus, geriatric patients metabolize APs by phase II reactions [12,14].
Reactions of phase I:
- -
- Oxidation;
- -
- Hydrolysis;
- -
- Reduction.
In these reactions, for subsequent conjugation, functional groups are added to the formed metabolites, which then become the active center in the phase II reaction [15].
Enzymes catalyzing this phase’s biotransformation are mostly from the cytochrome P450 (CYP) system, the flavin-containing monooxygenase system, monoamine oxidase, aldehyde and alcohol dehydrogenase, deaminases, esterases, amidases, and epoxide hydratases [16,17].
Oxidation reactions, which occur with CYP enzymes (mixed-function oxidases (MFOs) or mono-oxygenases) take place in the smooth endoplasmic reticulum (ER) of the cell. These reactions involve cytochrome P450 reductase, nicotinamide adenine dinucleotide phosphate (NADPH), and oxygen (O2). CYP enzymes also better metabolize APs with high fat solubility [17].
The CYP system is involved in numerous reactions, for example:
- -
- Other oxidations;
- -
- Dealkylation;
- -
- Deamination;
- -
- Sulfoxidation;
- -
- Oxidation [18].
NADPH–cytochrome P450 reductase catalyzes reduction reactions mostly in the ER or the cytosol. Being a membrane-bound enzyme, it transfers electrons from NADPH to heme proteins and CYP, including heme oxygenase from a flavin adenine dinucleotide (FAD)- and flavin mononucleotide (FMN)-containing enzyme NADPH-cytochrome P450 reductase [19]. The electron flow cheme is:
NADPH → FAD → FMN → P450 → O2
In reduction reactions, a substance receives a free-radical electron and then quickly loses it to oxygen, and a superoxide anion forms [20].
Hydrolytic reaction is a phase I reaction in which a water molecule joins and bond breakage subsequently happens, this reaction doesn’t occur in the ER [21].
2.2. Phase II of Antipsychotic Metabolism
Phase II (synthetic) reactions include conjugation reactions, adding highly polar groups (such as glutathione (GSH), sulfate, glycine, acetyl, or glucuronic acid, amino acids, or methyl) to the APs to increase renal elimination, which involves the enzyme-catalyzed combination of APs (or AP metabolites) with an endogenous substance. Through these reactions, the drug activity is decreased and polarity is increased. The functional group of substance is an active center in phase II reactions, this active center acts as the site of conjugation with the endogenous substance [22]. Carboxyl (–COOH), hydroxyl (–OH), amino (NH2), and thiol (–SH) groups that were attached in the previous reactions are now the sites on the APs for conjugation [23]. In this phase, fewer active products are formed that have higher molecular weight than previous substrates in comparison. In addition, through the attachment of large anionic groups like GSH, more polar metabolites are produced and reactive electrophiles are detoxified. Now these metabolites cannot actively move and diffuse through cell membranes [23]. Phase II reactions can take place on their own or after phase I. The synthesis of endogenous substances, the so-called “activated carriers” that are needed in the conjugation reaction (e.g., uridine diphosphate–glucuronate), requires energy [24].
Phase II reactions, in particular, glucuronidation, take place in the ER. To this end, uridine diphosphate glucuronic acid (UDPGA) is formed with the help of glucose, and the acid formed with the participation of glucuronyl transferase attaches the glucuronide to APs. APs can also be conjugated with other substances through transferases; these reactions often occur in the cytosol of the cell.
Numerous transferases in various combinations can metabolize most hydrophobic substances that have nucleophilic or electrophilic groups in phase II reactions [23,25].
Below are some conjugation enzymes of phase II biotransformation:
- -
- Acetylases;
- -
- Glucuronyl transferase;
- -
- Transacylases;
- -
- Sulfotransferase;
- -
- Ethylase;
- -
- Glutathione transferase;
- -
2.3. Phase III of Antipsychotic Metabolism
Phase III is the final point of AP transformation and its excretion. Often, phase II products are transported out of the cell via transport proteins of the ATP-binding cassette transporter family, where they undergo further metabolism or excretion. These proteins provide ATP-dependent transport of a wide range of hydrophobic anions. Anionic groups from previous reactions are now affinity tags for these membrane carrier proteins [27].
3. Oxidation of Antipsychotics in the Liver
The most important role in the metabolism of most APs is played by CYP monooxygenase (Figure 1 and Figure 2) (mixed-function oxidase) [28]. In mammals, at least 18 families of enzymes of the CYP system have been discovered so far. Individual enzymes of this system are involved in the biotransformation of certain APs, with a unique substrate specificity. This specificity may partially coincide in different enzymes of the CYP system (Table 1) [29]. Currently, at least 50 different P450 enzymes are known, but approximately 12 of them provide biotransformation of most APs. As mentioned above, the CYP family catalyzes phase I reactions. Nomenclature: the family number is indicated immediately after the term “CYP” with an Arabic numeral, the subfamily is named by a capital letter of the Latin alphabet, and the second Arabic numeral after the letter indicates a particular enzyme in a subfamily. As a result, the enzyme designation has the following form: CYP2D6, CYP3A4, CYP3A5, etc. [30].
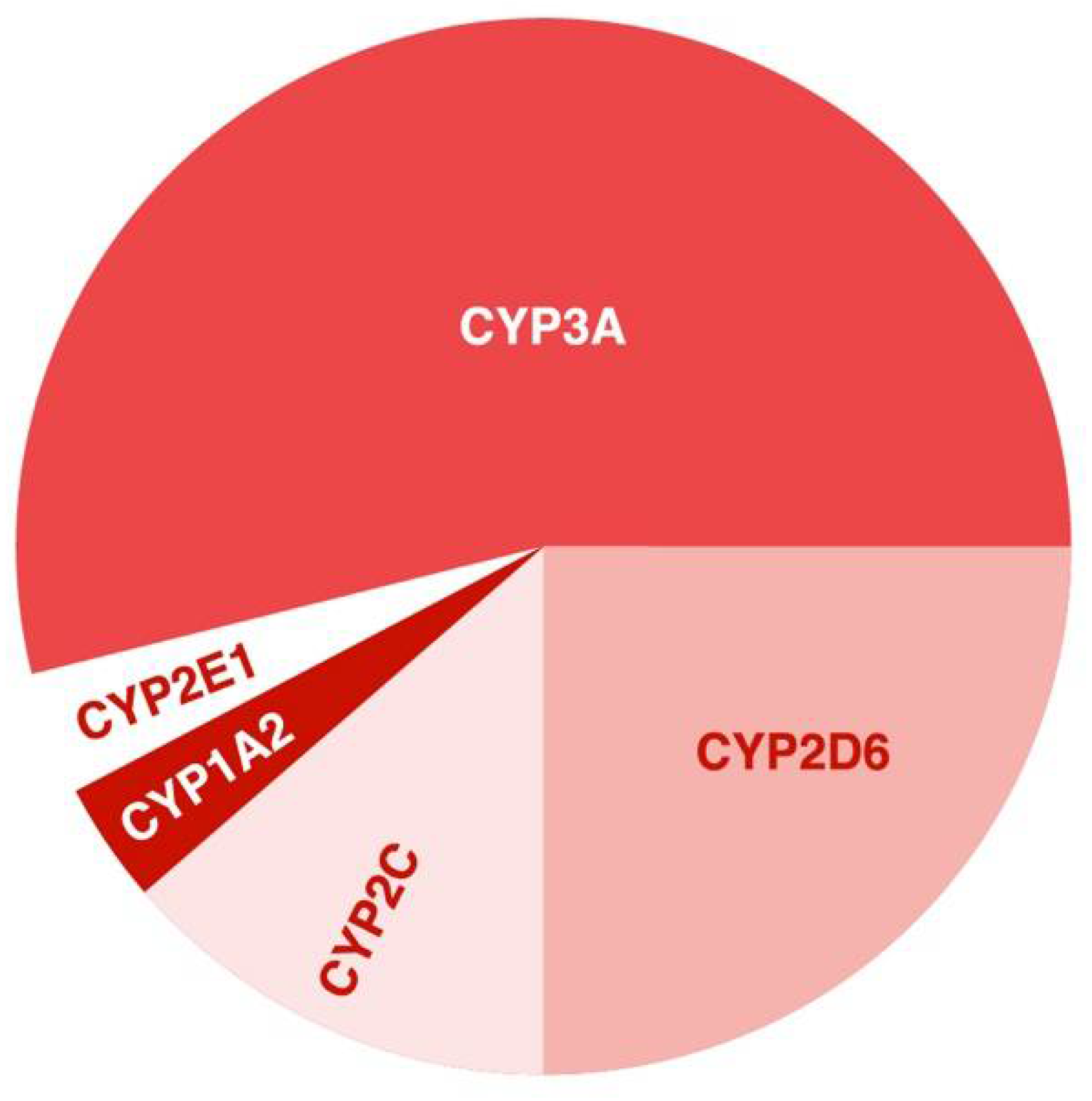
CYP2D6, CYP3A4, CYP3A5, CYP2C9, and CYP2C19 provide most of the activity (more than 50%) of P450; these enzymes prevail among liver enzymes (Figure 2) and are involved in the metabolism of most APs [31].
CYP participates in different reactions, for example catalyzes dealkylation at nitrogen, sulfur, and oxygen atoms; catalyzes aromatic and aliphatic hydroxylations; catalyzes reductions at nitrogen atoms; and ester and amide hydrolysis; heteroatom oxidations at nitrogen and sulfur atoms.
Most often, CYP enzymes are located in the liver, but they are also found in other organs and tissues of the human body—for example, in the small and large intestines, testicles or ovaries, duodenum, pancreas, kidneys, spleen, lymph node, and others [32]. The expression of some CYP enzymes is shown in Figure 3 [33]. In cells, the enzymes of the CYP system are located in the ER [17].
Phase I, involving CYP, has an oxidative and a reduction reaction. Synthesis of NADPH is dependent on cytochrome P450 reductase. The cofactor NADPH is involved in the reduction of oxygen to water in the general reaction where AP is oxidized.
Below is an aromatic hydroxylation reaction [34]:
Drug +O2 + NADPH + H+ → Drug-OH + NADP+ + H2O
CYP activity is variable and depends, among other things, on drug–drug interaction; APs and other drugs may modulate the work of particular CYP enzymatic pathways. Thus, the metabolism of concomitantly administered drugs may be changed. All drugs, including APs, can be divided into three groups related to the CYP system: substrates, inducers, and inhibitors of this system. Substrates are drugs metabolized under CYP enzyme catalytic activity [35].
P450 inhibitors are drugs that inhibit the biotransformation of drugs metabolized by a certain CYP enzyme; inhibition of drugs metabolized by the same CYP enzyme is also suppressed. There is competitive inhibition if drugs compete for the CYP enzyme, and non-competitive if a certain drug tightly binds to the CYP. When there is structural similarity between the substrate and the inhibitor at the molecular level, competitive inhibition occurs: the competitive inhibitor binds to the active site of the enzyme instead of the substrate, thus less substrate is bound to the enzymes. When the inhibitor and substrate have a dissimilar molecular structure, the inhibitor attaches to the enzyme and changes its structure and active site, thereby slowing down the reaction between the enzyme and the substrate [36]. Inhibition raises therapeutic drug levels (danger of toxicity) [35]. There are a lot of inhibitors among different drug groups—for example, Aps (haloperidol, olanzapine, clozapine, and others), ADs (fluvoxamine, clomipramine, duloxetine, and others), antiepileptic drugs (valproic acid, phenytoin, topiramate, and others), somatic drugs (isoniazid, cimetidine, ketoconazole, fluconazole, and others) [37], acute alcohol abuse, and grapefruit juice [36].
P450 inducers increase the amount of P450 enzymes in vivo. This process is associated with the activation of enzyme synthesis and lowers therapeutic drug level. A decrease in the therapeutic level of the drug, in particular APs, may occur due to the induction of the CYP enzymes, since the metabolism of drugs catalyzed by a certain enzyme is accelerated, as well as the metabolism of the inducer itself if it is metabolized by the same CYPs [38]. A significant number of drugs are inducers of various cytochrome enzymes, causing drug–drug interactions, for example, APs (clozapine, chlorpromazine, and others), antiepileptic drugs (phenytoin, carbamazepine, topiramate, and others), somatic drugs (griseofulvin, troglitazone, omeprazole [37], St. John’s wort [36], and others), chronic alcohol abuse [36], and environmental agents such as tobacco smoke [39].
When considering AP metabolism in the liver, we should also consider the extraction ratio and first-pass effect definitions.
The weight of a liver is 1.500 g, and the liver has high blood flow (1 mL/g/min), which provides massive excretion of drugs, in particular APs [40]. The amount of drugs remoted by the liver divided by the amount of drugs entering the organ is the extraction ratio; the extraction ratio is 1 when a drug is completely removed by the liver, if the bioavailability of the drug is 100%, then the hepatic extraction ratio is 0 and vice versa [41]. Hepatic clearance may be close to 1500 mL/min if APs are highly extracted by the liver [42]. The first pass effect is the process, when the bioavailability of some orally administered drugs is reduced because the fraction is removed during the first pass through the liver [43]. The pharmacokinetics of APs: absorption in the gastrointestinal tract, transport through the portal vein to the liver, after which APs reaches the general bloodstream. Thus, APs taken orally have the first-pass effect. Correction of the dosing regimen when patient has liver diseases is required, since in such cases a greater amount of APs reaches the systemic circulation, which may increase the therapeutic window. Every drug has a half-life, a fixed time it takes for a drug to lose half of its pharmacological action [41,44].
Some clinically relevant CYPs, such as CYP2C and CYP2D, have genetic polymorphisms, which influence metabolic variability in individuals. For example, different races and ethnic groups have different variability of certain enzymes. Since a genetically determined difference in the properties of CYP enzymes, such as Vmax or Km, affects the rate of AP metabolism, this should be taken into account when selecting therapy [45]. For example, the CYP3A subfamily is responsible for up to half of the total cytochrome P-450 in the liver [46]. CYP3A4 is the most abundant hepatic enzyme and is involved in the metabolism of over 50% of clinically important APs. APs or other drugs that are inhibitors or inducers of the CYP system can cause adverse drug reactions (ADRs) due to altered metabolism of these enzymes, which affects the concentration of metabolized drugs by influencing phase I reactions [47].
4. Function of Enzymes of Cytochrome P450 and Antipsychotic Metabolism
There may be serious drug-drug interactions between FGAs and different groups of drugs which may affect cytochrome P450 enzymes (Table 1) [48,49].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antipsychotics | Other Drugs | ||||
|---|---|---|---|---|---|
| Substrates | Inhibitors | Inducers | Substrates | Inhibitors | Inducers |
| CYP1A1 | |||||
| Partly: Haloperidol Olanzapine Perospirone | Clozapine | Acetaminophen Amiodarone R-Warfarin | Propofol | ||
| CYP1A2 | |||||
| Primary: | Promazine | Acetaminophen | Amiodarone | Carbamazepine | |
| Asenapine | Remoxipride | Alosetron | Cimetidine | Insulin | |
| Clozapine | Caffeine | Ciprofloxacin | Modafinil | ||
| Loxapine | Clomipramine | Citalopram | Nafcillin | ||
| Olanzapine | Duloxetine | Efavirenz | Omeprazole | ||
| Pimozide | Imipramine | Fluvoxamine | Rifampin | ||
| Thiothixene | Melatonin | Ribociclib | Rucaparib | ||
| Trifluoperazine | Mexiletine | Teriflunomide | |||
| Naproxen | Tobacco | ||||
| Partly: | Pirfenidone | ||||
| Chlorpromazine | Theophylline | ||||
| Haloperidol | Tizanidine | ||||
| Lumateperone | |||||
| Perphenazine | |||||
| Promazine | |||||
| Quetiapine | |||||
| Thioridazine Zotepine | |||||
| CYP2A6 | |||||
| Partly: | Cotinine | Methoxsalen | |||
| Clozapine | Nicotine | Pilocarpine | |||
| Promazine | Pilocarpine | Tryptamine | |||
| CYP2C8 | |||||
| Partly: | Amodiaquine | Abiraterone | |||
| Clozapine | Cerivastatin | Clopidogrel | |||
| Lumateperone | Dabrafenib | Deferasirox | |||
| Perospirone | Enzalutamide | Glitazones | |||
| Perphenazine | Olodaterol | Letermovir | |||
| Paclitaxel | Montelukast | ||||
| CYP2C9 | |||||
| Partly: | Amitriptyline | Amiodarone | Bosentan | ||
| Clozapine | Azilsartan | Capecitabine | Carbamazepine | ||
| Haloperidol | Capecitabine | Ceritinib | Enzalutamide | ||
| Olanzapine | Celecoxib | Efavirenz | Nevirapine | ||
| Perphenazine | Clopidogrel | Fenofibrate | Peginterferon alfa-2b | ||
| Promazine | Diclofenac | Fluconazole | Phenobarbital | ||
| Doxepin | Fluvastatin | Rifampin | |||
| Fluoxetine | Fluvoxamine | St. John’s wort | |||
| Fluvastatin | Isoniazid | Tocilizumab | |||
| CYP2C18 | |||||
| Partly: | Mephenytoin | Rifampicin | |||
| Perphenazine | Warfarin | ||||
| CYP2C19 | |||||
| Partly: | Clozapine | Amitriptyline | Armodafinil | Carbamazepine | |
| Clozapine | Olanzapine | Atomoxetine | Cetocanazole | Efavirenz | |
| Haloperidol | Brivaracetam | Cimetidine | Letermovir | ||
| Perphenazine | Carisoprodol | Citalopram | Prednisone | ||
| Promazine | Citalopram | Esomeprazole | Rifampicin | ||
| Pipotiazine | Clobazam | Felbamate | Ritonavir | ||
| Quetiapine | Clomipramine | Fluoxetine | St. John’s wort | ||
| Risperidone | Clopidogrel | Fluvoxamine | |||
| Thioridazine | Diazepam | Isoniazid | |||
| Doxepin | Modafinil | ||||
| Escitalopram | |||||
| CYP2D6 | |||||
| Primary: | Amoxapine | Atomoxetine | Amiodarone | Dexamethasone | |
| Aripiprazole Brexpiprazole Chlorpromazine Fluphenazine Haloperidol Iloperidone Loxapine Perphenazine Pimozide Risperidone Thioridazine Partly: Alimemazine Amoxapine Aripiprazole lauroxil Azenapine Cariprazine Clozapine Clozapine Flupentixol Levomepromazine Mesoridazine Methotrimeprazin Olanzapine Paliperidone Perospirone Pipotiazine Prochlorperazine Promazine Quetiapine Remoxipride Sertindol Trifluperazine Zuclopenthixol | Chlorpromazine Clozapine Fluphenazine Haloperidol Melperone Methotrimeprazine Olanzapine Perphenazine Pimozide Pipotiazine Risperidone Thioridazine Thiothixene | Carvedilol Citalopram Clomipramine Debrisoquine Desipramine Dexfenfluramine Dextromethorphan Donepezil Doxepin Duloxetine Letermovir Lidocaine Metoprolol Nebivolol Perphenazine Propranolol | Bupropion Celecoxib Cimetidine Clobazam Clomipramine Doxepin Duloxetine Fluoxetine Hydroxyzine Methadone Metoclopramide Paroxetine Quinidine Ritonavir Sertraline | Oritavancin Rifampin | |
| CYP2E1 | |||||
| Partly: | Fluphenazine | Acetaminopher | Disulfiram | Ethanol | |
| Clozapine Iloperidone | Methotrimeprazine Thioridazine | Aniline Chlorzoxazone Enflurane Ethanol | Quercetin Ribociclib | Isoniazid | |
| CYP3A4 | |||||
| Primary: Aripiprazole Brexpiprazole Cariprazine Haloperidol Loxapine Lumateperone Lurasidone Perphenazine Pimavanserin Pimozide Quetiapine Ziprasidone Partly: Alimemazine Asenapine Clozapine Fluspirilene Iloperidone Paliperidone Penfluridol Perospirone Pipotiazine Promazine Risperidone Sertindol Zotepine Zuclopenthixol | Clozapine Haloperidol Olanzapine Remoxipride | Chlorpromazine Clozapine | Alprazolam Bupivacaine Buspirone Disopyramide Eszopiclone Etonogestrel Flunisolide Grepafloxacin Indinavir Pantoprazole Ranolazine Terfenadine Voriconazole Zolpidem | Betamethasone Fluconazole Loratadine Quinine Voriconazole | Betamethasone Quinine Rifabutin Rofecoxib |
| CYP3A5 | |||||
| Primary: Aripiprazole Aripiprazole lauroxil Clozapine Haloperidol Iloperidone Olanzapine Paliperidone Partly: Pimavanserin Pimozide Quetiapine Risperidone | Remoxipride Reserpine | Clopidogrel Cyclosporine Indinavir Phenytoin Saquinavir Verapamil | Indinavir Saquinavir Verapamil | Carbamazepine Dexamethasone Phenytoin | |
| CYP3A7 | |||||
| Partly: Aripiprazole Aripiprazole lauroxil Haloperidol Iloperidone Pimozide Quetiapine | Remoxipride | Alprazolam Astemizole Diazepam Triazolam | Erythromycin Nelfinavir Saquinavir | Dexamethasone Phenytoin Triamcinolone | |
| CYP3A43 | |||||
| Olanzapine | Daclatasvir | Cobicistat | Dexamethasone | ||
| Remoxipride | Testosterone | Idelalisib | Rifampicin | ||
Many FGAs have multiple clearance pathways, such as haloperidol, perphenazine, chlorpromazine, thioridazine, and loxapine. That is why the effect of inducers and inhibitors of cytochrome enzymes on their metabolism is moderate [37].
Fluphenazine: Metabolized predominantly by CYP2D6, it is recommended to avoid co-administration of fluphenazine with drugs that strongly inhibit CYP2D6 [50].
Pimozide: Has an increased risk of QTc prolongation, so it is recommended not to prescribe CYP2D6 inhibitors with pimozide, although the latter has a moderate sensitivity to inhibition [51].
Chlorpromazine: has a pronounced sensitivity to CYP1A2 induction, with heavy smoking, an increase in the dosage of chlorpromazine, as well as thiothixene, is required, since smoking induces the CYP1A2 enzyme and reduces the concentration of CYP1A2 substrates in the blood serum [52,53,54].
Most SGAs are sensitive to inhibition or induction of CYP enzymes, which affects the change in the concentration of these drugs in the blood serum and requires dosage adjustment when administered simultaneously with drugs that affect the rate of cytochrome P450 enzymes, these data are presented in Table 1 [48,49].
Aripiprazole: When combined with other APs, there may be a decrease in the AP effect with an increase in the dosage of aripiprazole due to its partial agonism at D2 receptors. Since CYP3A4 and CYP2D6 are involved in the metabolism of aripiprazole, when this drug is combined with strong inhibitors of these enzymes, for example, with fluoxetine, a two-fold reduction in the dosage of aripiprazole is necessary, while when combined with strong inducers of CYP3A4 and CYP2D6 enzymes, for example, with carbamazepine, a twofold increase in dosage is necessary [55].
Asenapine: This AP is metabolized by CYP1A2 enzymes and glucuranization enzymes, so asenapine does not appear to be sensitive to CYP inducers or inhibitors. Undesirable drug-drug interactions may occur when combined with drugs that have similar ADRs: weight gain, sedation, parkinsonism [56].
Brexpiprazole: This APs is metabolized by CYP3A4 and CYP2D6. Brexpiprazole is sensitive to changes in CYP function, for example, rifampin as an inducer of CYP3A4 reduces serum levels of CYP3A4 substrates by 75%. Inhibitors of CYP3A4 and CYP2D6 enzymes double the serum concentration of brexpiprazole [57]. When combined with other APs, there may be a decrease in the AP effect with an increase in the dosage of Brexpiprazole due to its partial agonism at D2 receptors [58].
Cariprazine: This AP and its metabolites are catalyzed by the CYP3A4 enzyme; when combined with CYP3A4 inhibitors, a 50% dose reduction of brexpiprazole is necessary [59]. There are no studies evaluating interactions of cariprazine with CYP3A4 inducers.
Clozapine: This drug is metabolized primarily by CYP1A2 and CYP3A4, and partially by CYP2D6 [60]. Combination with strong inhibitors of CYP1A2, such as fluvoxamine or ciprofloxacin, requires a reduction in clozapine dosage to one-third of the initial dose. It is recommended not to prescribe clozapine with drugs that have similar adverse reactions [61]. It is also necessary to increase the dosage of clozapine twice for heavy smokers or reduce the dosage by 30–40% when quitting smokers [62].
Iloperidone: Metabolized by CYP2D6 and CYP3A4 enzymes, when combined with strong inhibitors of these enzymes, such as fluoxetine and paroxetine, a dose reduction of 50% is necessary [63].
Lumateperone: CYP3A4, CYP2C8 and CYP1A2 are involved in its metabolism. It is recommended not to prescribe a combination with strong or moderate inhibitors and inducers of CYP3A4. Also, the combination with valproic acid may increase the concentration of lumateperone due to the inhibition of uridine diphosphoglucuronate glucuronosyltransferase [64].
Lurasidone: Predominantly metabolized by CYP3A4. The co-administration of strong inhibitors or inducers of CYP3A4 such as rifampin or ketoconazole with lurasidone is contraindicated [65].
Olanzapine: This medication is dependent on CYP1A2 metabolism [66]. Co-administration with strong inhibitors or inducers of CYP1A2 can affect olanzapine serum levels; for example, cigarette smoking decreases its serum concentration.
Paliperidone: Metabolized in part with the participation of CYP3A4, efflux occurs with the participation of P-glycoprotein 1. Therefore, when combined with strong inducers such as carbamazepine and rifampicin, an increase in the dose of paliperidone is required, but if a strong inducer was canceled and paliperidone continued, a dose reduction of this AP is necessary [67].
Pimavanserin: Metabolized predominantly by CYP3A4 and partially by CYP3A5, dosage adjustment is necessary when combined with strong inhibitors of CYP3A4. Since pimavanserine can prolong the QT interval, the combination with drugs that have a similar effect is undesirable [68].
Quetiapine: Metabolized predominantly by CYP3A4, a five-fold increase in dose is recommended when combined with carbamazepine, and a dosage reduction to one-sixth when combined with strong inhibitors, such as ritonavir [69,70]. A list of strong inhibitors and inducers of CYP3A4 is presented in Table 1 [36,37,39,48,49].
Risperidone: AP is metabolized by CYP2D6, and serum concentrations of risperidone are increased when combined with fluoxetine, paroxetine, and bupropion as inhibitors of CYP2D6 [71].
Ziprasidone: Glutathione and aldehyde oxidase are predominantly involved in the metabolism of this AP, CYP3A4 - partially, so the change in the work of CYP3A4 is not clinically pronounced [72].
5. Discussion of Prospects for Translation of Basic Research into Real Clinical Practice
As mentioned above, it is extremely important for a practicing psychiatrist to know the oxidation pathway of APs, since most of them are metabolized in the liver. This is important both to prevent ADRs and to avoid unwanted drug–drug interactions, which will undoubtedly increase the effectiveness and safety of AP therapy [73]. At the same time, it is possible to study the activity of one or another cytochrome and changes in the oxidation of the enzyme due to changes in its activity only experimentally. In the organism of a patient suffering from mental disorders, we can only indirectly judge the change, taking into account modifiable and non-modifiable factors, including genetically determined changes.
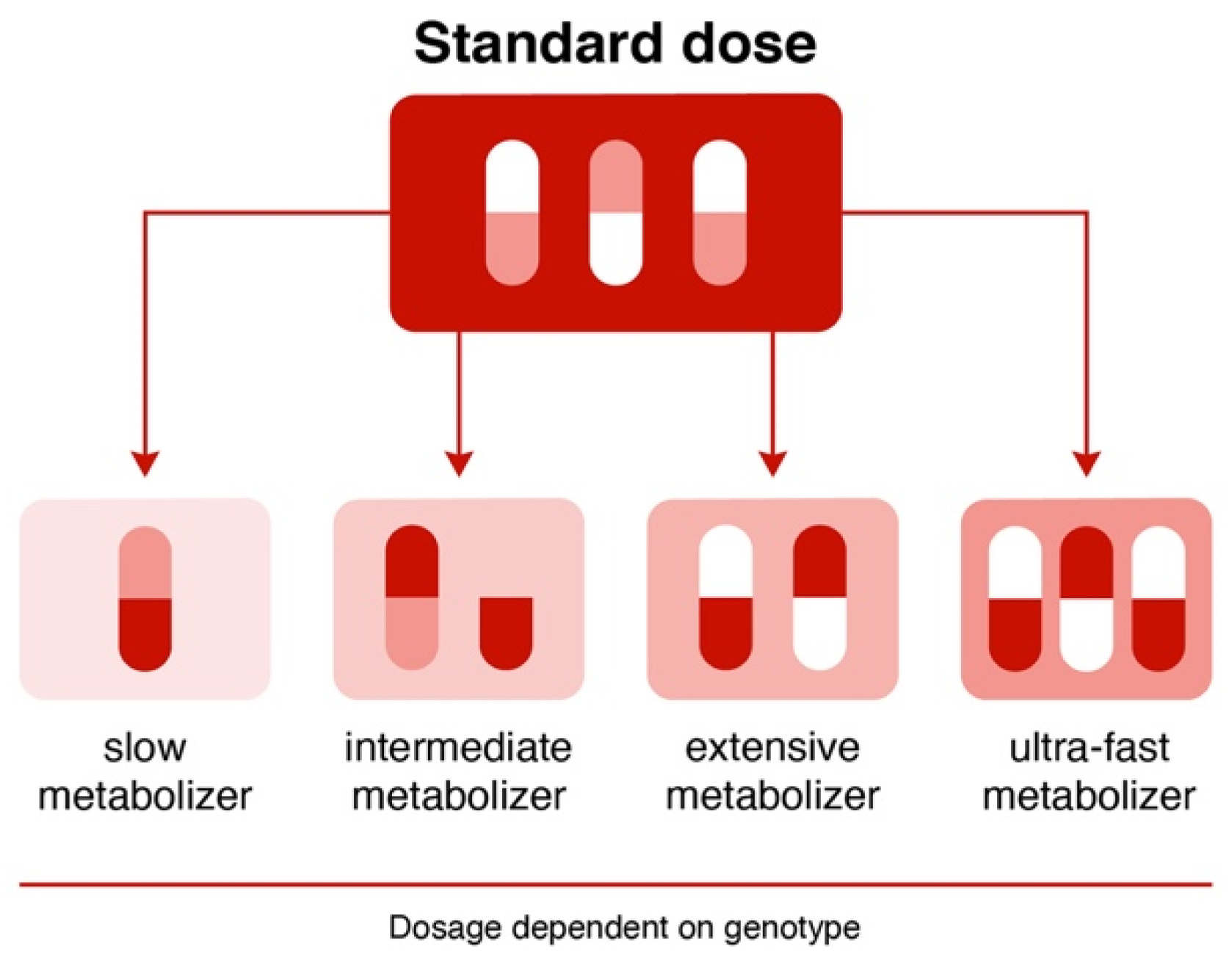
Currently, the rapid development of molecular genetics and fundamental and clinical pharmacogenetics indicate that the study of non-functional and low-functional single nucleotide variants (SNVs)/polymorphism of the genes encoding CYP enzymes can help in translating fundamental knowledge about the oxidation of APs into real clinical practice. Currently, depending on the genetically determined change in the degree of enzyme activity, four phenotypes are distinguished (Figure 4) [74].
In connection with this, introducing various pharmacogenetic panels is very promising—for example, the AmpliChip CYP450 pharmacogenetic test, which allows information to be obtained about the pharmacogenetic profile of a patients with mental disorders, depending on the carriage of allelic genotypes of two non-functional SNVs/polymorphisms of cytochrome P450 genes (CYP2D6 and CYP2C19). According to the test results, patients are divided into two phenotypes for the CYP2C19 gene: an extensive metabolizer and a poor metabolizer by testing for three SNVs, and into four phenotypes for the CYP2D6 gene by testing for 27 SNVs/polymorphisms, including seven duplications [75].
A good example of a system for evaluating the genetic contribution to AP metabolism in foreign practice is the GeneSight Psychotropic algorithm developed by a group of scientists based at the Mason Clinic (USA) [76]. The test is non-invasive and easy to use. GeneSight is based on a multi-gene multivariate genetic test that takes into account the characteristics of the genotype, phenotype, and information about the metabolism of the drug. The analysis is performed on allelic variants of 14 genes (CYP1A2, CYP2C9, CYP2C19, CYP3A4, CES1A1, CYP2B6, UGT1A4, UGT2B15, CYP2D6, HTR2A, HLA-A*3101, ADRA2A, HLA-B*1502, SLC6A4) [76]. The psychiatrist is provided with information already analyzed by the program based on the results of pharmacogenetic test (PGx). GeneSight contains a list of APs and Ads divided into three categories: “Use as directed,” “Use with caution,” and “Use with increased caution and with more frequent monitoring.” It also provides additional information that helps the psychiatrist decide whether to prescribe or cancel the drug in a particular patient [77].
Another PGx test, Genecept Assay, developed in the USA, makes it easier for the clinical pharmacologist to make decisions about prescribing APs and ADs. Genecept Assay makes it possible to predict the efficacy and safety of pharmacotherapy with these drugs in a wide range of mental disorders, including depression, obsessive-compulsive disorder, schizophrenia, attention-deficit/hyperactivity disorder, bipolar disorder, and others. SNVs are being studied in 20 genes encoding targets of AP and AD action, including 5HT2C, MC4R, DRD2, and COMT, and genes encoding isoenzymes of the cytochrome P450 system [78].
However, the data obtained for a cumulative assessment of the safety and efficacy of APs are not enough; it should be noted that 13 cytochrome P450 enzymes are involved in the oxidation of APs, and only a few of them are used in most real-life pharmacogenetic testing tools.
6. Limitation
The limitation of this entry paper is that it only took into account the oxidation pathway, although, undoubtedly, in order to predict and manage ADRs, it is necessary to take into account the role of other pathways, including oxidation not only in the liver, but also in brain neurons, in particular CYP1A1 and CYP1B1, which are expressed in the ER not only in the liver, but also in the brain [33].
In addition, the studies of P450 enzyme expression have shown that some of them are expressed not only in the liver, but also in other organs and systems. For example, CYP1A1 is expressed in the cerebellum, cerebral cortex, hippocampus, thyroid gland, parathyroid glands, adrenal glands, bronchi, lungs, tissues of the nasopharynx, oral mucosa, stomach, duodenum, rectum, liver, gallbladder, pancreas, kidneys, bladder, ovaries, testicles, epididymis, endometrium, placenta, tonsils, salivary glands, esophagus, prostate, fallopian tubes, cervix, heart muscle, skin, spleen, and lymph nodes [79]. This can probably cause the development of specific ADRs from certain organs and systems, and the translation of these pathways can help a practicing psychiatrist to suggest which organs and systems, when prescribing APs, need to be paid special attention when managing psychiatric patients. However, this approach has only just begun to be studied, and we have not found large studies.
7. Conclusions
Knowledge of the pathways of AP oxidation in the liver is very important from theoretical and practical points of view, since it can help to achieve an optimal balance between the efficacy and safety of APs in the practice of psychiatrists and other specialists.
In addition, it is important to remember that translating fundamental knowledge about oxidation into real practice is possible by expanding knowledge in the field of psychopharmacogenetics and by developing and introducing into clinical practice pharmacogenetic panels that would be useful to prescribe not at the stage of development of ADRs, but before the start of AP prescription.
However, it should be recognized that the solution of the tasks set is far from being discovered.
Author Contributions
Conceptualization, N.A.S.; methodology, R.F.N.; software, A.K.A.; validation, N.A.S., A.K.A. and R.F.N.; formal analysis, A.K.A.; investigation, A.K.A.; resources, A.K.A.; data curation, A.K.A.; writing—original draft preparation, N.A.S. and A.K.A.; writing—review and editing, R.F.N.; visualization, A.K.A.; supervision, N.A.S.; project administration, R.F.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Finkel, R.; Clark, M.A.; Cubeddu, L.X. Pharmacology, 4th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2008; p. 151. [Google Scholar]
- Lally, J.; MacCabe, J.H. Antipsychotic medication in schizophrenia: A review. Br. Med. Bull. 2015, 114, 1–179. [Google Scholar] [CrossRef] [PubMed]
- Grande, I.; Berk, M.; Birmaher, B.; Vieta, E. Bipolar disorder. Lancet 2016, 387, 1561–1572. [Google Scholar] [CrossRef]
- Caroff, S.N.; Hurford, I.; Lybrand, J.; Campbell, E.C. Movement Disorders Induced by Antipsychotic Drugs: Implications of the CATIE Schizophrenia Trial. Neurol. Clin. 2011, 29, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Sadock, B.J.; Sadock, V.A.; Ruiz, P. Kaplan and Sadock’s Comprehensive Textbook of Psychiatry, 9th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2009; pp. 4113–4119. [Google Scholar]
- Meltzer, H.Y. Update on typical and atypical antipsychotic drugs. An. Rev. Med. 2013, 64, 393–406. [Google Scholar] [CrossRef]
- Sheehan, J.J.; Sliwa, J.K.; Amatniek, J.C.; Grinspan, A.; Canuso, C.M. Atypical antipsychotic metabolism and excretion. Curr. Drug Metab. 2010, 11, 516–525. [Google Scholar] [CrossRef]
- Rourke, J.L.; Sinal, C.J. Biotransformation/Metabolism. Encycl. Toxicol. 2014, 1, 490–502. [Google Scholar]
- Shanu-Wilson, J.; Evans, L.; Wrigley, S.; Steele, J.; Atherton, J.; Boer, J. Biotransformation: Impact and Application of Metabolism in Drug Discovery. ACS Med. Chem. Lett. 2020, 11, 2087–2107. [Google Scholar] [CrossRef]
- Shen, W.W. The Metabolism of Atypical Antipsychotic Drugs: An Update. Ann. Clin. Psychiatry 1999, 11, 145–158. [Google Scholar] [CrossRef]
- Correia, M.A. Drug biotransformation. In Basic & Clinical Pharmacology, 14th ed.; Katzung, B.G., Ed.; McGraw Hill Education: New York, NY, USA, 2017; Volume 1, pp. 56–74. [Google Scholar]
- Josephy, D.P.; Guengerich, P.F.; Miners, J.O. “Phase I and Phase II” drug metabolism: Terminology that we should phase out? Drug Metab. Rev. 2005, 37, 575–580. [Google Scholar] [CrossRef]
- De Bruyn Kops, C.; Šícho, M.; Mazzolari, A.; Kirchmair, J. GLORYx: Prediction of the Metabolites Resulting from Phase 1 and Phase 2 Biotransformations of Xenobiotics. Chem. Res. Toxicol. 2021, 34, 286–299. [Google Scholar] [CrossRef]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, K. Drug Metabolism. Pharmacology 2009, 8, 131–173. [Google Scholar]
- Beedham, C. The role of non-P450 enzymes in drug oxidation. Pharm. World Sci. 1997, 19, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Danielson, P.B. The cytochrome P450 superfamily: Biochemistry, evolution and drug metabolism in humans. Curr. Drug Metab. 2002, 3, 561–597. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.V.; Flück, C.E. NADPH P450 oxidoreductase: Structure, function, and pathology of diseases. Pharmacol. Ther. 2013, 138, 229–254. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.; Møller, B.L. Plant NADPH-cytochrome P450 oxidoreductases. Phytochemistry 2010, 71, 132–141. [Google Scholar] [CrossRef]
- Klein, M.T.; Torry, L.A.; Wu, B.C.; Townsend, S.H.; Paspek, S.C. Hydrolysis in supercritical water: Solvent effects as a probe of the reaction mechanism. J. Supercrit. Fluids 1990, 3, 222–227. [Google Scholar] [CrossRef]
- Jancova, P.; Anzenbacher, P.; Anzenbacherova, E. Phase II drug metabolizing enzymes. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2010, 154, 103–116. [Google Scholar] [CrossRef]
- Jancova, P.; Siller, M. Phase II Drug Metabolism. Top. Drug Metab. 2012, 35–60. [Google Scholar]
- McCarver, D.G. The Ontogeny of Human Drug-Metabolizing Enzymes: Phase II Conjugation Enzymes and Regulatory Mechanisms. J. Pharmacol. Exp. Ther. 2002, 300, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Al-Zoughool, M.; Talaska, G. 4-Aminobiphenyl N-glucuronidation by liver microsomes: Optimization of the reaction conditions and characterization of the UDP-glucoronosyltransferase isoforms. J. Appl. Toxicol. 2006, 26, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, G.C.; Loose, D.S. Pharmacology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 12–15. [Google Scholar]
- Omiecinski, C.J.; Vanden Heuvel, J.P.; Perdew, G.H.; Peters, J.M. Xenobiotic Metabolism, Disposition, and Regulation by Receptors: From Biochemical Phenomenon to Predictors of Major Toxicities. Toxicol. Sci. 2010, 120, S49–S75. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R. Cytochrome P450 diversity in the tree of life. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 141–154. [Google Scholar] [CrossRef]
- Nasyrova, R.F.; Neznanov, N.G. Pharmacogenetics of antipsychotics. In Clinical Psychopharmacogenetics; Nasyrova, R.F., Kravtsov, V.V., Dobrodeeva, V.S., Schneider, N.A., Neznanov, N.G., Eds.; DEAN: Saint Petersburg, Russia, 2019; pp. 93–174. (In Russian) [Google Scholar]
- Uno, Y.; Iwasaki, K.; Yamazaki, H.; Nelson, D.R. Macaque cytochromes P450: Nomenclature, transcript, gene, genomic structure, and function. Drug Metab. Rev. 2011, 43, 346–361. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.P.; Laszlo, K. Rapid Review Pharmacology, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 7–9. [Google Scholar]
- Nelson, D.R. Cytochrome P450 Homepage. Hum. Genom. 2009, 4, 59–65. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 15 January 2022).
- Guengerich, F.P. Mechanisms of Cytochrome P450-Catalyzed Oxidations. ACS Catal. 2018, 7, 10964–10976. [Google Scholar] [CrossRef]
- Rendic, S.; Di Carlo, F.J. Human cytochrome P450 enzymes: A status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab. Rev. 1997, 29, 413–580. [Google Scholar] [CrossRef]
- Le, T.; Bhushan, V.; Sochat, M.; Vaidyanathan, V.; Schimansky, S.; Abrams, J.; Kallianos, K. First Aid for the USMLE Step 1, 30th ed.; McGraw Hill Education: New York, NY, USA, 2020; pp. 230, 252. [Google Scholar]
- Drugbak Online. Available online: https://go.drugbank.com/ (accessed on 18 January 2022).
- Hukkanen, J. Induction of cytochrome P450 enzymes: A view on human in vivo findings. Expert Rev. Clin. Pharmacol. 2012, 5, 569–585. [Google Scholar] [CrossRef]
- Drug Interactions Flockhart Table. Available online: https://drug-interactions.medicine.iu.edu/MainTable.aspx (accessed on 18 January 2022).
- Eshmuminov, D.; Leoni, F.; Schneider, M.A.; Becker, D.; Muller, X.; Onder, C.; Hefti, M.; Schuler, M.J.; Dutkowski, P.; Graf, R.; et al. Perfusion settings and additives in liver normothermic machine perfusion with red blood cells as oxygen carrier. A systematic review of human and porcine perfusion protocols. Transpl. Int. 2018, 31, 956–969. [Google Scholar] [CrossRef]
- Pond, S.M.; Tozer, T.N. First-pass elimination basic concepts and clinical consequences. Clin. Pharmacokinet. 1984, 9, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.; Benet, L.Z.; Graham, G.G. Clearance concepts in pharmacokinetics. J. Pharmacokinet. Biopharm. 1973, 1, 123–136. [Google Scholar] [CrossRef] [PubMed]
- First Pass Effect. StatPearls. Available online: https://www.ncbi.nlm.nih.gov/books/NBK551679/ (accessed on 21 February 2022).
- Kaplan Medical. Pharmacokinetics. In USMLE Step 1 Lecture Notes 2019: Pharmacology; Davis, C., Harris, S.R., Eds.; Kaplan Publishing: Berkshire, UK, 2019; Volume 1, p. 7. [Google Scholar]
- Inger, J.; Mangus, I.S. Genetic polymorphism and toxicology--With emphasis on cytochrome p450. Toxicol. Sci. 2011, 120, 1–13. [Google Scholar]
- Medsafe: New Zealand Medicines and Medical Devices Safety Authority. Drug Metabolism–The Importance of Cytochrome P450 3A4. Available online: https://www.medsafe.govt.nz/profs/puarticles/march2014drugmetabolismcytochromep4503a4.htm (accessed on 22 April 2022).
- Werk, A.N.; Cascorbi, I. Functional gene variants of CYP3A4. Clin. Pharmacol. Ther. 2014, 96, 340–348. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 20 January 2022).
- US Food and Drug Administration. Clinical Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry, January 2020. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions (accessed on 20 January 2022).
- Fluphenazine: Drug Information. Available online: https://www.uptodate.com/contents/fluphenazine-drug-information?topicRef=14773&source=see_link (accessed on 20 January 2022).
- Pimozide: Drug Information. Available online: https://www.uptodate.com/contents/pimozide-drug-information?topi69cRef=14773&source=see_link (accessed on 20 January 2022).
- Stimmel, G.L.; Falloon, I.R. Chlorpromazine plasma levels, adverse effects, and tobacco smoking: Case report. Clin. Psychiatry 1998, 44, 420. [Google Scholar]
- Pantuck, E.J.; Pantuck, C.B.; Anderson, K.E.; Conney, A.H.; Kappas, A. Cigarette smoking and chlorpromazine disposition and actions. Clin. Pharmacol. Ther. 1982, 31, 533–538. [Google Scholar] [CrossRef]
- Ereshefsky, L.; Saklad, S.R.; Watanabe, M.D.; Davis, C.M.; Jann, M.W. Thiothixene pharmacokinetic interactions: A study of hepatic enzyme inducers, clearance inhibitors, and demographic variables. J. Clin. Psychopharmacol. 1991, 11, 296. [Google Scholar] [CrossRef]
- Aripiprazole (Oral and Long-Acting Injectable [Abilify Maintena]): Drug Information. Available online: https://www.uptodate.com/contents/aripiprazole-oral-and-long-acting-injectable-abilify-maintena-drug-information?topicRef=14776&source=see_link (accessed on 20 January 2022).
- Asenapine: Drug Information. Available online: https://www.uptodate.com/contents/asenapine-drug-information?topicRef=14776&source=see_link (accessed on 20 January 2022).
- Rexulti (Brexpiprazole): Otsuka America Pharmaceutical, Inc. 2015. Available online: http://www.otsuka-us.com/Products/Documents/Rexulti.PI.pdf (accessed on 20 January 2022).
- Citrome, L.; Stensbøl, T.B.; Maeda, K. The preclinical profile of brexpiprazole: What is its clinical relevance for the treatment of psychiatric disorders? Expert Rev. Neurother. 2015, 15, 1219–1229. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration Safety Communication: Vraylar Package Insert. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/204370lbl.pdf (accessed on 20 January 2022).
- Clozaril (Clozapine): Novartis, Inc. 2014. Available online: https://www.pharma.us.novartis.com/product/pi/pdf/Clozaril.pdf (accessed on 20 January 2022).
- Clozapine-Clozapine Tablet. Remedyrepack Inc. Highlights of Prescribing Information. Available online: https://dailymed.nlm.nih.gov/dailymed/fda/fdadrugxsl.cfm?setid=4bb4e035-7dbc-4582-8e46-3f128f6c2790&type=display (accessed on 22 April 2022).
- Anderson, G.D.; Chan, L.N. Pharmacokinetic Drug Interactions with Tobacco, Cannabinoids and Smoking Cessation Products. Clin. Pharmacokinet. 2016, 55, 1353–1368. [Google Scholar] [CrossRef]
- Fanapt (Iloperidone): Vanda Pharmaceutical, Inc. 2014. Available online: https://www.fanapt.com/product/pi/pdf/fanapt.pdf (accessed on 20 January 2022).
- Lumateperone: Drug Information. Available online: https://www.uptodate.com/contents/lumateperone-drug-information?topicRef=14776&source=see_link (accessed on 20 January 2022).
- Latuda (Lurasidone): Sunovion Pharmaceuticals, Inc. 2013. Available online: http://www.latuda.com/LatudaPrescribingInformation.pdf (accessed on 20 January 2022).
- Zyprexa (Olanzapine). Eli Lilly and Company, Inc. 2015. Available online: http://pi.lilly.com/us/zyprexa-pi.pdf (accessed on 20 January 2022).
- Invega (Paliperidone): Janssen Pharmaceuticals, Inc. 2014. Available online: http://www.invega.com/prescribing-information (accessed on 20 January 2022).
- Nuplazid (Pimavanserin): Full Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/207318lbl.pdf (accessed on 20 January 2022).
- Seroquel (Quetiapine): AstraZeneca 2013. Available online: http://www1.astrazeneca-us.com/pi/seroquel.pdf (accessed on 20 January 2022).
- Seroquel XR (Quetiapine Extended Release): AstraZeneca 2013. Available online: http://www.azpicentral.com/seroquel-xr/seroquelxr.pdf#page=1 (accessed on 20 January 2022).
- Risperdal (Risperidone): Janssen Pharmaceuticals, Inc. 2014. Available online: http://www.janssenpharmaceuticalsinc.com/assets/risperdal.pdf (accessed on 20 January 2022).
- Geodon (Ziprasidone): Pfizer, Inc. 2014. Available online: http://labeling.pfizer.com/ShowLabeling.aspx?id=584 (accessed on 20 January 2022).
- Javaid, J.I. Clinical pharmacokinetics of antipsychotics. J. Clin. Pharmacol. 1994, 34, 286–295. [Google Scholar] [CrossRef]
- Belle, D.J.; Singh, H. Genetic factors in drug metabolism. Am. Fam. Physician 2008, 77, 1553–1560. [Google Scholar] [PubMed]
- Pouget, J.G.; Shams, T.A.; Tiwari, A.K.; Muller, D.J. Pharmacogenetics and outcome with antipsychotic drugs. Dialogues Clin. Neurosci. 2014, 16, 555–566. [Google Scholar] [CrossRef] [PubMed]
- GeneSight Test. Available online: https://genesight.com/product/ (accessed on 22 April 2022).
- Genesight Test. Available online: https://genesight.com/ (accessed on 20 January 2022).
- Genesept Assay. Available online: https://www.dynacare.ca/corporate-clients/wellness-featured-services/mental-health-solution.aspx (accessed on 20 January 2022).
- The Human Protein Atlas: CYP1A1. Available online: https://www.proteinatlas.org/ENSG00000140465-CYP1A1/tissue (accessed on 20 January 2022).
Figure 1.
Antipsychotic metabolism phases.
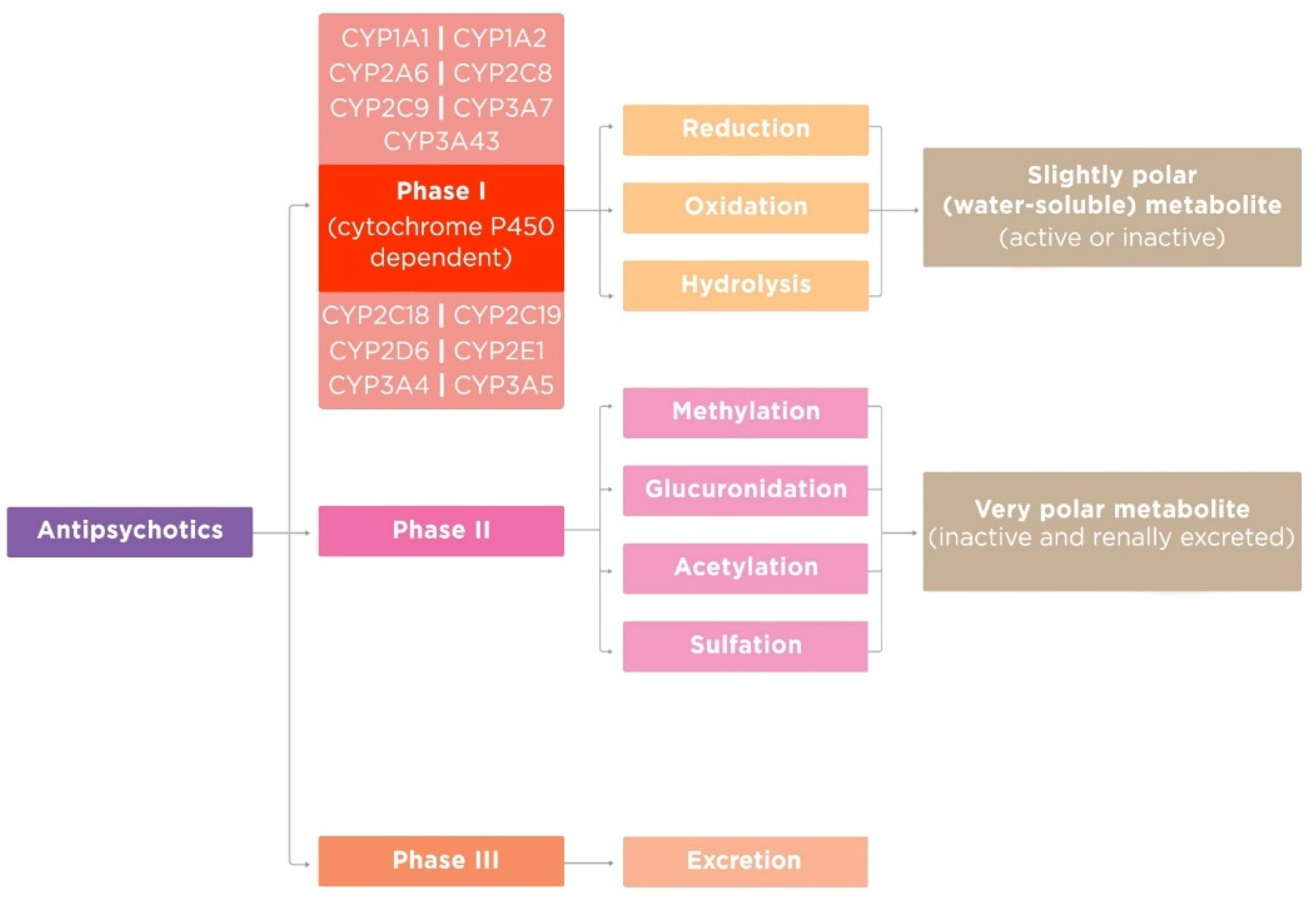
Figure 2.
Estimated percentage of antipsychotics metabolized by the major cytochrome P450 enzymes ([31], modified by the authors).
Figure 2.
Estimated percentage of antipsychotics metabolized by the major cytochrome P450 enzymes ([31], modified by the authors).

Figure 3.
The expression of some CYP enzymes in the human body: (a) CYP3A4; (b) CYP2D6; (c) CYP2C19 ([33], modified by the authors).
Figure 3.
The expression of some CYP enzymes in the human body: (a) CYP3A4; (b) CYP2D6; (c) CYP2C19 ([33], modified by the authors).
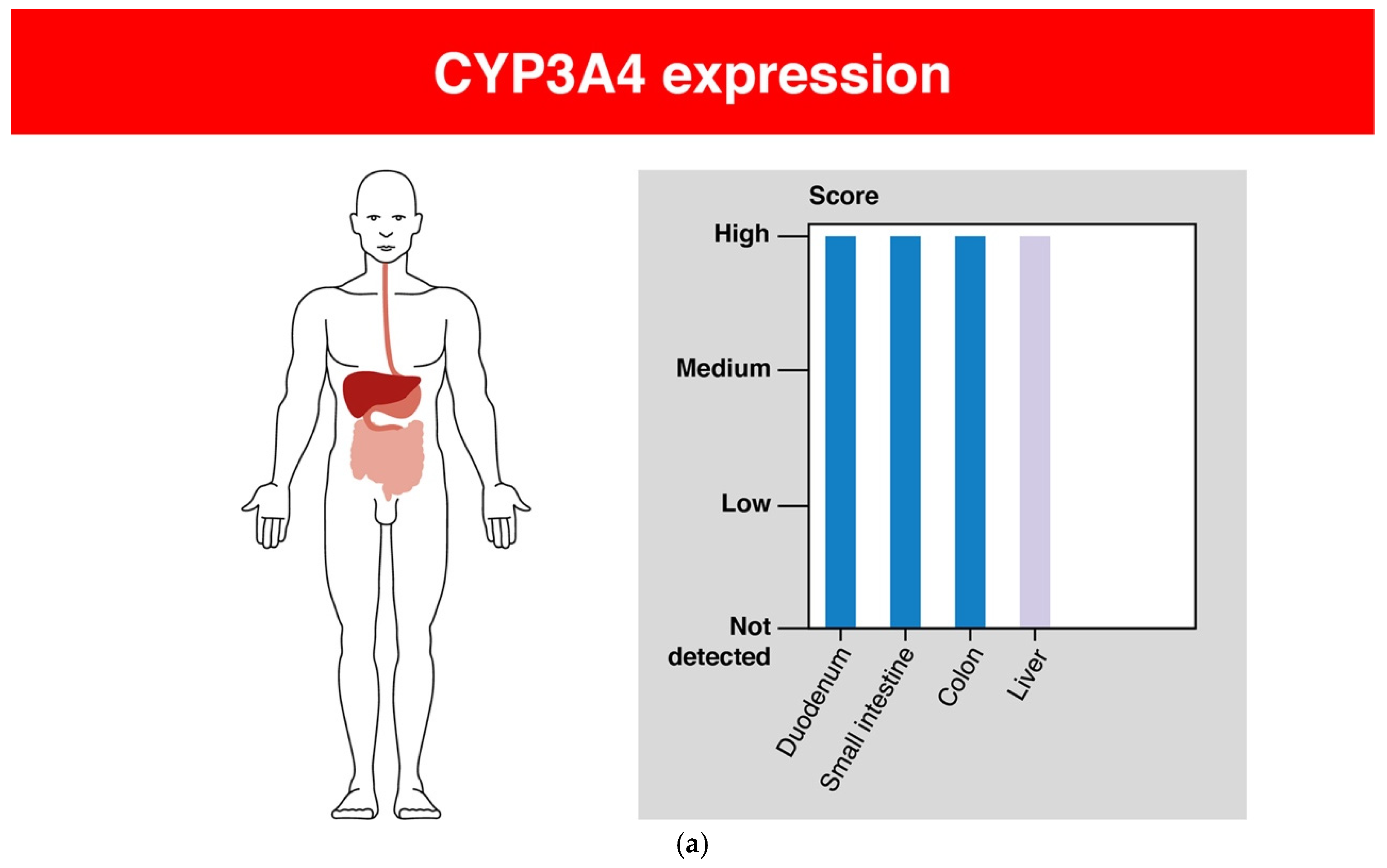

Figure 4.
Pharmacogenetic phenotypes ([74], modified by the authors).
Figure 4.
Pharmacogenetic phenotypes ([74], modified by the authors).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shnayder, N.A.; Abdyrakhmanova, A.K.; Nasyrova, R.F. Oxidation of Antipsychotics. Encyclopedia 2022, 2, 974-989. https://doi.org/10.3390/encyclopedia2020064
AMA Style
Shnayder NA, Abdyrakhmanova AK, Nasyrova RF. Oxidation of Antipsychotics. Encyclopedia. 2022; 2(2):974-989. https://doi.org/10.3390/encyclopedia2020064
Chicago/Turabian StyleShnayder, Natalia A., Aiperi K. Abdyrakhmanova, and Regina F. Nasyrova. 2022. "Oxidation of Antipsychotics" Encyclopedia 2, no. 2: 974-989. https://doi.org/10.3390/encyclopedia2020064