
SARS-CoV-2 Lineage P.4 Detection in Southeast Brazil: A Retrospective Genomic and Clinical Overview
 , , , , , , , , , , ,
, , , , , , , , , , ,  , , ,
, , ,  , ,
, ,  , add
Show full author list
, add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. SARS-CoV-2 Samples
2.2. SARS-CoV-2 Gamma//VOC Detection by Real-Time PCR
2.3. SARS-CoV-2 Sequencing
2.4. Data Processing and Identification of SARS-CoV-2 Lineages
2.5. Evolutionary Analyses
2.6. Clinical Data and Statistical Analysis
3. Results
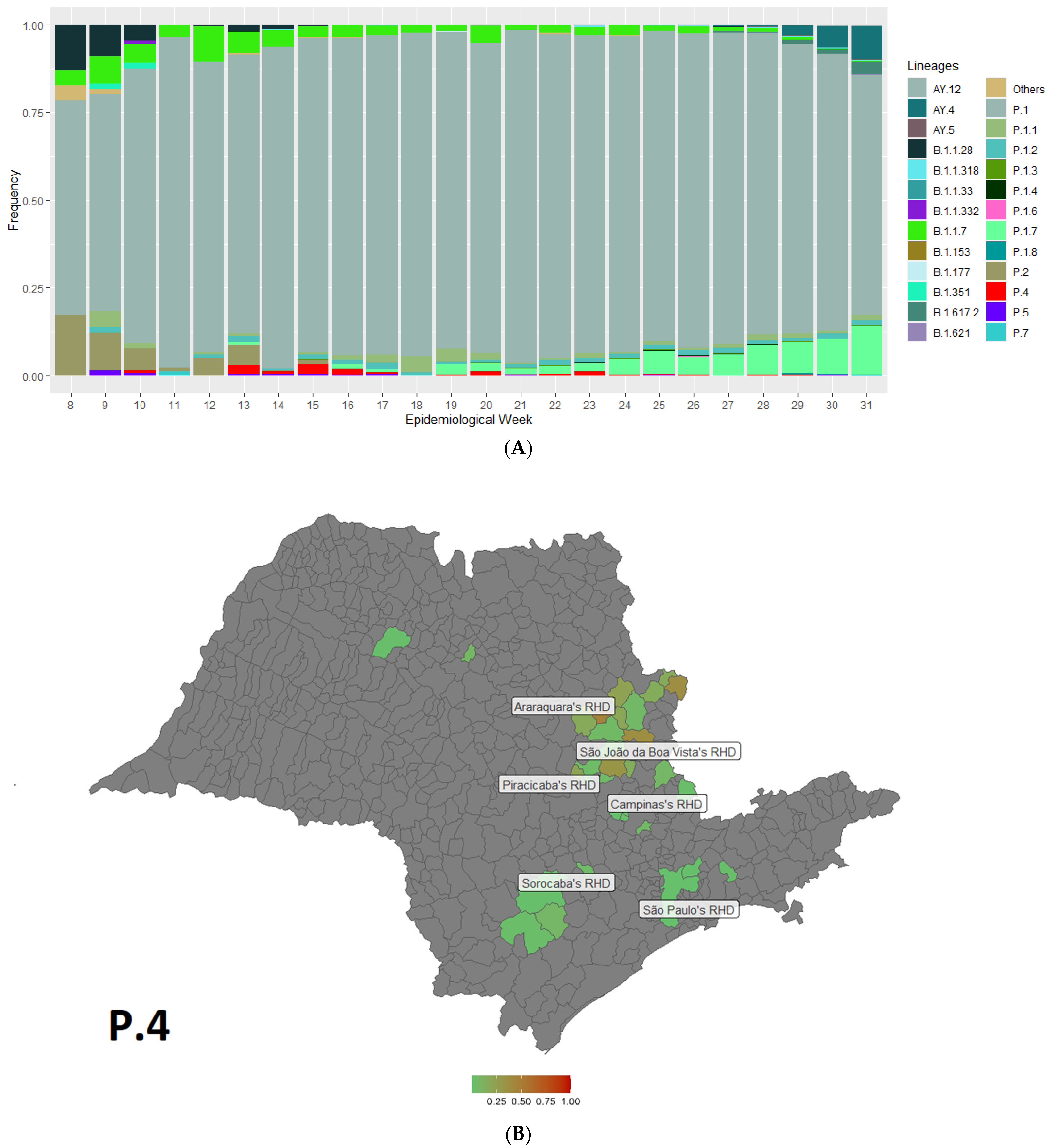
3.1. Detection of P.4 SARS-CoV-2 Lineage
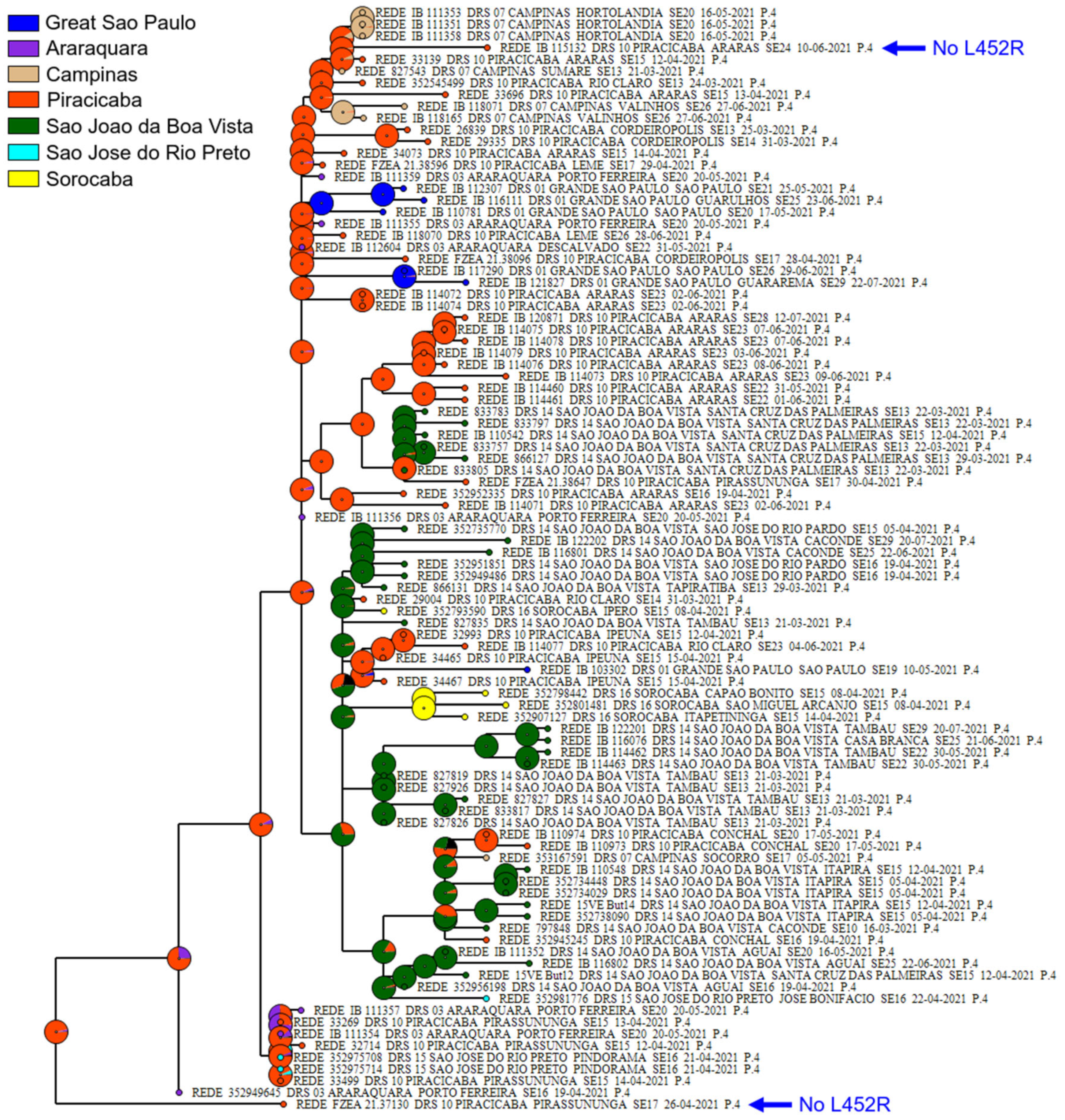
3.2. Phylogenetic Analysis of P.4 Genomes
3.3. Targeted Detection in the City of Araras-SP and Clinical Outcome of P4 in Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef]
- Iacobucci, G. COVID-19: New UK variant may be linked to increased death rate, early data indicate. BMJ 2021, 372, n230. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Naveca, F.G.; Nascimento, V.; de Souza, V.C.; de Lima Corado, A.; Nascimento, F.; Silva, G.; Costa, Á.; Duarte, D.; Pessoa, K.; Mejía, M.; et al. COVID-19 in Amazonas, Brazil, was driven by the persistence of endemic lineages and P.1 emergence. Nat. Med. 2021, 27, 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. Delta coronavirus variant: Scientists brace for impact. Nature 2021, 595, 17–18. [Google Scholar] [CrossRef]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2—What Do They Mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef]
- Snijder, E.J.; Bredenbeek, P.J.; Dobbe, J.C.; Thiel, V.; Ziebuhr, J.; Poon, L.L.M.; Guan, Y.; Rozanov, M.; Spaan, W.J.M.; Gorbalenya, A.E. Unique and Conserved Features of Genome and Proteome of SARS-coronavirus, an Early Split-off From the Coronavirus Group 2 Lineage. J. Mol. Biol. 2003, 331, 991–1004. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef]
- Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef]
- Elena, S.F.; Sanjuán, R. Adaptive value of high mutation rates of RNA viruses: Separating causes from consequences. J. Virol. 2005, 79, 11555–11558. [Google Scholar] [CrossRef]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92, 14. [Google Scholar] [CrossRef] [PubMed]
- Castonguay, N.; Zhang, W.; Langlois, M.-A. Meta-Analysis and Structural Dynamics of the Emergence of Genetic Variants of SARS-CoV-2. Front. Microbiol. 2021, 1, 1637. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, V.B.; Ferrareze, P.A.G.; Zimerman, R.A.; Cybis, G.B.; Thompson, C.E. Mutation hotspots and spatiotemporal distribution of SARS-CoV-2 lineages in Brazil, February 2020-2021. Virus Res. 2021, 304, 198532. [Google Scholar] [CrossRef] [PubMed]
- Bittar, C.; Possebon, F.S.; Ullmann, L.S.; Geraldini, D.B.; da Costa, V.G.; de Almeida, L.G.P.; Paulo, P.R.; Nascimento-Júnior, N.M.; Cilli, E.M.; Artico Banho, C.; et al. The Emergence of the New P.4 Lineage of SARS-CoV-2 With Spike L452R Mutation in Brazil. Front. Public Health 2021, 9, 1465. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Sant’Anna, F.H.; Varela, A.P.M.; Prichula, J.; Comerlato, J.; Comerlato, C.B.; Roglio, V.S.; Pereira, G.F.M.; Moreno, F.; Seixas, A.; Wendland, E.M. Emergence of the novel SARS-CoV-2 lineage VUI-NP13L and massive spread of P.2 in South Brazil. Emerg. Microbes Infect. 2021, 10, 1431–1440. [Google Scholar] [CrossRef]
- Tchesnokova, V.; Kulasekara, H.; Larson, L.; Bowers, V.; Rechkina, E.; Kisiela, D.; Sledneva, Y.; Choudhury, D.; Maslova, I.; Deng, K.; et al. Acquisition of the L452R mutation in the ACE2-binding interface of Spike protein triggers recent massive expansion of SARS-CoV-2 variants. J. Clin. Microbiol. 2021, 59, e00921-21. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136.e11. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.; Max Carvalho, L.; Pybus, O. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Yu, Y.; Harris, A.J.; Blair, C.; He, X. RASP (Reconstruct Ancestral State in Phylogenies): A tool for historical biogeography. Mol. Phylogenet. Evol. 2015, 87, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wu, T.; Liu, Q.; Yang, Z. The SARS-CoV-2 outbreak: What we know. Int. J. Infect. Dis. 2020, 94, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant. Cell 2021, 184, 3426. [Google Scholar] [CrossRef]
- Mor, O.; Mandelboim, M.; Fleishon, S.; Bucris, E.; Bar-Ilan, D.; Linial, M.; Nemet, I.; Kliker, L.; Lustig, Y.; Israel National Consortium for SARS-CoV-2 Sequencing; et al. The Rise and Fall of a Local SARS-CoV-2 Variant with the Spike Protein Mutation L452R. Vaccines 2021, 9, 937. [Google Scholar] [CrossRef] [PubMed]
- Barona-Gómez, F.; Delaye, L.; Díaz-Valenzuela, E.; Plisson, F.; Cruz-Pérez, A.; Díaz-Sánchez, M.; García-Sepúlveda, C.A.; Sanchez-Flores, A.; Pérez-Abreu, R.; Valencia-Valdespino, F.J.; et al. Phylogenomics and population genomics of SARS-CoV-2 in Mexico during the pre-vaccination stage reveals variants of interest B.1.1.28.4, B.1.1.222 or B.1.1.519 and the nucleocapsid mutation S194L associated with symptoms. Microb. Genom. 2021, 7, 000684. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chong, K.C.; Wong, M.C.S.; Boon, S.S.; Huang, J.; Wang, M.H.; Ng, R.W.Y.; Lai, C.K.C.; Chan, P.K.S. A global analysis of replacement of genetic variants of SARS-CoV-2 in association with containment capacity and changes in disease severity. Clin. Microbiol. Infect. 2021, 27, 750–757. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Zhou, D.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Antibody evasion by the P.1 strain of SARS-CoV-2. Cell 2021, 184, 2939–2954.e9. [Google Scholar] [CrossRef]
- Hoffmann, M.; Arora, P.; Groß, R.; Seidel, A.; Hörnich, B.F.; Hahn, A.S.; Krüger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell 2021, 184, 2384–2393.e12. [Google Scholar] [CrossRef]
- Coutinho, R.M.; Marquitti, F.M.D.; Ferreira, L.S.; Borges, M.E.; da Silva, R.L.P.; Canton, O.; Portella, T.P.; Poloni, S.; Franco, C.; Plucinski, M.M.; et al. Model-based estimation of transmissibility and reinfection of SARS-CoV-2 P.1 variant. Commun. Med. 2021, 1, 48. [Google Scholar] [CrossRef]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; da Silva Candido, D.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, abh2644. [Google Scholar] [CrossRef]
- Giovanetti, M.; Fonseca, V.; Wilkinson, E.; Tegally, H.; San, E.J.; Althaus, C.L.; Xavier, J.; Nanev Slavov, S.; Viala, V.L.; Ranieri Jerônimo Lima, A.; et al. Replacement of the Gamma by the Delta variant in Brazil: Impact of lineage displacement on the ongoing pandemic. Virus Evol. 2022, 8, veac024. [Google Scholar] [CrossRef] [PubMed]
- Alcantara, L.C.J.; Nogueira, E.; Shuab, G.; Tosta, S.; Fristch, H.; Pimentel, V.; Souza-Neto, J.A.; Coutinho, L.L.; Fukumasu, H.; Sampaio, S.C.; et al. SARS-CoV-2 epidemic in Brazil: How the displacement of variants has driven distinct epidemic waves. Virus Res. 2022, 315, 198785. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Non-P.4 | P.4 | p-Value | |
|---|---|---|---|
| AGE, years (n = 251) | 228 (90.8%) | 23 (9.2%) | 0.0032 |
| ≤9 | 4 (1.6%) | 0 (0%) | |
| 10–19 | 19 (7.6%) | 1 (0.420%) | |
| 20–29 | 55 (21.96%) | 4 (1.6%) | |
| 30–39 | 52(20.7%) | 7 (2.8%) | |
| 40–49 | 44 (17.5%) | 2 (0.8%) | |
| 50–59 | 42 (16.7%) | 5 (2.0%) | |
| 60–69 | 9 (3.6%) | 3 (1.2%) | |
| 70–79 | 3 (1.2%) | 0 (0%) | |
| ≥80 | 0 (0%) | 1 (0.4%) | |
| SEX (n = 251) | 228 (90.84%) | 23 (9.16%) | 0.4968 |
| Female | 108 (43.03%) | 13 (5.18%) | |
| Male | 120 (47.81%) | 10 (3.98%) | |
| DISEASE STAGE (n = 249) | 228 (91.6%) | 21 (8.4%) | 0.0502 |
| Severe illness | 2 (0.8%) | 0 (0%) | |
| Moderate illness | 6 (2.4%) | 1 (0.4%) | |
| Mild illness | 220 (88.3%) | 20 (8.0%) | |
| PATIENT HOSPITALIZED (n = 249) | 228 (91.6%) | 21 (8.4%) | 0.0200 |
| No | 220 (88.3%) | 20 (8.0%) | |
| Yes | 8 (3.2%) | 1 (0.4%) | |
| DEATH (n = 249) | 228 (91.6%) | 21 (8.4%) | 0.0800 |
| No | 226 (90.8%) | 21 (8.4%) | |
| Yes | 2 (0.8%) | 0 (0%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poleti, M.D.; Lesbon, J.C.C.; de Mattos Oliveira, E.C.; Patané, J.S.L.; Clemente, L.G.; Viala, V.L.; Ribeiro, G.; Pinheiro, J.F.P.; Giovanetti, M.; Alcantara, L.C.J.; et al. SARS-CoV-2 Lineage P.4 Detection in Southeast Brazil: A Retrospective Genomic and Clinical Overview. COVID 2022, 2, 1768-1777. https://doi.org/10.3390/covid2120127
Poleti MD, Lesbon JCC, de Mattos Oliveira EC, Patané JSL, Clemente LG, Viala VL, Ribeiro G, Pinheiro JFP, Giovanetti M, Alcantara LCJ, et al. SARS-CoV-2 Lineage P.4 Detection in Southeast Brazil: A Retrospective Genomic and Clinical Overview. COVID. 2022; 2(12):1768-1777. https://doi.org/10.3390/covid2120127
Chicago/Turabian StylePoleti, Mirele Daiana, Jéssika Cristina Chagas Lesbon, Elisângela Chicaroni de Mattos Oliveira, José Salvatore Leister Patané, Luan Gaspar Clemente, Vincent Louis Viala, Gabriela Ribeiro, Jéssica Fernanda Perissato Pinheiro, Marta Giovanetti, Luiz Carlos Junior Alcantara, and et al. 2022. "SARS-CoV-2 Lineage P.4 Detection in Southeast Brazil: A Retrospective Genomic and Clinical Overview" COVID 2, no. 12: 1768-1777. https://doi.org/10.3390/covid2120127
APA StylePoleti, M. D., Lesbon, J. C. C., de Mattos Oliveira, E. C., Patané, J. S. L., Clemente, L. G., Viala, V. L., Ribeiro, G., Pinheiro, J. F. P., Giovanetti, M., Alcantara, L. C. J., de Lima, L. P. O., Martins, A. J., dos Santos Barros, C. R., Marqueze, E. C., de Souza Todão Bernardino, J., Moretti, D. B., Brassaloti, R. A., de Lello Rocha Campos Cassano, R., Mariani, P. D. S. C., ... Fukumasu, H. (2022). SARS-CoV-2 Lineage P.4 Detection in Southeast Brazil: A Retrospective Genomic and Clinical Overview. COVID, 2(12), 1768-1777. https://doi.org/10.3390/covid2120127