1. Introduction
The global topical drug delivery market is growing fast due to the increasing prevalence of chronic skin diseases [
1]. New and better technologies for topical drug delivery are also expected to boost the growth of this market [
2]. Animal models have been used worldwide to test the efficacy and toxicity of topical drugs. However, animal experimentation raises important ethical and regulatory concerns [
3,
4]. In addition, there is a lack of accuracy of the animal-to-human extrapolation, with approximately 90% of the drug candidates that appear safe and effective in animal studies failing to win approval when tested in humans [
5,
6,
7]. These factors resulted in an increasing need to replace and/or complement animal testing with physiologically relevant reconstructed human skin models suitable for topical drug delivery and disease modeling [
8,
9].
Techniques for culturing three-dimensional (3D) skin cells have been developed, resulting in significant advancements in skin tissue engineering [
9,
10]. However, current commercially available human skin models still lack complexity and present weaker barrier properties than in vivo human skin [
11]. Organ-on-a-chip (OoC) technology aims to surpass the limitations of conventional cell-based culture platforms and increase the predictive power of in vitro models by creating customized cellular microenvironments with precise fluidic, mechanical, and structural control [
12]. Moreover, integrating sensors in OoC allows real-time monitoring of tissue viability and function [
13].
In the last few years, several skin-on-a-chip (SoC) platforms have been reported and their physiological relevance has been shown. However, most of these platforms have been limited to maintaining transferred skin tissues (e.g., skin biopsies) to increase their longevity and/or establish a co-culture with other organ models [
14,
15,
16,
17,
18,
19,
20]. These studies provide valuable information regarding the potential of SoC devices for clinical and testing purposes. However, the integration of models previously developed off-chip limits the potential benefits of the dynamic culture. More recently, skin models generated on-chip have been reported. Wufuer et al. developed a SoC consisting of 3 cell layers (keratinocytes, fibroblasts, and endothelial cells) separated by two porous membranes to simulate inflammation and edema [
21]. Ramadan et al. further explored the potential of the SoC devices by including an immune component, describing the interaction between the HaCaT cell line and a monocytic cell line in a bi-channel microfluidic device [
22]. Although these studies replicated some important features of healthy and diseased skin, they included monolayer systems that do not replicate the 3D environment of in vivo human skin.
Only a handful of publications have reported OoC devices for 3D skin tissue formation inside the platform. Mori et al. fabricated a culture device composed of anchoring structures and nylon wires across the device’s connectors to produce a full-thickness human skin model (FTSm), including the dermis and the epidermis [
23]. This study was an important first step in developing 3D FTSm inside the platform to mimic in vivo vascularization. However, the limitations of using collagen to develop the dermal compartment were apparent. Collagen contraction made it necessary to increase the device’s complexity and compromised the model’s reproducibility, frequently resulting in clogged channels. Lee et al. developed 3D FTSm inside an SoC device composed of two polydimethylsiloxanes (PDMS) fluidic chambers [
24]. The group also reported skin contraction during the differentiation process, resulting in detachment of the scaffold. Moreover, the stratum corneum of the SoC models was less homogeneous than the controls.
Both the above-mentioned studies highlight the limitations of using natural hydrogels to generate the dermal compartment inside an OoC device. These systems reported fibroblast-mediated contraction and matrix degradation leading to distortion and disruption of the skin model. Moreover, by including dermal compartments comprising exclusively of animal-derived collagen type I, they are not fully representative of the in vivo extracellular matrix (ECM), which includes multiple types of collagens, fibrous proteins, and proteoglycans. Recently, Sriram et al. used a combination of fibrin and polyethylene glycol (PEG) to produce a stable dermal component capable of sustaining the epidermis without signs of contraction [
25]. The group produced an SoC with improved epidermal differentiation and barrier function.
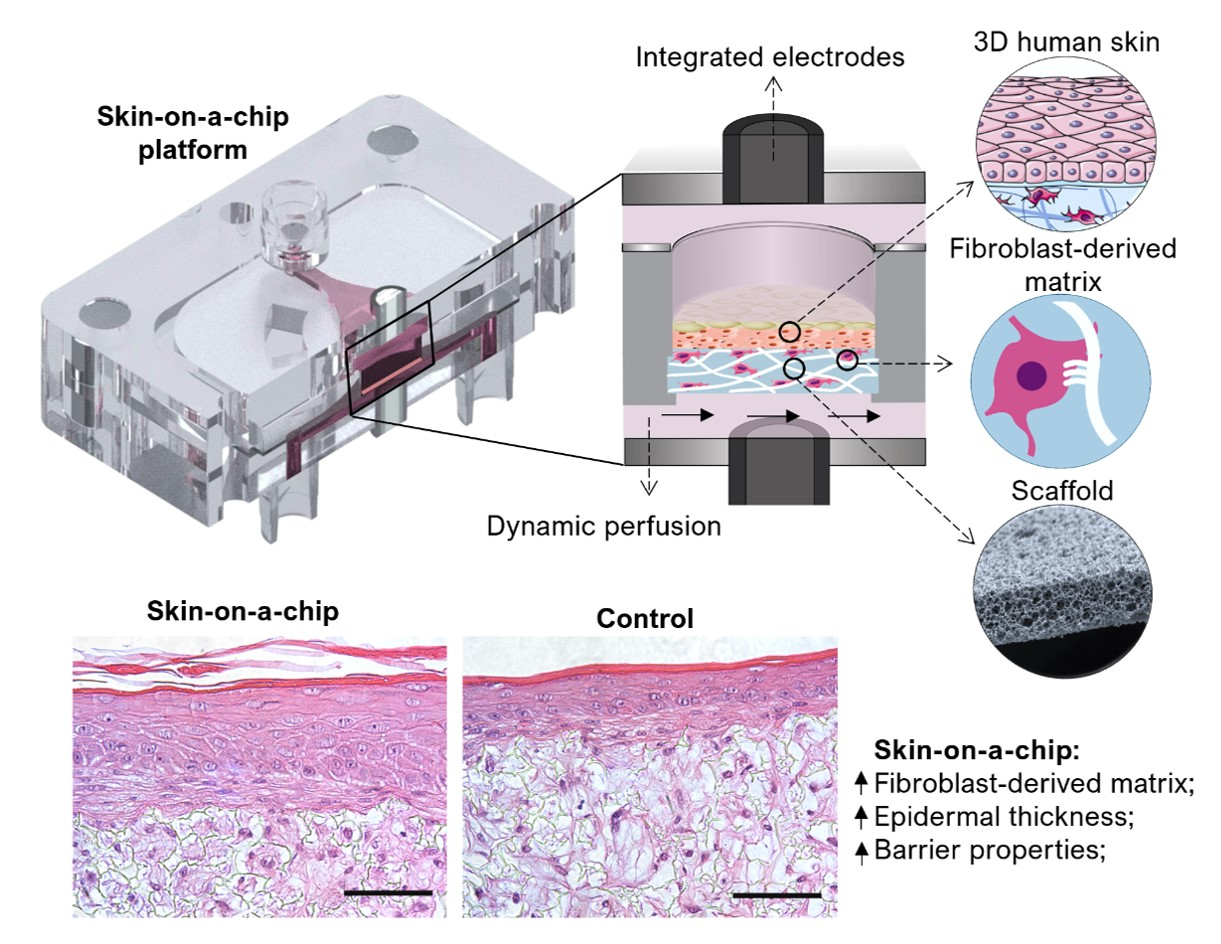
Here, we present an alternative approach to generating a 3D full-thickness SoC with a stable dermal compartment representing the in-vivo microenvironment. Recently, our group developed an OoC with integrated electrodes for the development and real-time monitoring of biological barriers [
26]. As a proof of concept, transepithelial resistance (TEER) was measured during the formation of an epidermis inside the chip. Here, we study the impact of dynamic perfusion on both dermis and epidermis formation by performing histological and functional analyses. In the described approach, an inert porous scaffold is integrated into the device, where dermal cells are seeded and stimulated to produce their own endogenous ECM (fibroblast-derived matrix, FDM). This process results in a mechanically stable structure and excludes animal-derived hydrogels. Furthermore, the SoC platforms described in the literature are irreversibly sealed, usually through plasma bonding, making it difficult the complete tissue retrieval for endpoint assays (e.g., histological analysis and immunofluorescence). The described SoC includes a removable culture insert and interchangeable modules (e.g., for permeation studies).
2. Materials and Methods
2.1. Fabrication and Assembly of the SoC Device
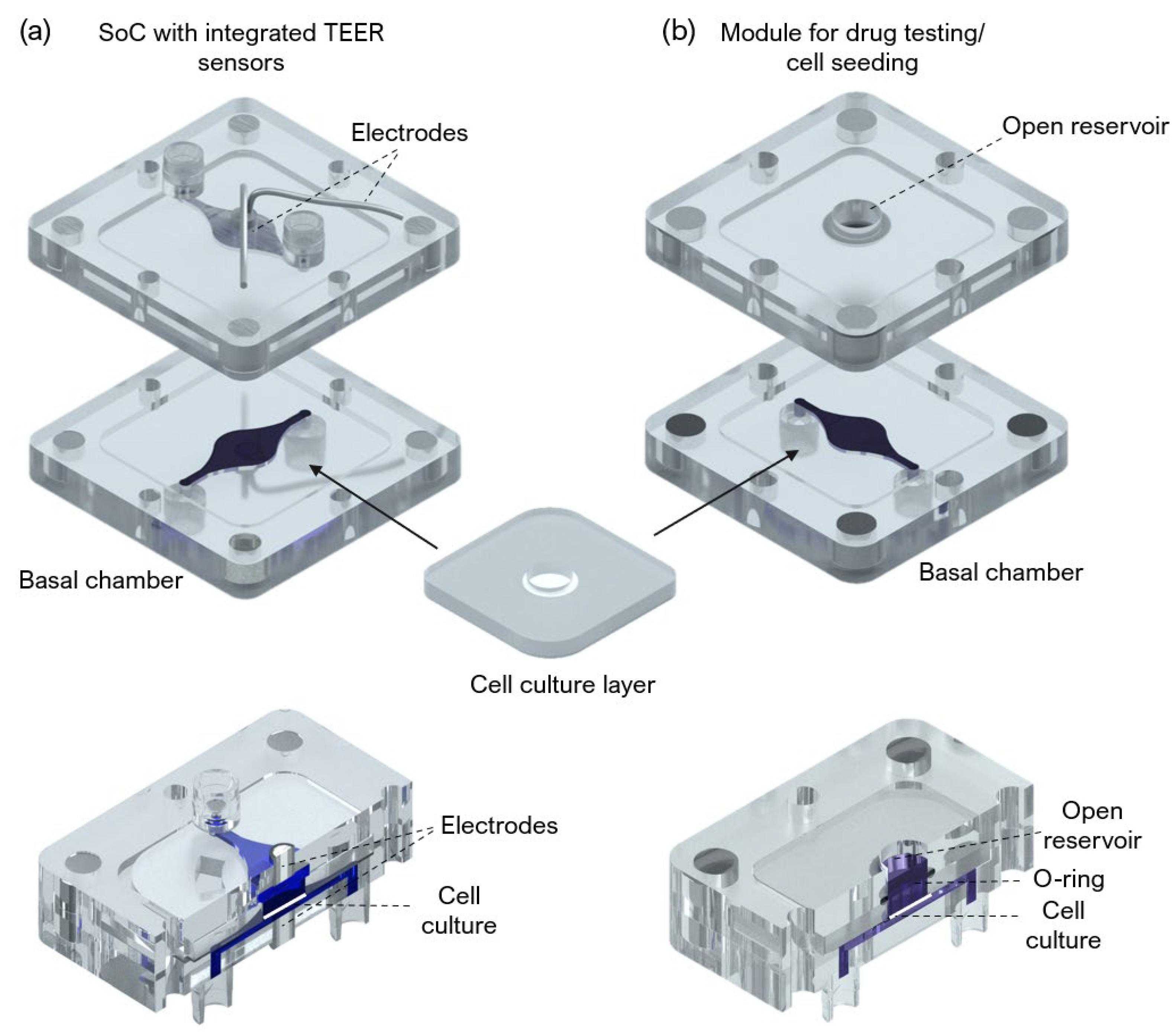
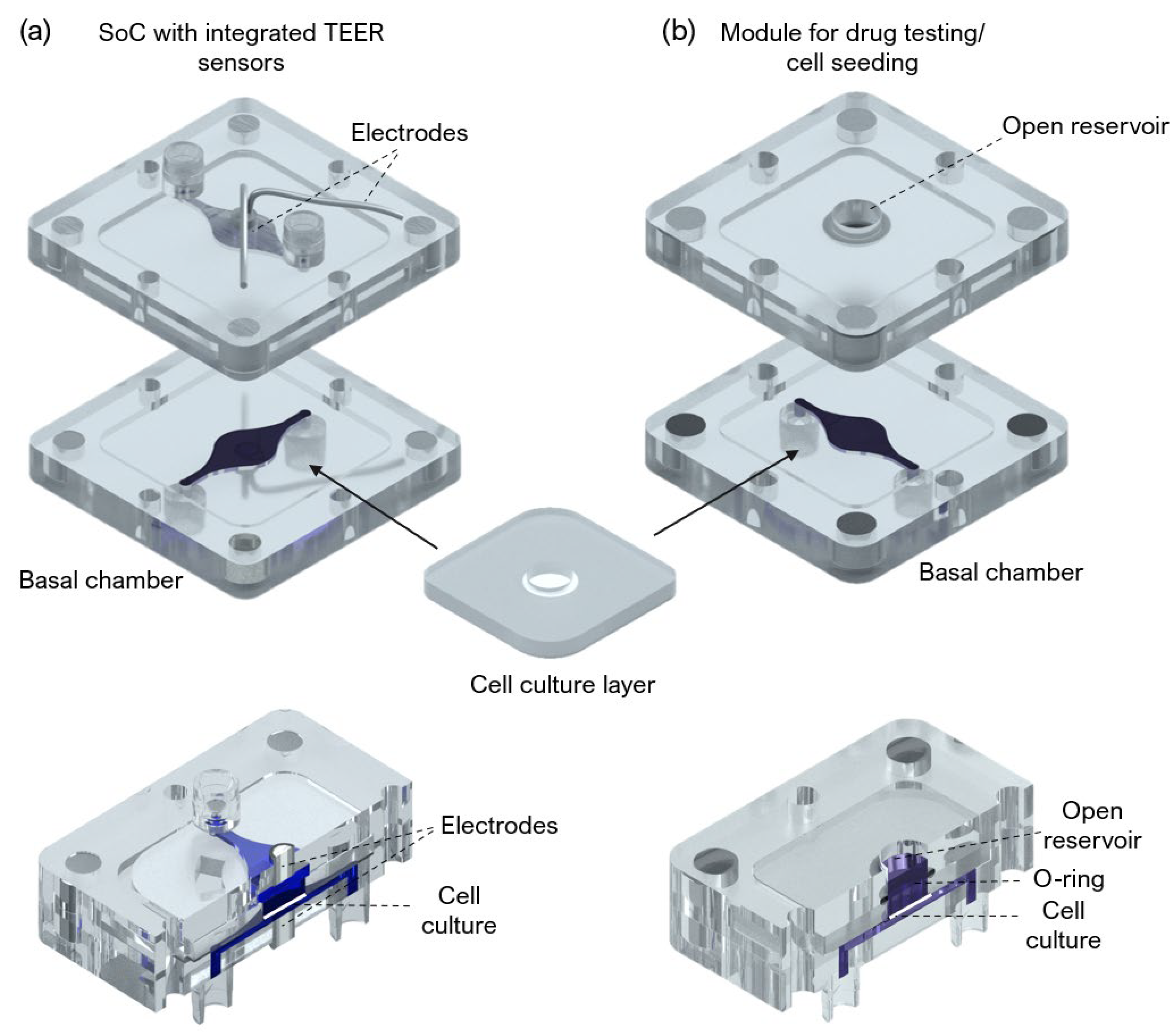
A SoC device with a modular architecture and a “user-friendly” approach was designed and fabricated to produce an FTSm and in situ drug testing. The device is composed of 3 main layers: two fluidic compartments (apical and basal) and a central insert layer for cell culture. The design of the SoC was optimized to ensure stability during long-term skin culture while providing a flexible and modular geometry compatible with different studies. A closed schematic and exploded view of the SoC device are shown in
Figure 1a,b, respectively.
The protocol for the fabrication of the device was previously described by our group [
26]. Briefly, the apical and basal chambers were fabricated through a combination of computer numerically controlled (CNC) micromachining and laser cutting (Xometry Europe GmbH, Ottobrunn, Germany). The apical and basal layers were constructed in polycarbonate (31.5 × 31.5 × 4 mm, length x width x height) and included a central cavity designed to accept the cell culture layer (21 × 21 × 1 mm, length x width x height). The dimensions of the fluidic channels were 0.9 × 0.6 mm (width × heigh), and the central circular perfusion chambers were 5 × 0.6 mm (diameter × heigh). For the access ports, mini female luer interfaces (microfluidic ChipShop, Jena, Germany) were glued to the inlets and outlets in both the upper and lower polycarbonate housings.
To obtain the PDMS layer for cell culture that could fit in the polycarbonate cavities, dedicated molds were fabricated using CNC machining and laser cutting. The molds were filled with degassed PDMS prepolymer mixed with a curing agent (Sylgard 184, Dow Corning, Midland, MI, USA) at a 10:1 (
w/w) base to catalyst ratio and placed in an oven for 12 h at 65 °C. Polystyrene scaffolds (Alvetex
®, REPROCELL Europe Ltd., Glasgow, UK) were cut in a circular shape 6 mm diameter and sandwiched between the PDMS insert layer and a double-sided adhesive tape (PCR tape, ThermalSeal RTS
®, Sigma-Aldrich, St. Louis, MO, USA) with 21 × 21 mm and an open central region of 5 mm for cell culture. The PCR adhesive tape was chosen due to its biocompatibility for cell culture, transparency, and good sealing properties. To better seal the PDMS insert against the polycarbonate housing and avoid leakage, the PCR tape was covered with a thin PDMS layer. The chips were sterilized using UV radiation. The lids, insets, tubing, and fittings were sterilized in an autoclave before assembling them onto the chip on the setup day. The final assembled culture platform is shown in
Figure 1c, with blue dye used to identify the distinct basal and apical compartments.
Figure 1d shows the PDMS cell culture layer with a central polystyrene scaffold.
Figure 1d shows two SoC devices placed in dedicated support.
An SoC device with integrated electrodes for TEER measurement and a module compatible with drug testing and direct cell seeding were also designed and fabricated (
Figure A1). The TEER module includes Ag/AgCl-sintered pellet electrodes concentric placed in both apical and basal perfusion layers as described by Zoio et al. [
26]. A module for in situ drug testing and/or cell seeding consisted of a top polycarbonate housing with embedded magnets and a central reservoir, fabricated using CNC machining (Xometry Europe GmbH, Ottobrunn, Germany). A 6 mm diameter O-ring was added to ensure a leakage-free seal around the culture insert. The chips were sterilized using UV radiation.
2.2. Computational Fluid Dynamic Simulations
To characterize the hydrodynamics of the developed SoC, 3D models of the fluidic compartments and scaffold domain were generated with CAD (AutoCAD, Waltham, MA, USA) and imported to COMSOL Multiphysics™ (COMSOL Multiphysics GmbH, Gottingen, Germany) for finite element analysis (FEA).
The fluid was assumed as a homogeneous, incompressible Newtonian fluid. The density of the culture medium was assumed to be the same as water, 993.2 kg/m
3, and the dynamic viscosity was 7.8 × 10
−3 kg/m/s at a temperature of 37 °C. The boundary conditions were defined based on the inlet conditions for the flow rate (2 μL/min) and ambient pressure outlet conditions. COMSOL’s Free and Porous Media Interface was used for modeling a combination of porous media and free flow domain. The Navier-Stokes equation (incompressible flows) was used to model the free flow in the apical and basal chambers, and the Brinkman equations were applied to the porous domain (the scaffold layer). At the interface between free and porous media flow, the implemented boundary conditions between these domains enforce continuity for the velocity and the pressure. For all solid walls, no-slip boundary conditions were set. For all models, the “extra fine” mesh (under COMSOL calibration for general physics) was chosen, automatically assigning mesh sizes.
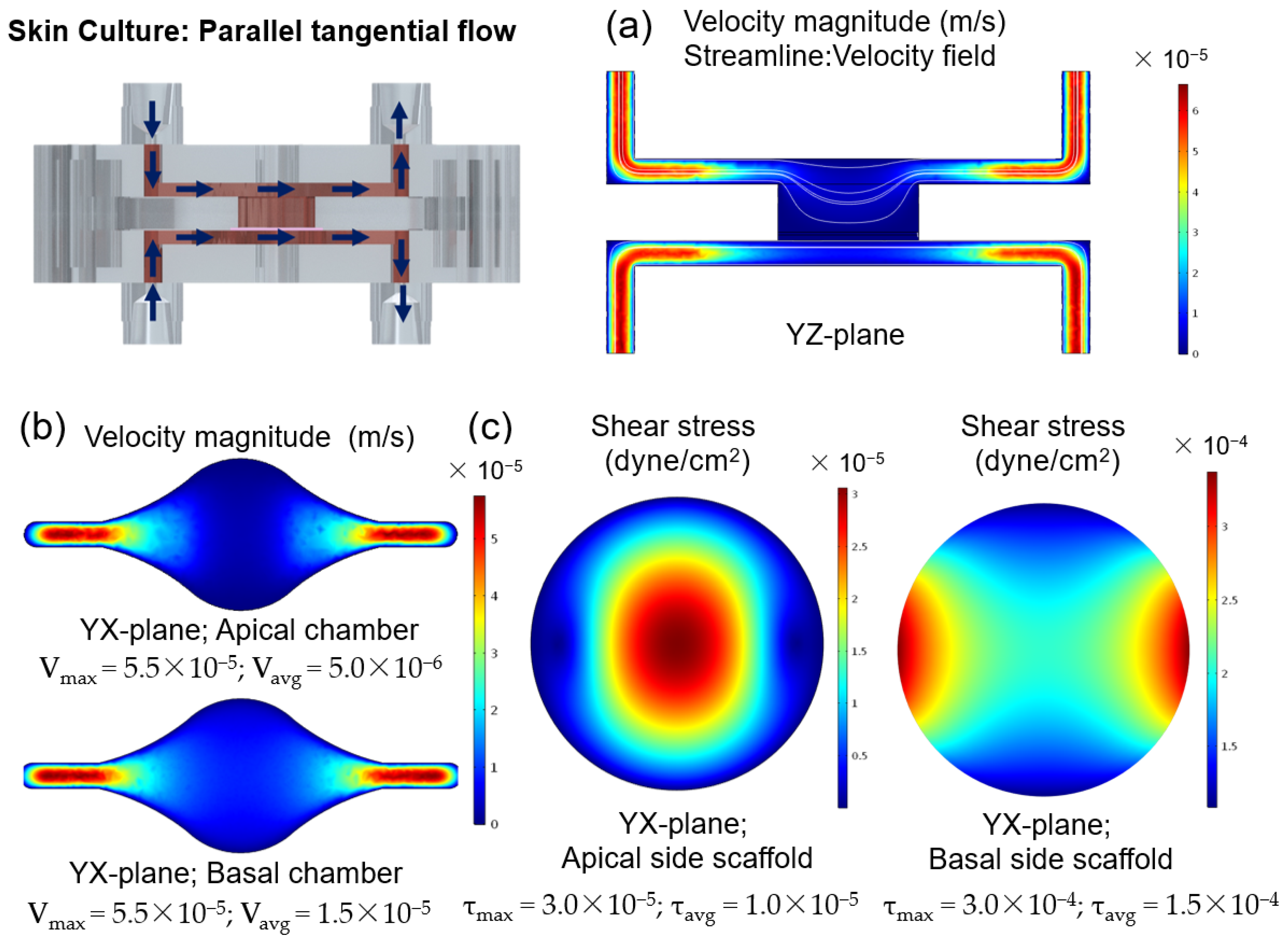
Figure A2 shows the results obtained by performing FEA using the designed SoC under double tangential flow.
2.3. Primary Cells and Cell Maintenance
Primary human dermal fibroblasts isolated from neonatal foreskin (HDFn, CellnTec; Stauffcherstr, Switzerland) were maintained in Fibroblast Growth medium (FGM) composed of Iscove’s Modified Dulbecco’s Medium (IMDM, Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, Waltham, MA, USA), at 37 °C in a 5% CO2 humidifier, following supplier’s instructions. HDFns were used for up to 10 passages and subcultured at a 90% confluency.
Primary human epidermal keratinocytes isolated from neonatal foreskin (HEKn, Gibco, Waltham, MA, USA) were maintained in keratinocyte growth medium (KGM) composed of EpiLife medium (Gibco, Waltham, MA, USA), supplemented with 0.06 mM calcium and keratinocyte growth factor (HKGS, Gibco, Waltham, MA, USA), at 37 °C in a 5% CO2 humidified incubator. The KGM medium was replaced every other day until the cells reached 50% confluency. At this point, the medium was replaced every day. 6th passage HEKns were used, subcultured at 70–80% confluency. All cultures were routinely monitored for contaminations.
2.4. Generation of the Dermal Equivalents
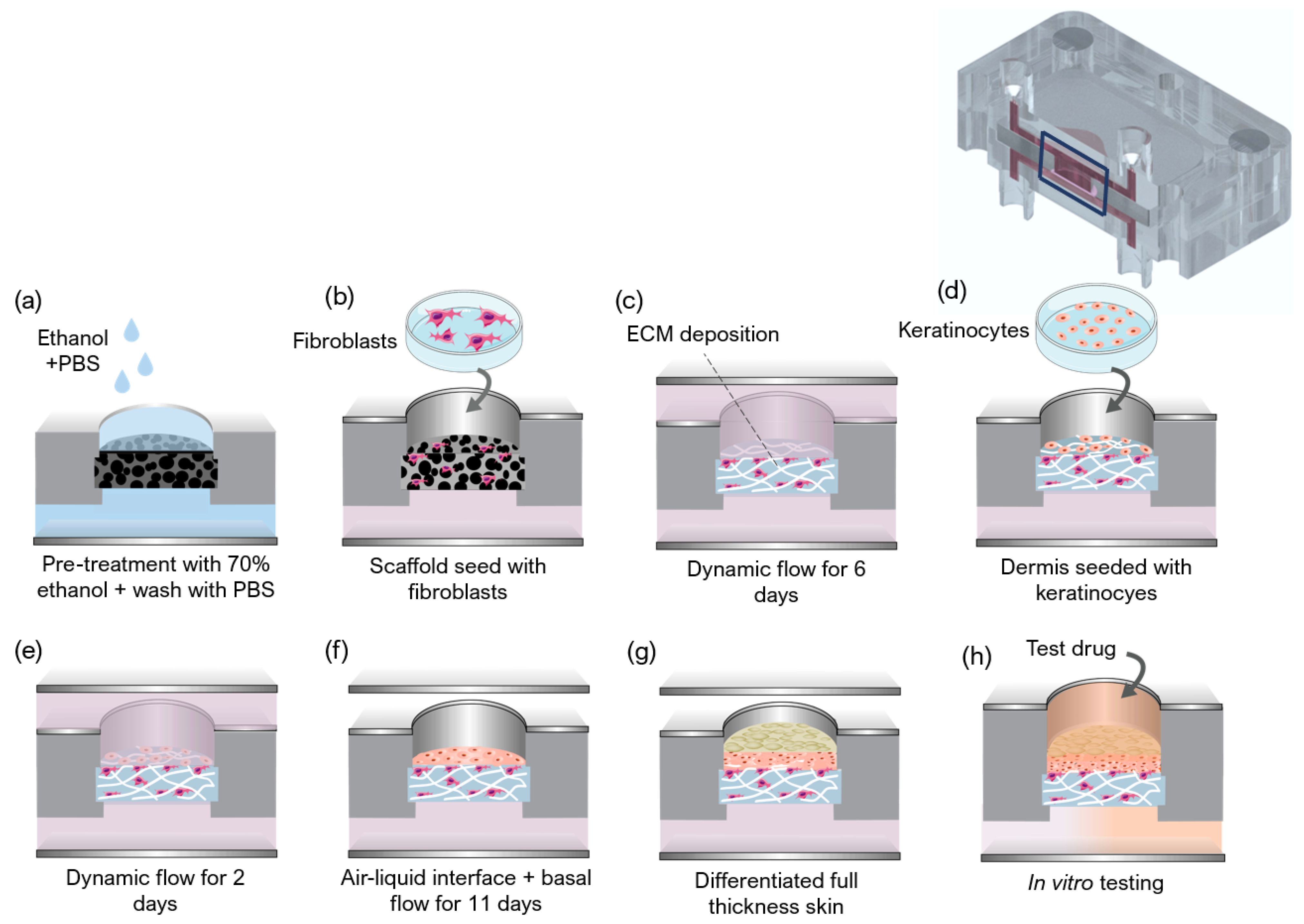
The protocol used to generate a full-thickness and fully human skin model inside the OoC is depicted in
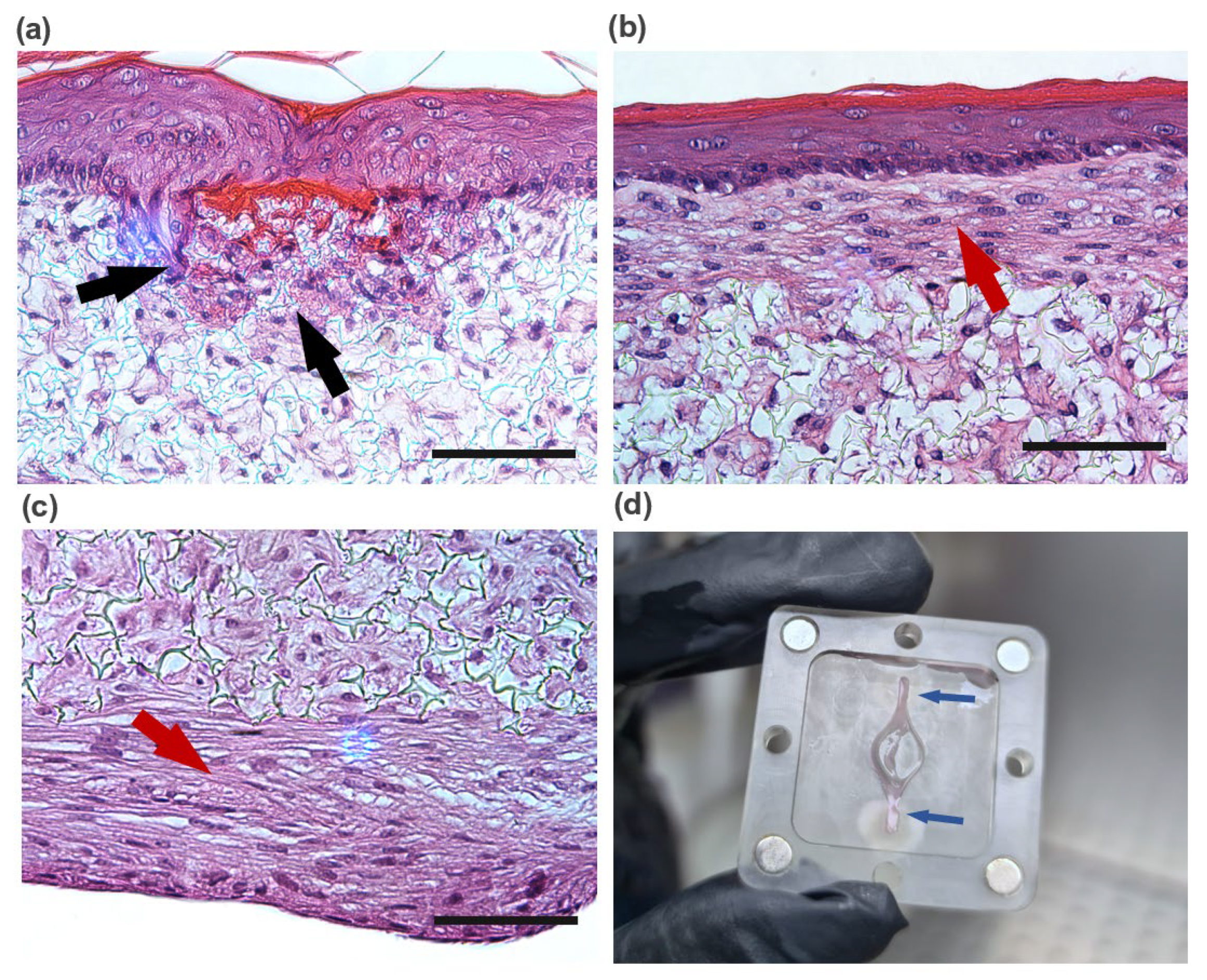
Figure 2. The process can be divided into three main steps: the development of a mature dermis; the culture of an epidermal cell monolayer on top of the dermis under submerged conditions; and the culture of the epidermis at the air-liquid interface (ALI) until a fully-differentiated skin is produced.
The formation of the fully-human dermal equivalent was achieved by seeding HDFns within the Alvetex
® scaffolds (REPROCELL Europe Ltd., Glasgow, UK) and by stimulating them to proliferate and secrete endogenous dermal FDM (
Figure 2a–c) [
27]. The scaffolds were pre-treated with 70% ethanol to render its hydrophilicity followed by two washes with phosphate-buffered saline (PBS) (
Figure 2a). Different concentrations of HDFns (5 × 10
6, 10 × 10
6, 20 × 10
6 cells/mL) were seeded directly on the scaffolds in 30 μL FGM medium and incubated at 37 °C, in a humidified atmosphere of 5% CO
2 for 1.5 h (
Figure 2b). After the cells adhered to the scaffold’s structure, the cell culture layer was introduced into the device, between the apical and basal chambers. The single tangential flow configuration was achieved by connecting the inlet ports of the lower compartments to 3-stop Tygon
® LMT-55 Tubing (Ismatec; Cole-Parmer GmbH, Wertheim, Germany) mounted on a multichannel peristaltic pump (LABV1 with an MC10 pump head, Baoding Shenchen Precision Pump Co., LTD, Baoding, China). The tubing was connected to a media reservoir to supply the culture media to the SoC. A filter (Whatman GmbH, Dassel, Germany) placed at the reservoir allowed permanent gas exchange. The medium was pumped from the reservoir through the inlets of the basal chambers and left the SoC through the basal outlets. The inlets and outlets of the apical chamber were closed with mini-luer plugs (microfluidic ChipShop, Jena, Germany). The dermal equivalent in the culture chamber of the device was perfused with FGM + 100 μg/mL ascorbic acid 2-phosphate (Sigma-Aldrich, St. Louis, MO, USA) at a flow rate of 1.5 μL/min for 6 days (
Figure 2c).
Dermal equivalents were maintained on fabricated supports, which could fit in 6-well plates, and kept under static conditions as controls. After the cell adhesion period, 6 mL of FGM + 100 μg/mL ascorbic acid 2-phosphate was applied to the bottom of each well to flood the cell culture insert. The controls were maintained for 6 days, the medium changed after 3 days, to study the formation of the dermal equivalent without perfusion.
2.5. Generation of the FTSm
The FTSm was generated by seeding HEKn cells onto the dermal equivalents in KGM containing high calcium concentration (1.5 mM). HEKns were harvested by trypsinization, and 30 µL of cell suspension 5.0 × 10
6 cells/mL was pipetted directly on top of the mature dermis (
Figure 2d). This was followed by a 3-h incubation period for cell adhesion at 37 °C in a 5% CO
2 incubator. After this period, double-sided perfusion was established using the perfusion module (
Figure 2e). For this, the inlet ports on the top and lower compartments were connected to the tubing mounted on the peristaltic pump and connected to a media reservoir with supplemented KGM medium. The medium was pumped from the reservoir through the inlets of the apical and basal chambers and left the SoC through the apical and basal outlets. With this configuration, the tissue in the culture chamber of the device was double-sided perfused with KGM at a flow rate of 2 μL/min. The formation of a differentiated epidermis was achieved by pumping the medium only through the basal chamber while pumping air through the apical chamber. The ALI medium was composed of KGM containing 1.5 mM calcium, supplemented with 10 ng/mL KGF (Keratinocyte growth factor, Gibco, Waltham, MA, USA) and 100 μg/mL ascorbic acid 2-phosphate A flow rate of 1.5 μL/ml was established for medium and airflow, maintained up to a further 11 days.
The controls were maintained under submerged conditions in KGM containing high calcium concentration (1.5 mM), using the fabricated supports 6-well plates, without perfusion. After 48 h, the models were raised as well. For the controls, the FTSm were raised to ALI by removing the medium in the upper compartment and culture in 4 mL KGM containing 1.5 mM calcium, supplemented with 10 ng/m and 100 μg/mL ascorbic acid 2-phosphate. This volume resulted in the basal side of the skin tissue being in contact with the media and the upper surface to remain exposed to the air. The incubation continued at 37 °C in 5% CO2 for 11 days, changing the media every two days, to allow the formation of an FTSm.
2.6. Histochemistry and Immunofluorescence Analysis
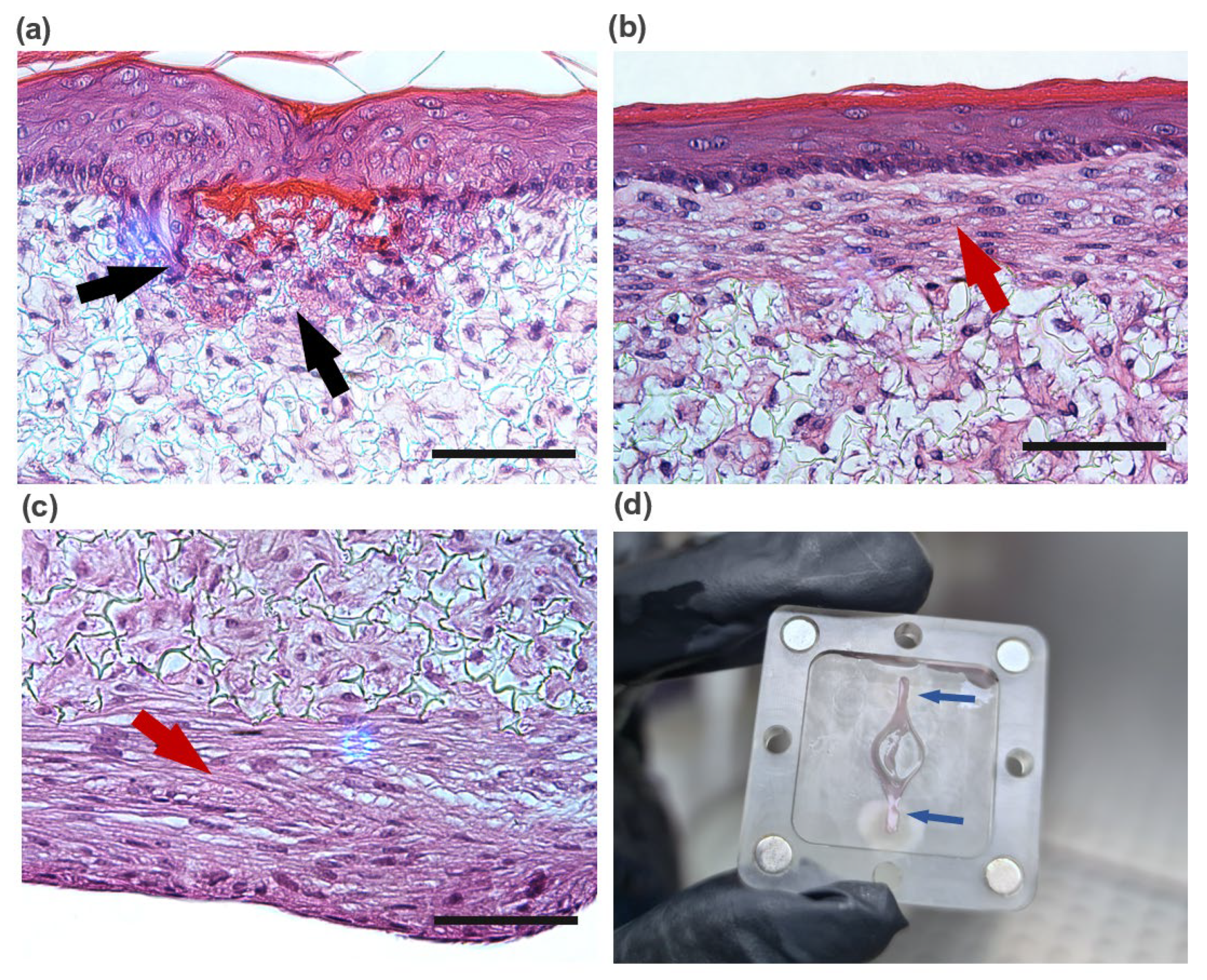
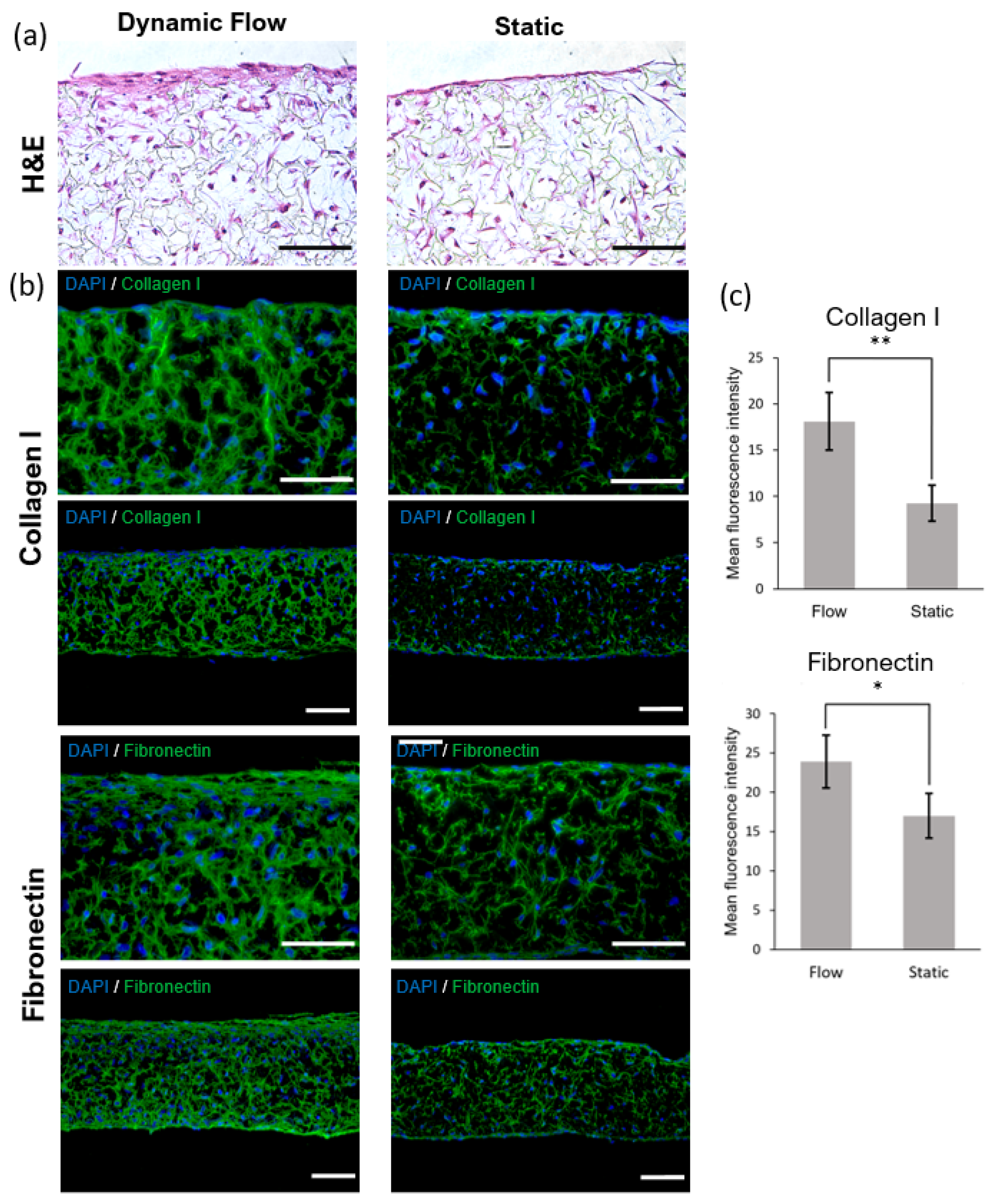
The reconstructed tissues were fixed in 10% neutral buffered formalin (Sigma) immediately after being taken out of the chip. The samples were embedded in paraffin to allow for transverse sectioning. Paraffin-embedded sections (5 µm-thick) were de-paraffined and re-hydrated for morphological evaluation by staining with haematoxylin and eosin (H&E) through standard methods. Immunofluorescence analysis was performed to detect significant markers related to dermal maturation and epidermal differentiation. Skin sections were deparaffinized using HistoChoice® (Sigma-Aldrich) and incubated in a dry chamber at 65 °C for 30 min. Then, skin sections were hydrated for 5 min in a gradient of ethanol solutions (100%, 96%, and 70%). The antigen retrieval was performed by heating in citrate buffer (pH = 6). For anti-collagen IV antibody, the antigen retrieval was followed by proteinase K [20 µg/mL in Tris-EDTA (TE) Buffer] incubation for 30 s at 37 °C with 10 min of cooling at room temperature. Immunofluorescence staining of sections was performed using the following primary antibodies: 1:500 anti-collagen IV (Abcam, Cambridge, UK), 1:10 anti-cytokeratin 10 (Progen), 1:1000 anti-cytokeratin 14 (Abcam), 1:100 anti-cytokeratine 15 (Sigma-Aldrich), 1:250 anti-fibronectin (Abcam), 1:50 anti-filaggrin (Invitrogen, Waltham, MA, USA) and 1:100 anti-collagen I (Abcam) diluted in a solution with PBS-BSA 1% with overnight incubation in a humidified chamber at 4 °C. Secondary antibodies, 1:1000 Alexa Fluor® 488 Goat Anti-Rabbit IgG (Invitrogen) or 1:1000 FITC Goat Anti-Mouse IgG (Sigma-Aldrich), diluted in a solution with PBS-BSA 1%, were then added with an incubation of 2 h. Nuclei were stained with 1 μg/mL of 4,6-diamidino-2-Phenylindole (DAPI, Invitrogen), and slides were mounted using Vectashield (Vector Laboratories). Images were obtained using the Nikon Eclipse TE2000-S fluorescence microscope (Nikon Instruments, Melville, NY, USA) and analysed with the ImageJ software.
2.7. TEER Measurements
TEER was measured using the SoC with integrated tetrapolar electrodes and an EVOM volt-ohmmeter (WPI Europe, Friedberg, Germany). The volt-ohmmeter supplies an AC square-wave current of ±10 µA at 12.5 Hz through the outer current-carrying electrodes and measures the voltage drop across the inner voltage-sensing electrodes. This technique was used to measure the TEER of 3 batches of FTSm generated on-chip, and 3 batches of FTSm generated off-chip (controls). TEER was recorded after 11 days of culture at ALI, following completion of the FTSm. For the controls, the cell culture layer was removed from its original supports and transferred to an SoC module with integrated electrodes. The ALI was interrupted by pumping PBS into the basal and apical chambers to bridge an electrical connection between top and bottom sensors. TEER was recorded for 5 min to achieve stabilisation at room temperature. TEER was also measured for a blank scaffold to take into account the contribution of the geometry of the fluidic chambers and subtracted from each recording. The values in Ω·cm2 were obtained by multiplying the obtained electrical resistance by the skin surface area.
2.8. Permeation Assays
Permeation assays were conducted following TEER measurement for 3 batches of FTSm generated on-chip, and 3 baches of FTSm generated off-chip. The drug testing module was used to perform in situ permeation experiments. The culture media reservoir was replaced with a reservoir containing the receiver solution (PBS) supplied in the basal compartment. The perfusates samples flowing out from the basal chamber were collected directly in a 96-well plate, according to predetermined time intervals. The donor compartment was filled with 100 µL of fluorescein isothiocyanate (FITC)—dextran with an average molecular weight of 4.4 kDa (Sigma-Aldrich) at a 2.5 mg/mL concentration in PBS. The donor compartment was covered with PCR tape to minimize evaporation. The perfusate was collected into a different well every 30 min for 6 h after applying the donor solution. This was achieved by pumping the receiver solution at an 8 µL/min flow rate. The concentrations of the skin-permeated FITC-dextran were determined by fluorescence spectroscopy (SLM 8100, SLM Instruments Inc., Rochester, NY, USA) at excitation and emission wavelengths of 495 and 519 nm, respectively. The concentrations were calculated using a calibration curve and converted in cumulative amounts that permeated per unit skin surface area. The slope of the linear portion of the area-normalized cumulative amount profiles were used to calculate the permeability coefficient, following Fick’s first law.
2.9. Statistical Analysis
Quantitative data are presented as a mean ± standard deviation (SD). The statistical significance was calculated using the one-way ANOVA. Statistical differences are noted as *, ** or ***, corresponding to p < 0.05, p < 0.01 and p < 0.001, respectively.
4. Discussion
In this study, we present an SoC device to develop a full-thickness and fully human skin construct. The device includes a flexible architecture based on a modular and reusable approach. Conventional OoC are irreversibly sealed, usually through plasma bonding, making it difficult to completely remove the tissue for analysis [
28]. The reversible-sealing feature allows easy removal of the skin tissue for histological and immunohistochemistry analysis without damaging the tissue and direct cell seeding. Furthermore, the device is fabricated using rapid prototyping and excludes soft-lithography and plasma bonding, making it compatible with mass production [
29]. Importantly, the described design made it possible to integrate a thick scaffold (200 µm) for in vivo-like 3D cell culture generation of the dermal compartment.
One of the major challenges for generating a biomimetic skin model is the development of a dermal compartment with in vivo-like composition and mechanical environment while assuring its stability and reproducibility during culture and testing [
30]. Typical FTSm generated inside an OoC device uses animal-derived hydrogels to recreate the dermal compartment [
23,
24,
31,
32,
33,
34,
35]. These constructs are not fully representative of the in vivo ECM, which comprises multiple types of collagens, fibrous proteins, and proteoglycans but suffer from fibroblast-mediated contraction and matrix degradation, limiting their lifespan, reproducibility, and applicability [
36]. In particular, the collagen detachment inside the SoC platform leads to various documented problems, including the inability to maintain a leakage-free fluid-tissue-air barrier and the inability to perform permeation assays in situ [
37,
38]. For SoC applications, the materials used for the dermis construct should mimic the biophysical properties of the in vivo skin and remain stable during cell culture. This was achieved in previous studies by adding synthetic or natural polymers to the hydrogels, for example, by combining fibrinogen with PEG [
25]. However, the rheological behaviour of hydrogels makes them difficult to be manipulated and introduced in OoC devices in a reproducible manner [
39].
Here, we propose an alternative approach by combining an inert scaffold integrated on the chip with the concept of a fibroblast-derived matrix, in which the dermal cells are stimulated to produce their own endogenous ECM components (FDM). Our group previously optimized this technique for the culture of a pigmented FTSm, under static conditions [
27]. We showed that the developed model could maintain its architecture and function for up to 50 days at ALI. In this study, we evaluate the impact of the dynamic flow on the development of a dermis construct and FTSm.
There is abundant evidence that mechanical forces are important for the architecture and physiology of multiple tissues such as the lung, bone, and vascular tissues [
40]. Furthermore, studies showed the effect of interstitial flown on fibroblasts, especially regarding its alignment [
41,
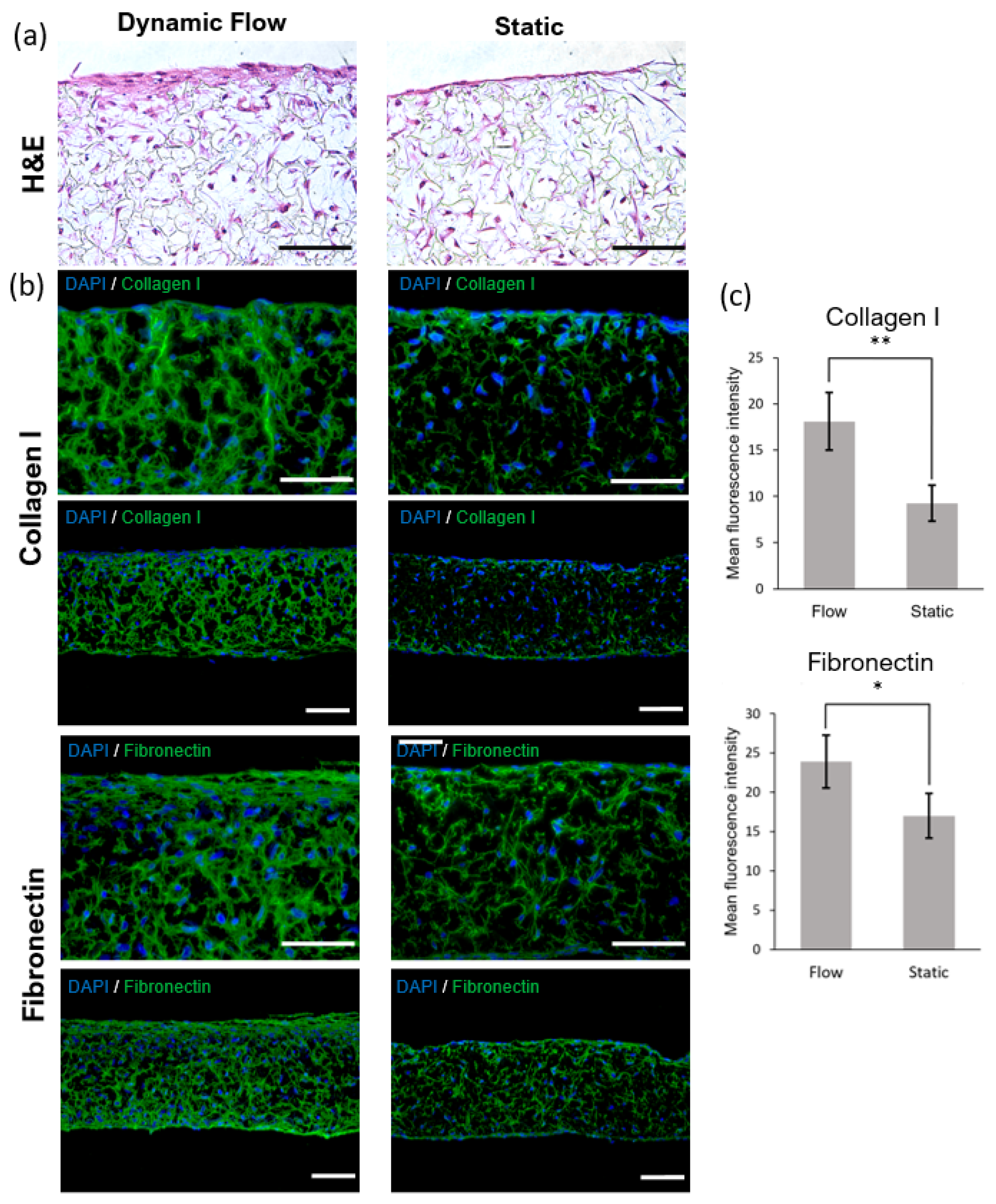
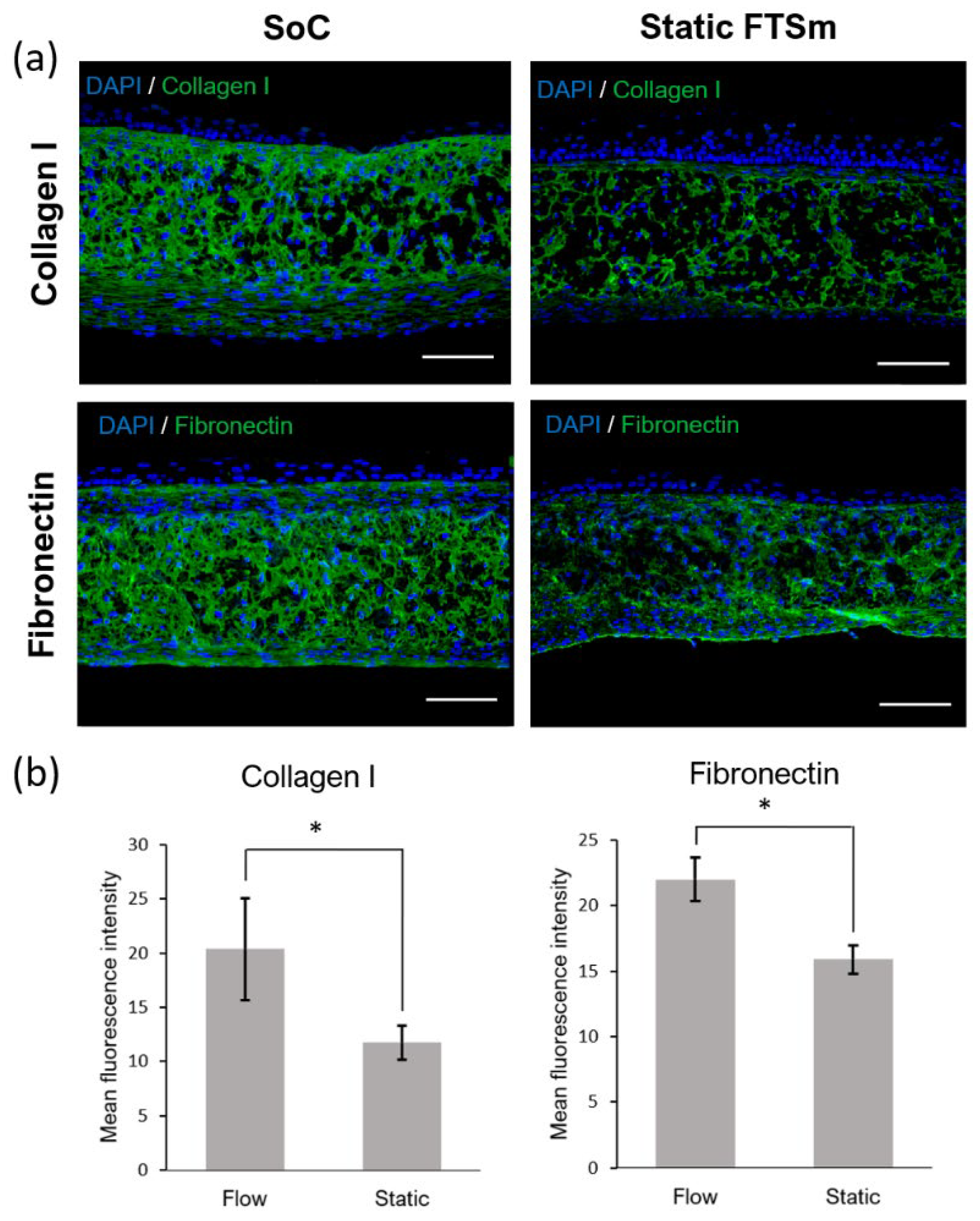
42]. The histological analysis of the generated dermal compartment showed fibroblasts populate the scaffold and generate FDM proteins, giving rise to a mature in vivo-like dermal structure. Compared to the static culture, developing a dermal compartment using dynamic flow resulted in increased deposition of FDM (fibronectin and collagen type I) onto the scaffold structure. The effect of the dynamic flow on FDM deposition could be seen in the case of dermal compartments cultured for 6 days on-chip and in FTSm cultured for approximately 20 days on-chip. It can be hypothesized that this phenomenon results from increased interstitial flow on-chip. In a porous medium such as the dermal compartment, the pore walls impede the momentum transport of the fluid outside the individual pores. Consequently, shear stresses in the fluid are often negligible. However, the dynamic flow can increase the convection necessary for transporting nutrients through the interstitial space, mimicking important components of the microcirculation [
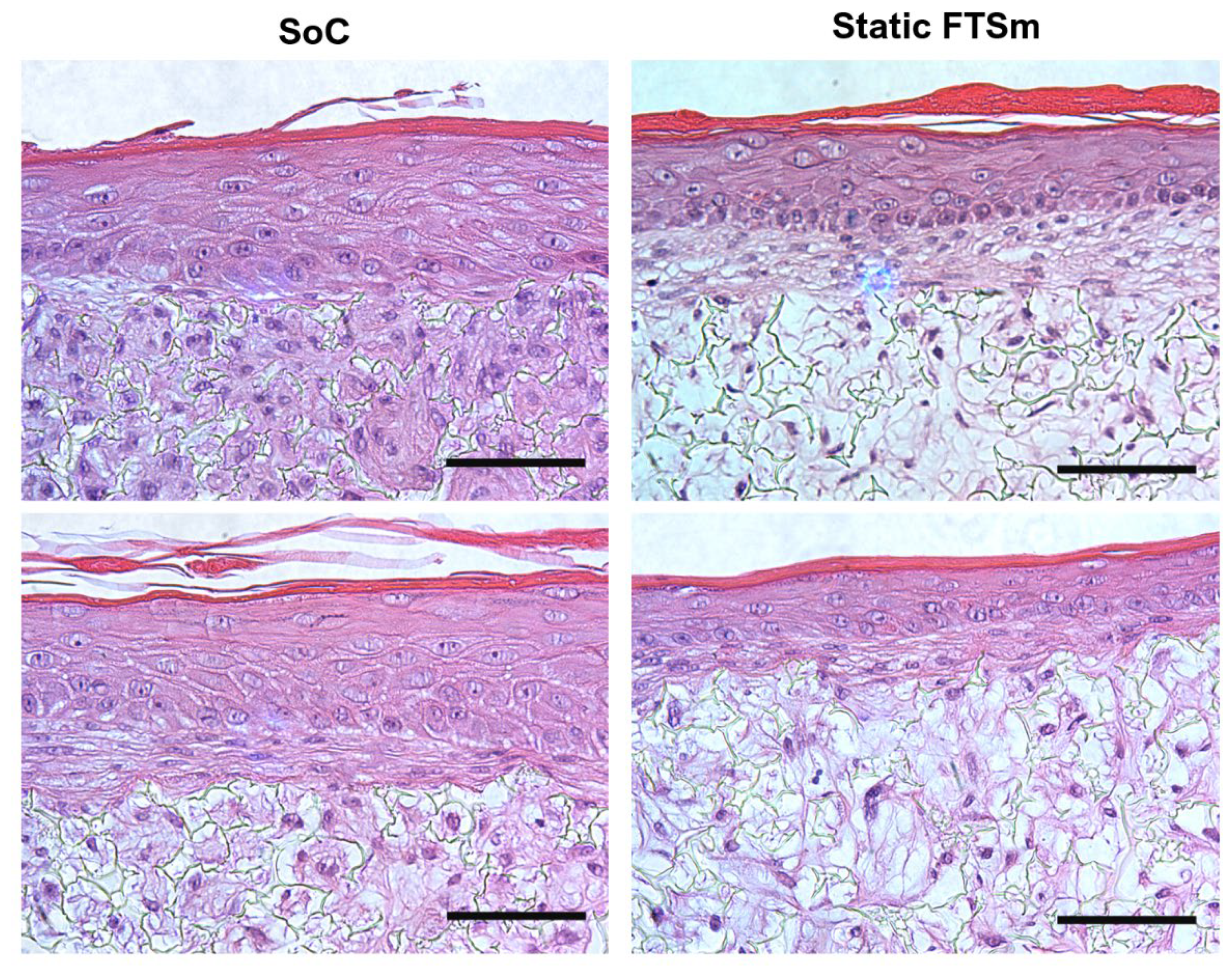
43]. Future experiments should be performed to determine interstitial flow velocity using Darcy’s law which relates velocity to the pressure gradient and the hydraulic conductivity of the dermal compartment. The described dermis-on-a-chip could be a relevant stand-alone model to study in vivo-like FDM deposition and remodelling. The tissue development using a porous polymeric scaffold can overcome important limitations of current SoC models due to its mechanical stability and robustness. The biologic variability is reduced since this approach does not require exogenous animal-derived collagen hydrogels. Moreover, since no hydrogels are introduced in the model, the manipulation of the device could be easily performed in a reproducible manner with syringe/peristaltic pumps.
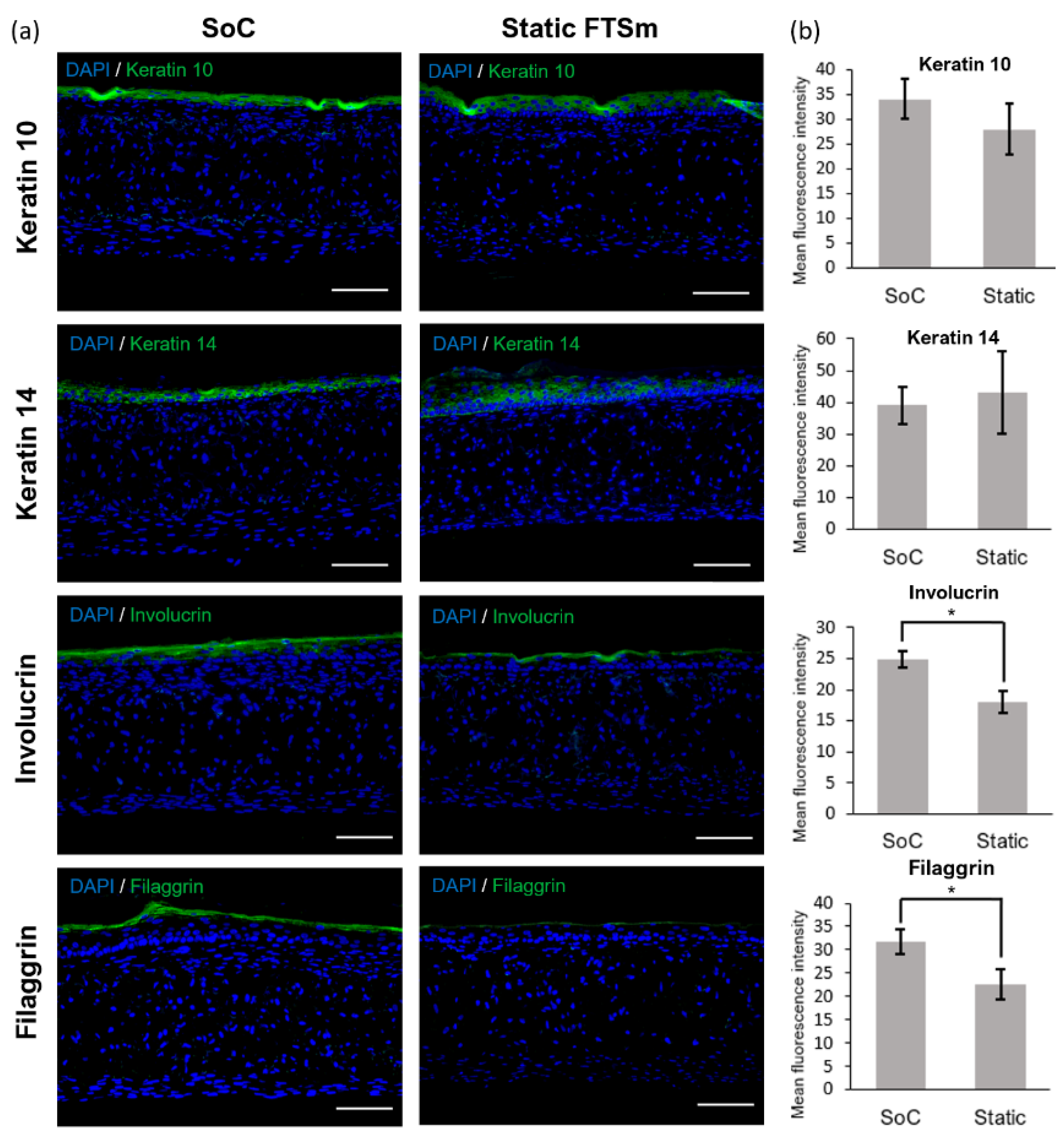
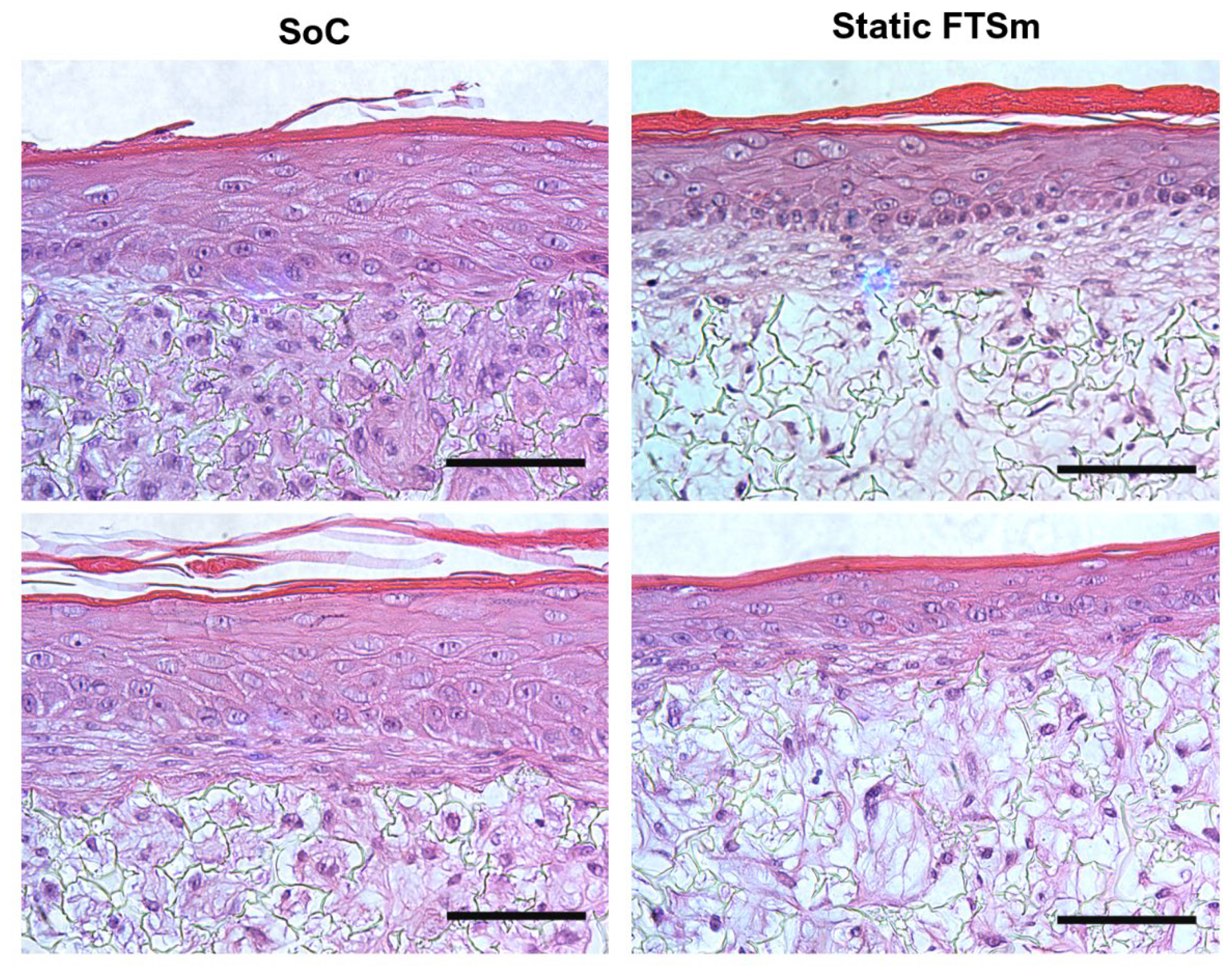
The mature dermal matrix was then used to recreate an FTSm with added perfusion. Conventional skin models still have important limitations, including increased permeability and different architecture than native human skin [
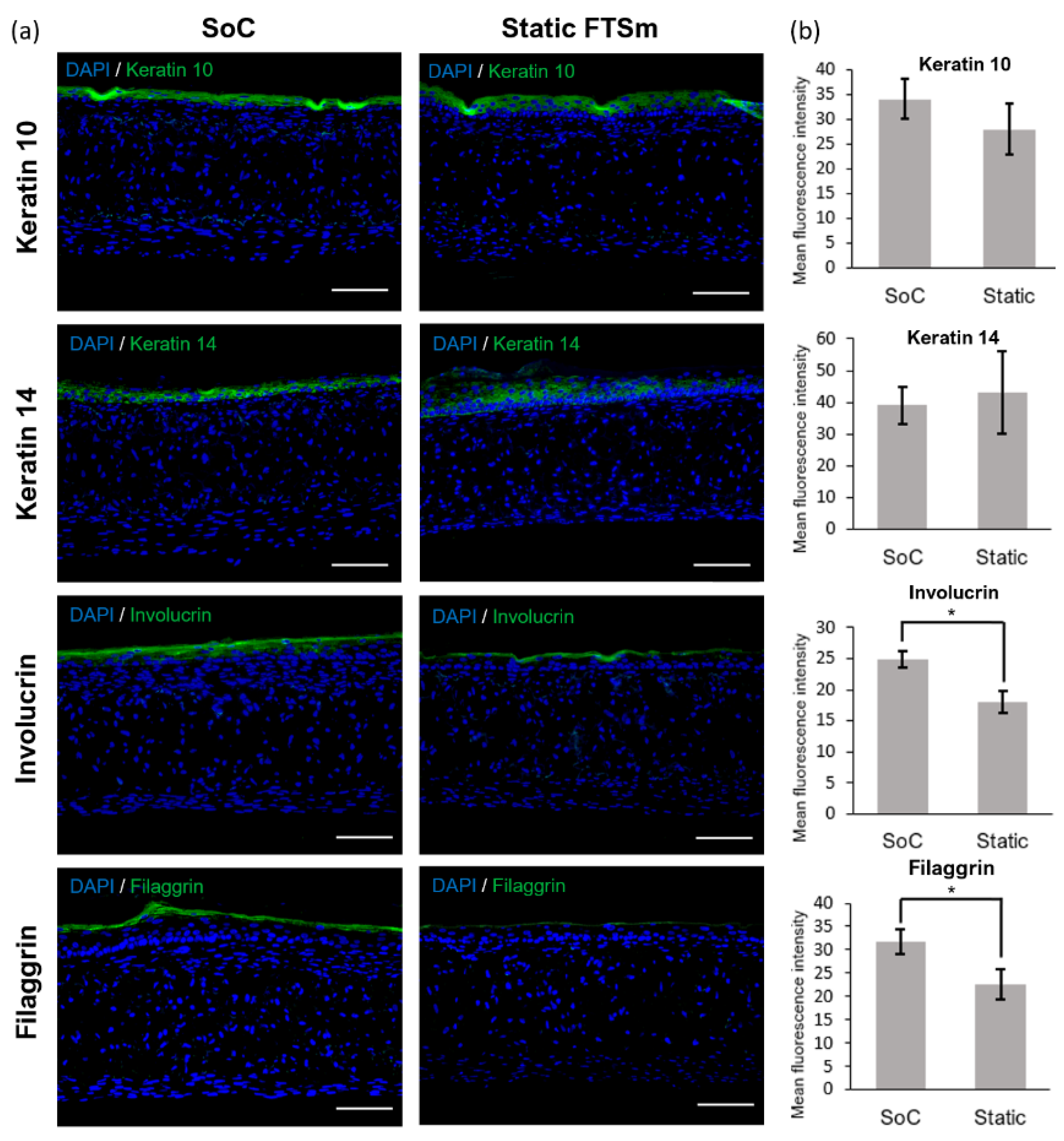
44]. One proposed explanation is the lack of in vivo-like mechanical forces and dynamic flow under static conditions. Using the developed OoC platform, we generated a mature, pluristratified, and orthokeratinized epidermal construct with increased thickness and barrier function compared to the static models. The SoC models presented an organized cell morphology with basal keratinocytes undergoing sequential differentiation and stratification, forming keratinocytes with a morphology characteristic of the stratum spinosum and stratum granulosum. Finally, the keratinocytes underwent terminal differentiation into anuclear, flattened corneocytes within the stratum corneum layers, similar to the in vivo skin. Immunofluorescence analysis further showed the presence of markers of basal, suprabasal, and terminally differentiated cells expressed in the corresponding layers. Compared to the static model, the SoC showed increased filaggrin expression, a skin barrier protein and differentiation marker, and a trend for increased involucrin expression. This observation is similar to others reported in the literature [
25,
34].
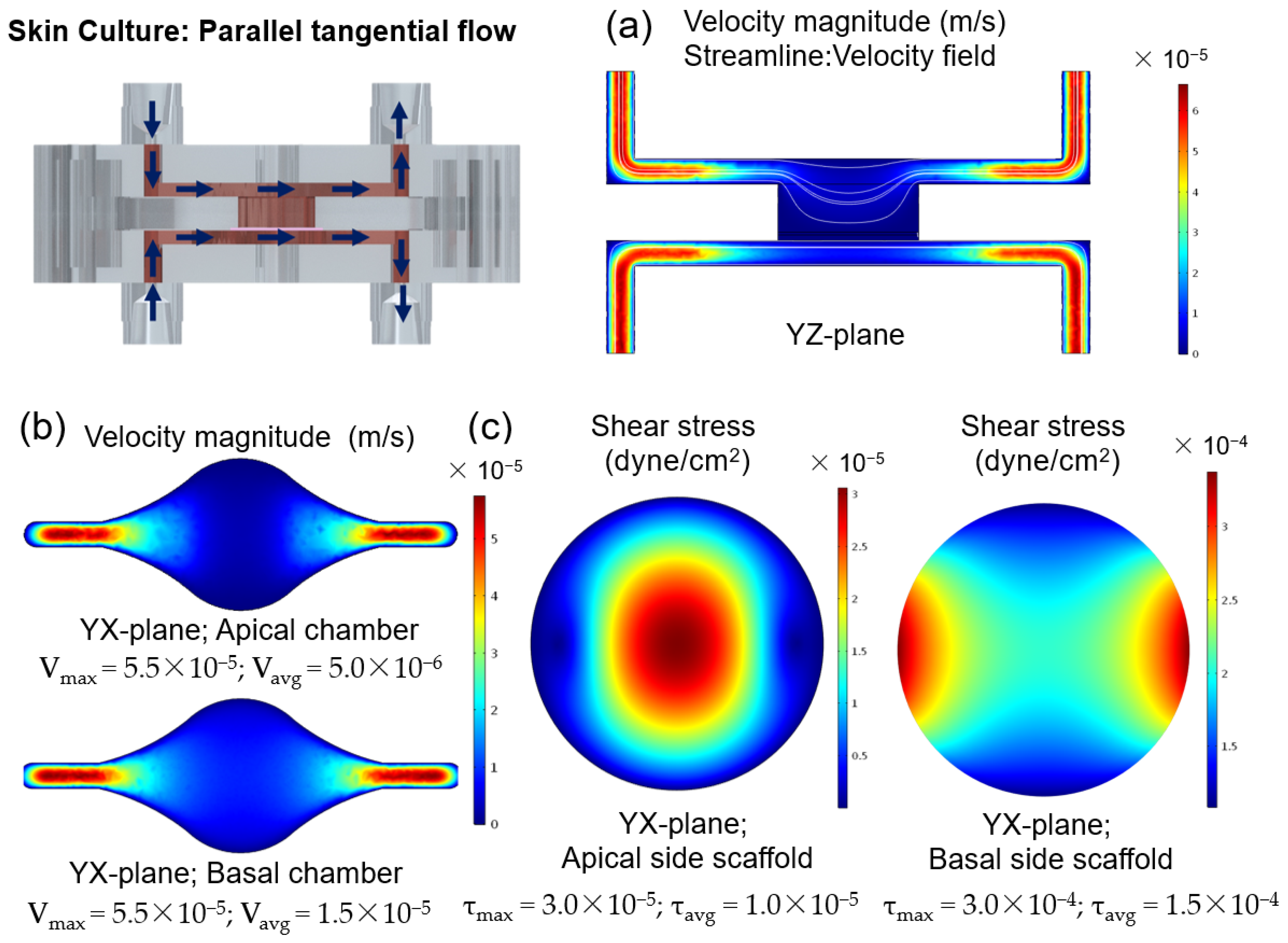
Shear stresses with a magnitude of 0.06 dyn/cm
2 impact keratinocyte morphology leading to morphological variation and cytoskeletal reorganization, and a magnitude of 6 dyn/cm
2 results in cellular disruption [
45]. It has been hypothesized that shear stresses acting on the epidermal layer under submerged conditions could contribute to the positive effect of dynamic perfusion on skin constructs. To study this effect, simulations were performed to calculate the shear stress and to study the hydrodynamics of the SoC during the 48 h under submerged conditions, with double tangential perfusion. The overall chamber design protects the cells from the effects of the shear stress and, combined with small flow rates, results in low shear stress with a maximum magnitude of 3 × 10
−5 dyn/cm
2 at the centre of the apical chamber. These low values are not expected to significantly impact the cell morphology and behaviour. Thus, the particular role of shear stress for skin optimization seems to be reduced using the described device and protocol. It can be hypothesized that the differences between the SoC model and the controls result from the dynamic transport and interstitial flow inside the chip, which can enhance the transport of nutrients and induce various morphogenetic effects [
43]. Furthermore, the peristaltic flow may induce mechanical stretching, generating a thicker epidermis [
46].
It is relevant to note that, in this study, the static FTsm present lower thickness and lower filaggrin expression than the models generated in our previous studies using primary HEKns at a 3rd passage [
27]. This is possibly a consequence of the higher keratinocyte passage (6th passage) used. Further studies should be performed to fully characterize the impact of the cell passage on skin differentiation.
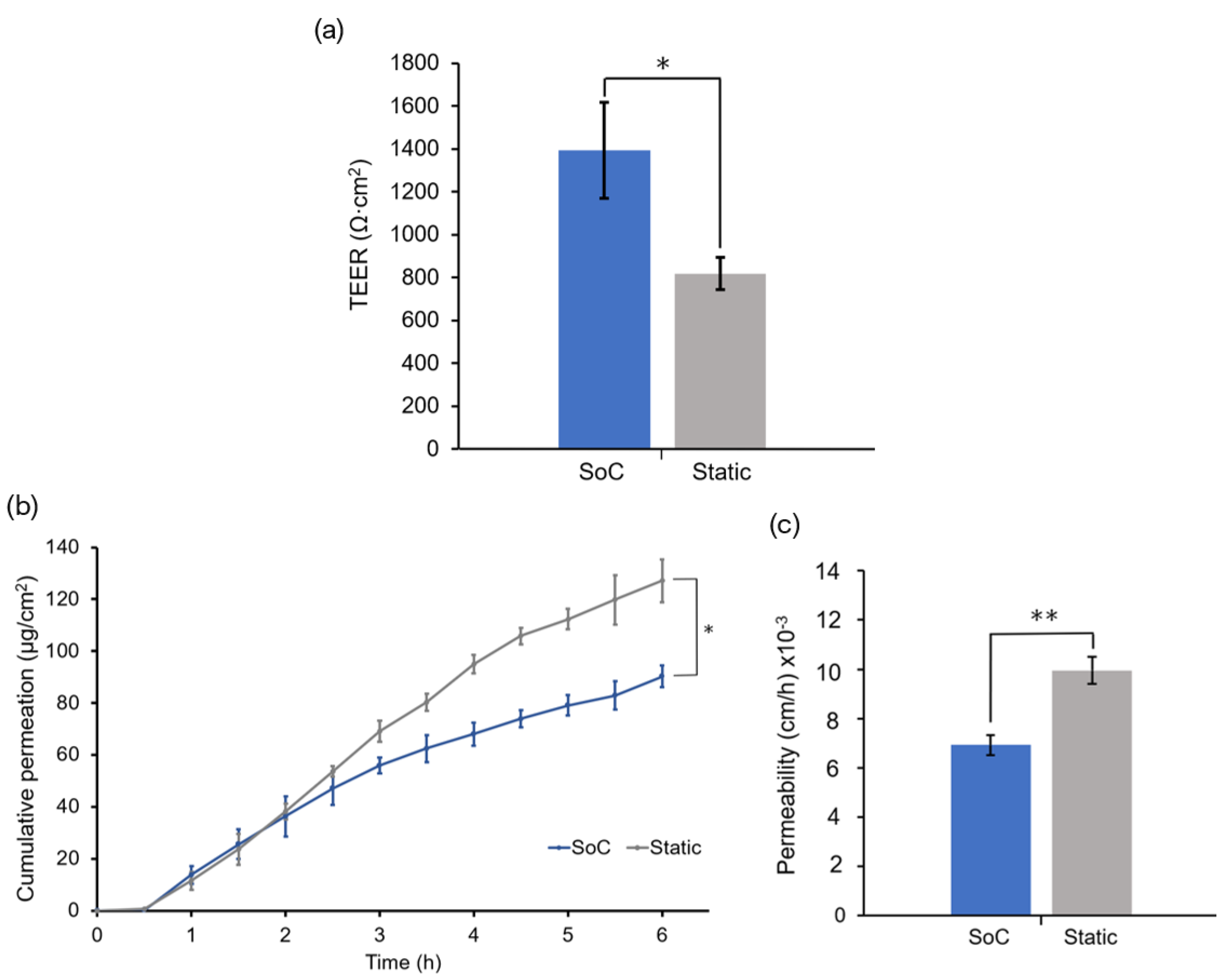
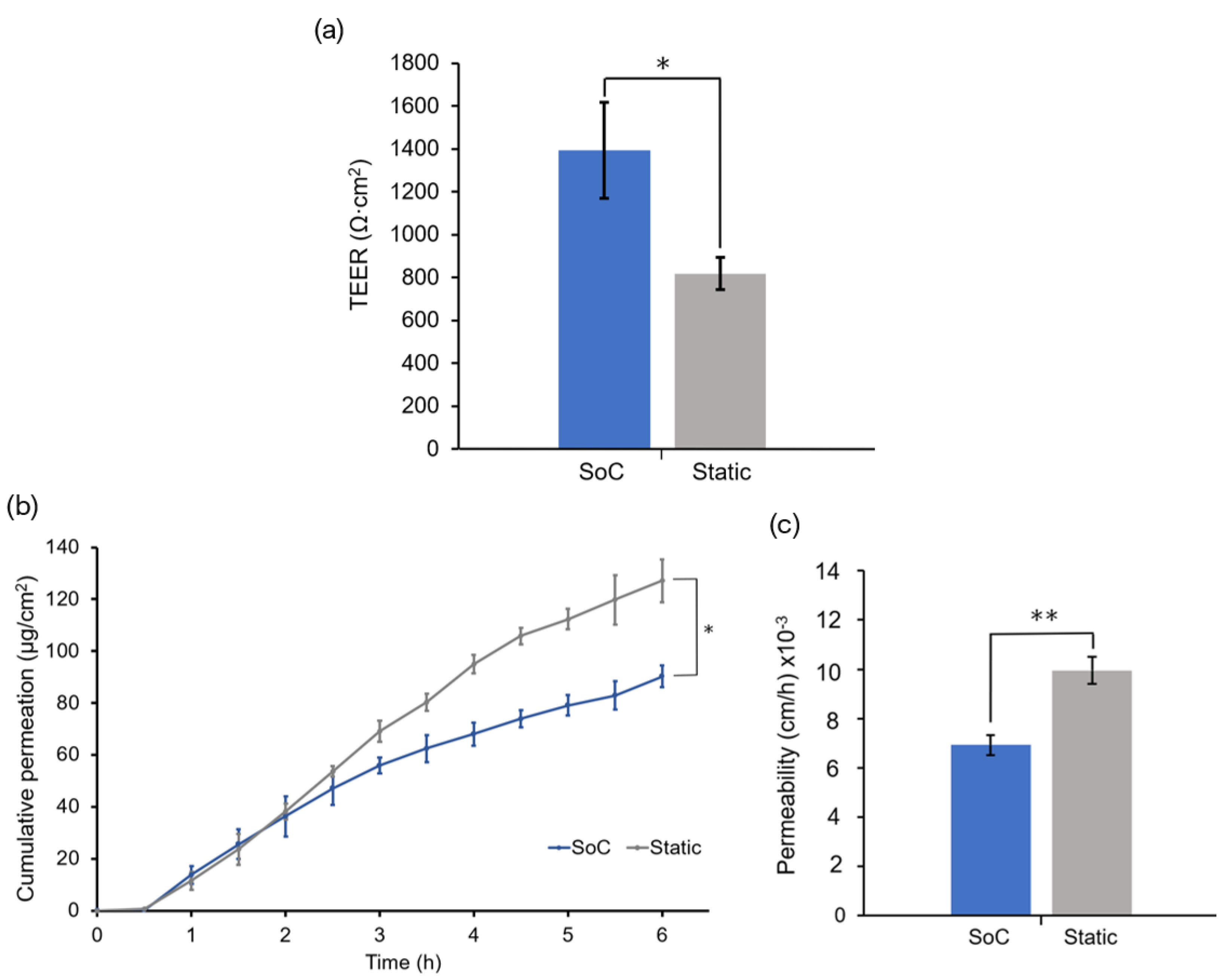
The skin barrier function was also evaluated by performing TEER measurements. In a previous study, we showed that the OoC with integrated sensors could monitor the skin barrier in real-time, which is useful for following skin development and evaluating the toxicity of topical drugs [
26]. In this study, we used TEER to compare the barrier of the SoC models with the static controls. The mean TEER of the skin tissues generated on-chip was significantly higher than those generated off-chip, without perfusion. This result correlates with the increased expression of filaggrin and involucrin in SoC models, both proteins that contribute to the integrity of the epidermal barrier. The increased SoC barrier function could also be seen by its significantly lower FITC-dextran permeability when compared to the controls. The permeation studies could be performed directly on-chip using the drug testing module, avoiding Franz diffusion cell experiments. The non-contractile nature of the developed dermal compartment avoided common artifacts reported in the literature, such as incorrect sealing compromising the permeation results [
34].





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}