
Multi-Omics Analysis of Mouse Fecal Microbiome Reveals Supplier-Dependent Functional Differences and Novel Metagenome-Assembled Genomes

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice and Sample Collection
2.2. Power Analysis
2.3. DNA Extraction
2.4. RNA Extraction
2.5. Metagenomic Library Preparation
2.6. Metatranscriptomic Library Preparation
2.7. Meta-Omic Preprocessing, Assembly, Binning, and Analyses
2.8. Phylogenomics, Pangenome Construction and Differential Analyses
2.9. Data Analyses and Figures
3. Results
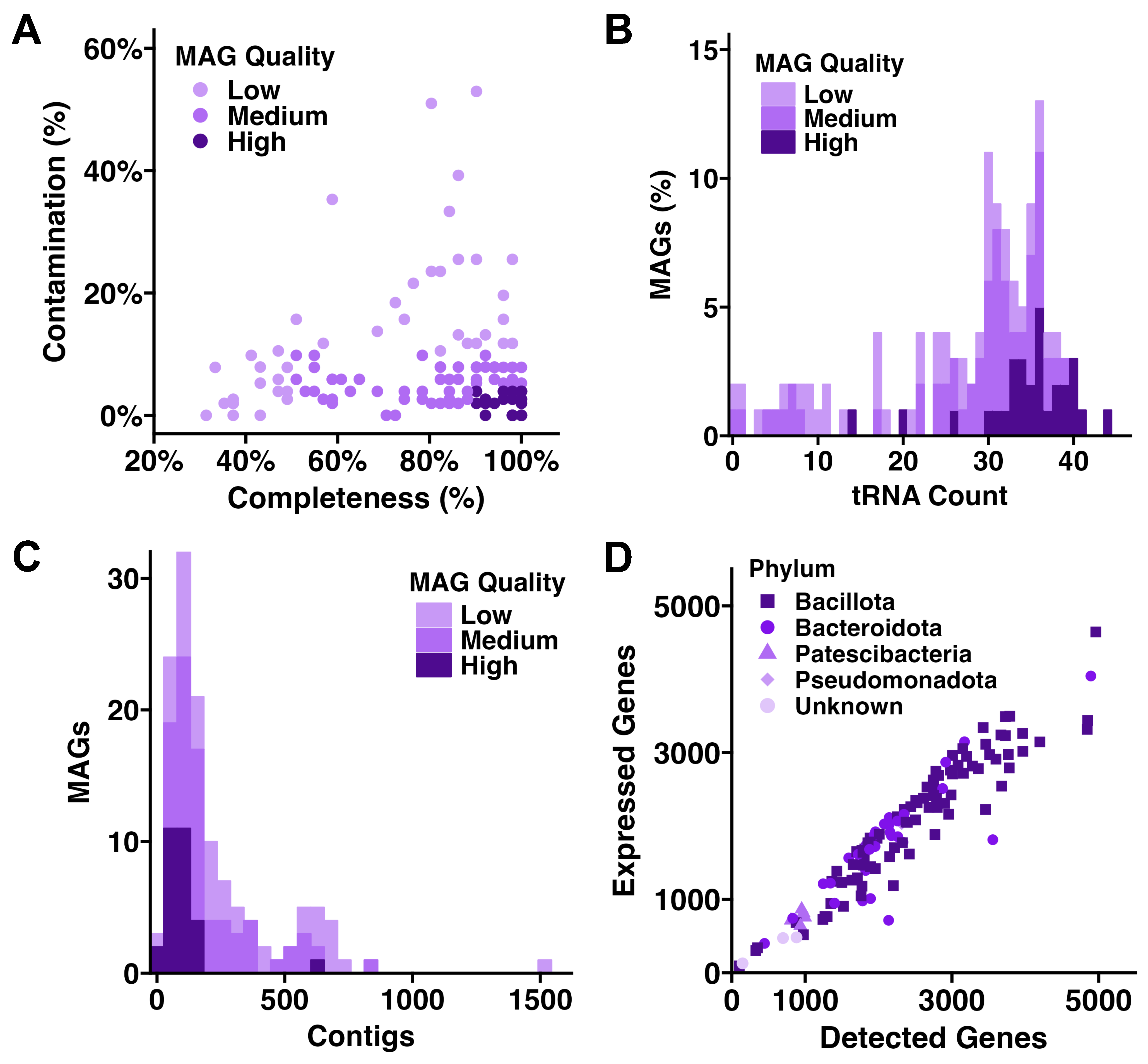
3.1. Metagenomic, Metatranscriptomic, and Taxonomic Summary
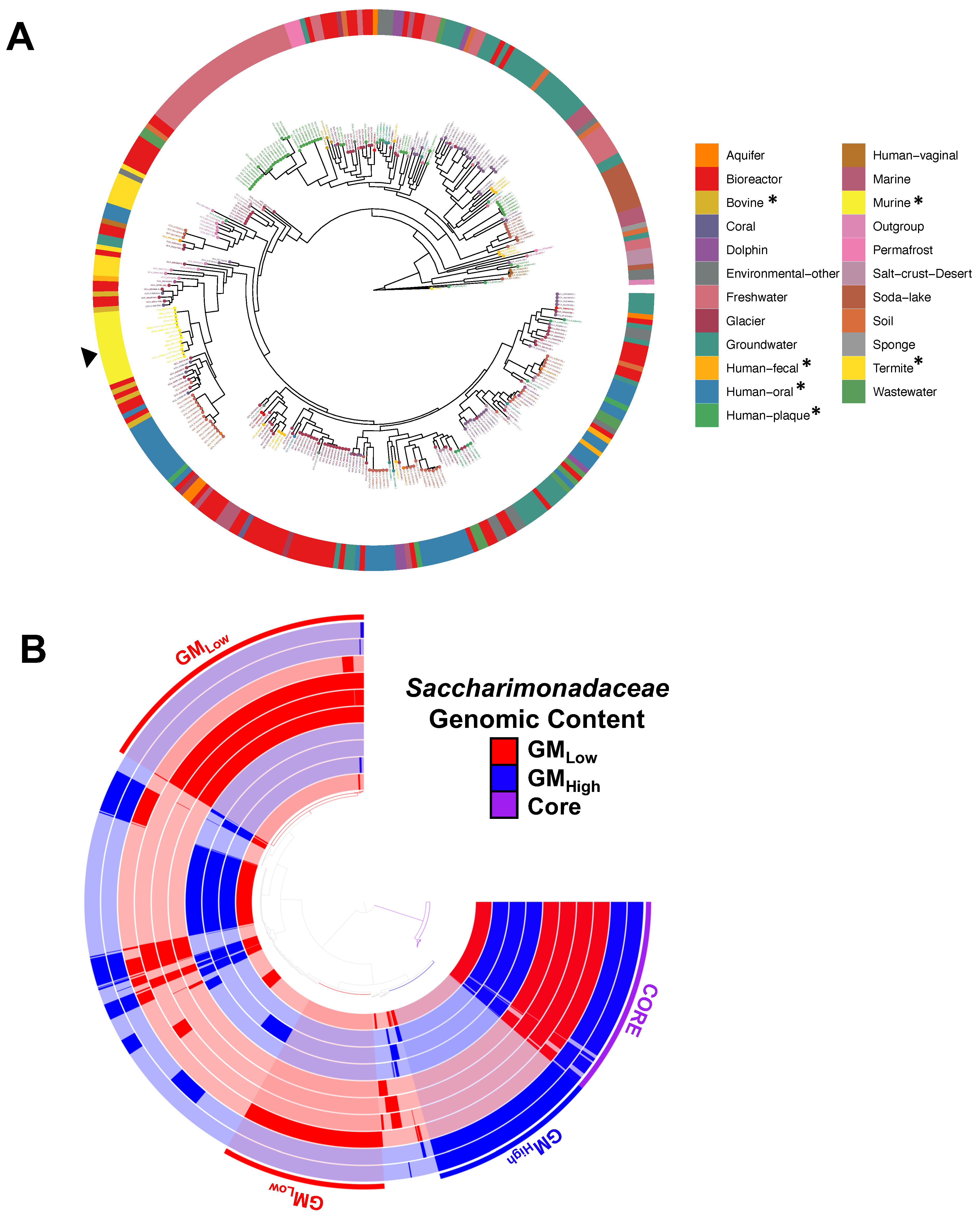
3.2. Candidate Phyla Radiation Taxa Demonstrate Strain-Level Differences Between Vendors
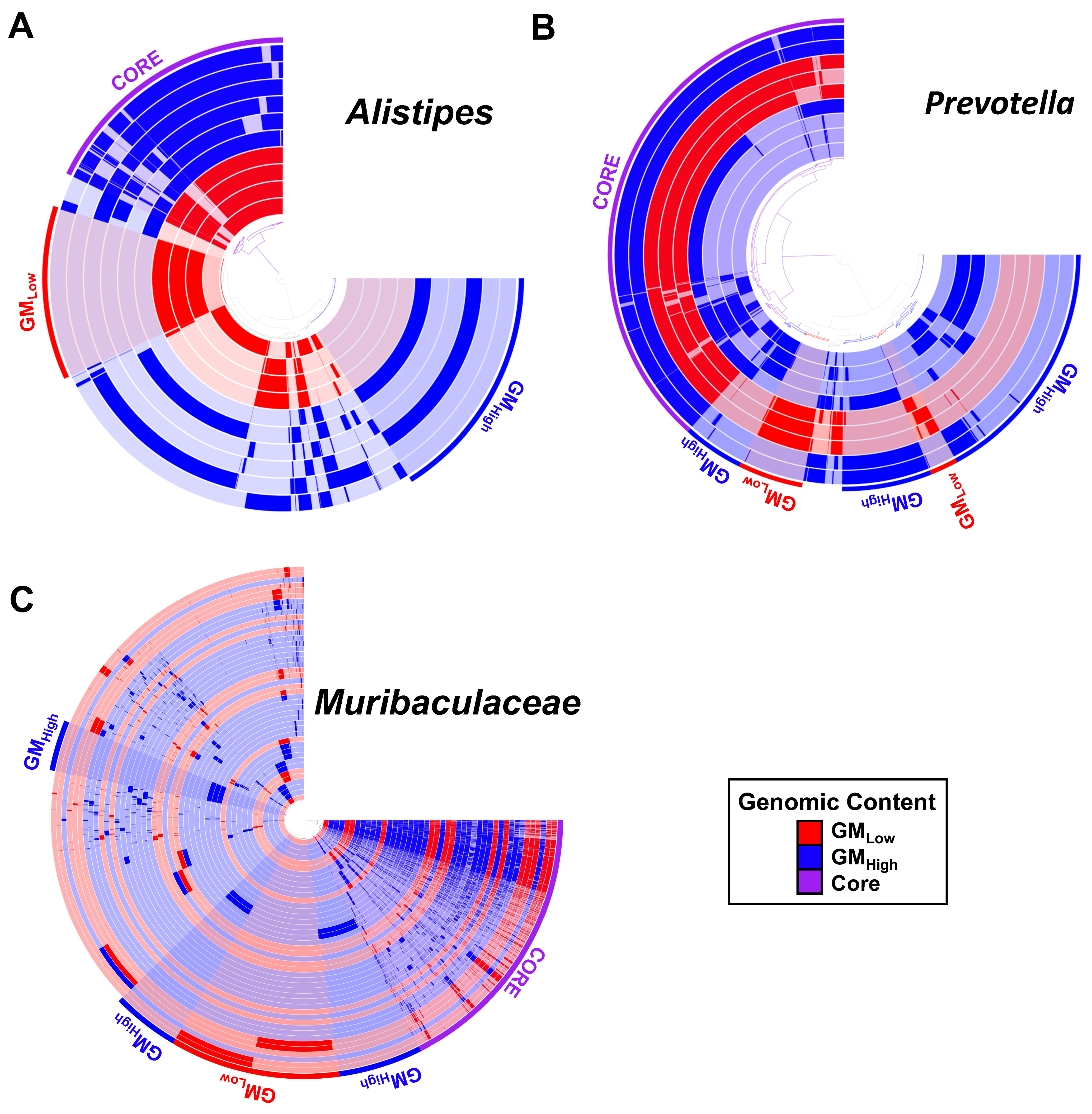
3.3. Distinct Source-Dependent MAGs Within Multiple Taxonomies
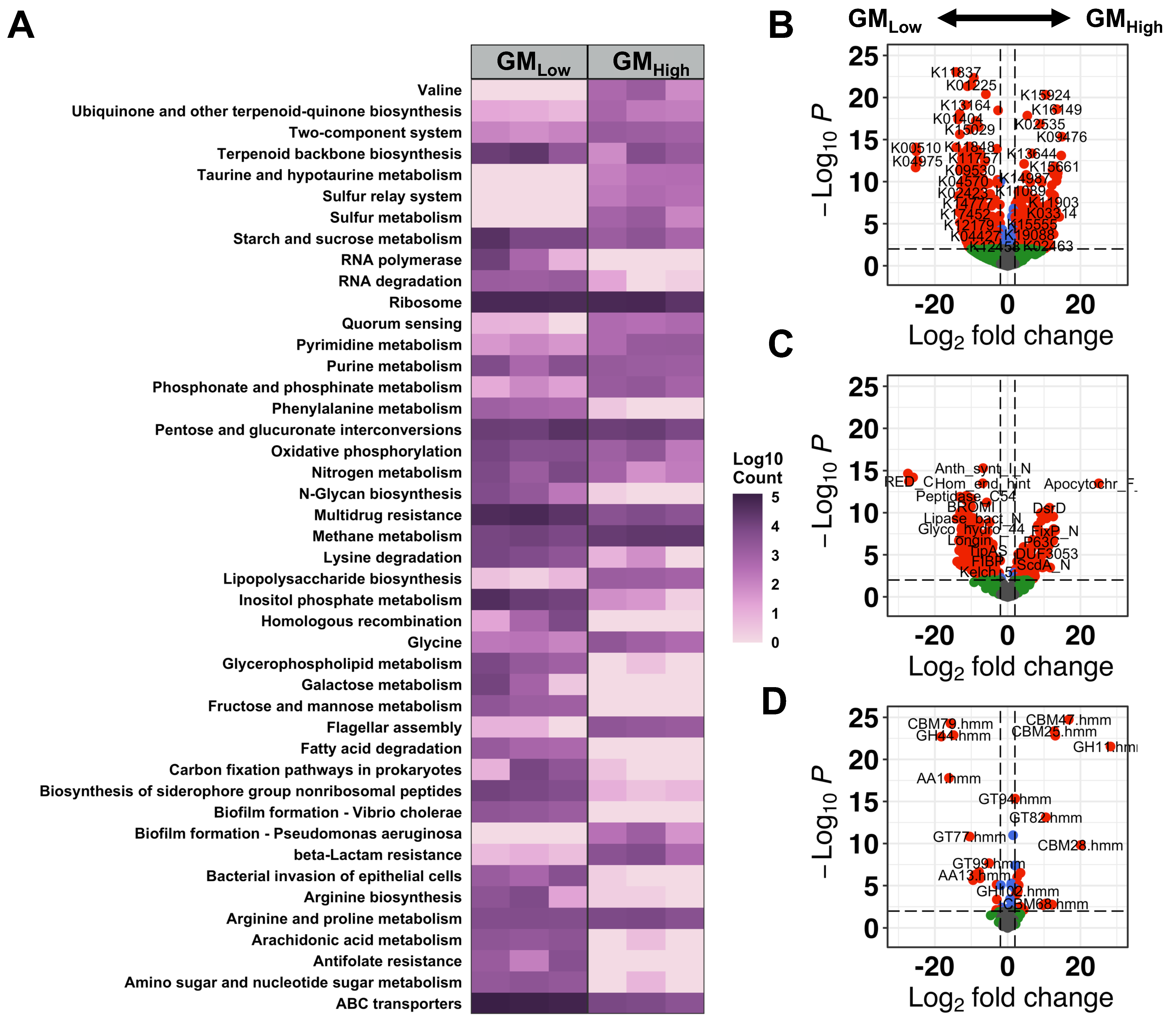
3.4. Functional Differences Between Source-Dependent GM
3.5. Supplier-Origin GMs Indicate Variable Levels of Enzymatic Activity Associated with Eukaryotes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Korem, T.; Zeevi, D.; Zmora, N.; Weissbrod, O.; Bar, N.; Lotan-Pompan, M.; Avnit-Sagi, T.; Kosower, N.; Malka, G.; Rein, M.; et al. Bread Affects Clinical Parameters and Induces Gut Microbiome-Associated Personal Glycemic Responses. Cell Metab. 2017, 25, 1243–1253.e5. [Google Scholar] [CrossRef] [PubMed]
- Zeevi, D.; Korem, T.; Zmora, N.; Israeli, D.; Rothschild, D.; Weinberger, A.; Ben-Yacov, O.; Lador, D.; Avnit-Sagi, T.; Lotan-Pompan, M.; et al. Personalized Nutrition by Prediction of Glycemic Responses. Cell 2015, 163, 1079–1094. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The Commensal Microbiome Is Associated with Anti-PD-1 Efficacy in Metastatic Melanoma Patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Chatelier, E.L.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut Microbiome Influences Efficacy of PD-1-Based Immunotherapy against Epithelial Tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut Microbiome Modulates Response to Anti-PD-1 Immunotherapy in Melanoma Patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Franklin, C.L.; Ericsson, A.C. Microbiota and Reproducibility of Rodent Models. Lab Anim. 2017, 46, 114–122. [Google Scholar] [CrossRef]
- Ericsson, A.C.; Gagliardi, J.; Bouhan, D.; Spollen, W.G.; Givan, S.A.; Franklin, C.L. The Influence of Caging, Bedding, and Diet on the Composition of the Microbiota in Different Regions of the Mouse Gut. Sci. Rep. 2018, 8, 4065. [Google Scholar] [CrossRef]
- Sofi, M.H.; Gudi, R.; Karumuthil-Melethil, S.; Perez, N.; Johnson, B.M.; Vasu, C. PH of Drinking Water Influences the Composition of Gut Microbiome and Type 1 Diabetes Incidence. Diabetes 2014, 63, 632–644. [Google Scholar] [CrossRef]
- Wolf, K.J.; Daft, J.G.; Tanner, S.M.; Hartmann, R.; Khafipour, E.; Lorenz, R.G. Consumption of Acidic Water Alters the Gut Microbiome and Decreases the Risk of Diabetes in NOD Mice. J. Histochem. Cytochem. 2014, 62, 237–250. [Google Scholar] [CrossRef]
- Hart, M.L.; Ericsson, A.C.; Franklin, C.L. Differing Complex Microbiota Alter Disease Severity of the IL-10−/− Mouse Model of Inflammatory Bowel Disease. Front. Microbiol. 2017, 8, 792. [Google Scholar] [CrossRef]
- Zhao, Y.; Tarbell, K.V. Comment on Sofi et al. PH of Drinking Water Influences the Composition of Gut Microbiome and Type 1 Diabetes Incidence. Diabetes 2014;63:632-644. Diabetes 2015, 64, e19. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, Q.; Li, D.; Onyema, O.O.; Mei, Z.; Manafi, A.; Banerjee, A.; Mahgoub, B.; Stoler, M.H.; Barker, T.H.; et al. Vendor-specific Microbiome Controls Both Acute and Chronic Murine Lung Allograft Rejection by Altering CD4+Foxp3+ Regulatory T Cell Levels. Am. J. Transpl. 2019, 19, 2705–2718. [Google Scholar] [CrossRef] [PubMed]
- Ericsson, A.C.; Davis, J.W.; Spollen, W.; Bivens, N.; Givan, S.; Hagan, C.E.; McIntosh, M.; Franklin, C.L. Effects of Vendor and Genetic Background on the Composition of the Fecal Microbiota of Inbred Mice. PLoS ONE 2015, 10, e0116704. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.S.; de Vries, L.; Kot, W.; Hansen, L.H.; Castro-Mejia, J.L.; Vogensen, F.K.; Hansen, A.K.; Nielsen, D.S. Mouse Vendor Influence on the Bacterial and Viral Gut Composition Exceeds the Effect of Diet. Viruses 2019, 11, 435. [Google Scholar] [CrossRef]
- Hufeldt, M.R.; Nielsen, D.S.; Vogensen, F.K.; Midtvedt, T.; Hansen, A.K. Variation in the Gut Microbiota of Laboratory Mice Is Related to Both Genetic and Environmental Factors. Comp. Med. 2010, 60, 336–347. [Google Scholar]
- Orcutt, R.P.; Gianni, F.J.; Judge, R.J. Development of an “Altered” Schaedler Flora for NCI Gnotobiotic Rodents. Microecol. Ther. 1987, 17, 59. [Google Scholar]
- Schaedler, R.W.; Dubos, R.; Costello, R. The Development of the Bacterial Flora in the Gastrointestinal Tract of Mice. J. Exp. Med. 1965, 122, 59–66. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Chien, C.C.; Paster, B.J.; Ericson, R.L.; Orcutt, R.P.; Schauer, D.B.; Fox, J.G. Phylogeny of the Defined Murine Microbiota: Altered Schaedler Flora. Appl. Environ. Microb. 1999, 65, 3287–3292. [Google Scholar] [CrossRef]
- Mandal, R.K.; Denny, J.E.; Waide, M.L.; Li, Q.; Bhutiani, N.; Anderson, C.D.; Baby, B.V.; Jala, V.R.; Egilmez, N.K.; Schmidt, N.W. Temporospatial Shifts within Commercial Laboratory Mouse Gut Microbiota Impact Experimental Reproducibility. BMC Biol. 2020, 18, 83. [Google Scholar] [CrossRef]
- Hoy, Y.E.; Bik, E.M.; Lawley, T.D.; Holmes, S.P.; Monack, D.M.; Theriot, J.A.; Relman, D.A. Variation in Taxonomic Composition of the Fecal Microbiota in an Inbred Mouse Strain across Individuals and Time. PLoS ONE 2015, 10, e0142825. [Google Scholar] [CrossRef]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A Core Gut Microbiome in Obese and Lean Twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, E.M.; Nguyen, H.; Heasley, K.T.; Saechao, C.H.; Gil, L.M.; Rogers, A.W.L.; Miller, B.M.; Rolston, M.R.; Lopez, C.A.; Litvak, Y.; et al. Endogenous Enterobacteriaceae Underlie Variation in Susceptibility to Salmonella Infection. Nat. Microbiol. 2019, 4, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.L.; Ericsson, A.C.; Lloyd, K.C.K.; Grimsrud, K.N.; Rogala, A.R.; Godfrey, V.L.; Nielsen, J.N.; Franklin, C.L. Development of Outbred CD1 Mouse Colonies with Distinct Standardized Gut Microbiota Profiles for Use in Complex Microbiota Targeted Studies. Sci. Rep. 2018, 8, 10107. [Google Scholar] [CrossRef]
- de Nies, L.; Busi, S.B.; Tsenkova, M.; Halder, R.; Letellier, E.; Wilmes, P. Evolution of the Murine Gut Resistome Following Broad-Spectrum Antibiotic Treatment. Nat. Commun. 2022, 13, 2296. [Google Scholar] [CrossRef]
- Ericsson, A.C.; Hart, M.L.; Kwan, J.; Lanoue, L.; Bower, L.R.; Araiza, R.; Lloyd, K.C.K.; Franklin, C.L. Supplier-Origin Mouse Microbiomes Significantly Influence Locomotor and Anxiety-Related Behavior, Body Morphology, and Metabolism. Commun. Biol. 2021, 4, 716. [Google Scholar] [CrossRef] [PubMed]
- Narayanasamy, S.; Jarosz, Y.; Muller, E.E.L.; Heintz-Buschart, A.; Herold, M.; Kaysen, A.; Laczny, C.C.; Pinel, N.; May, P.; Wilmes, P. IMP: A Pipeline for Reproducible Reference-Independent Integrated Metagenomic and Metatranscriptomic Analyses. Genome Biol. 2016, 17, 260. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An Adaptive Binning Algorithm for Robust and Efficient Genome Reconstruction from Metagenome Assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An Automated Binning Algorithm to Recover Genomes from Multiple Metagenomic Datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef]
- Hickl, O.; Queirós, P.; Wilmes, P.; May, P.; Heintz-Buschart, A. Binny: An Automated Binning Algorithm to Recover High-Quality Genomes from Complex Metagenomic Datasets. bioRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of Genomes from Metagenomes via a Dereplication, Aggregation and Scoring Strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef]
- Krinos, A.; Hu, S.; Cohen, N.; Alexander, H. EUKulele: Taxonomic Annotation of the Unsung Eukaryotic Microbes. J. Open Source Softw. 2021, 6, 2817. [Google Scholar] [CrossRef]
- Queirós, P.; Delogu, F.; Hickl, O.; May, P.; Wilmes, P. Mantis: Flexible and Consensus-Driven Genome Annotation. Gigascience 2021, 10, giab042. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam Protein Families Database: Towards a More Sustainable Future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes Database (CAZy): An Expert Resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An Advanced Analysis and Visualization Platform for ‘omics Data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.D. GToTree: A User-Friendly Workflow for Phylogenomics. Bioinformatics 2019, 35, 4162–4164. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Heintz-Buschart, A.; May, P.; Laczny, C.C.; Lebrun, L.A.; Bellora, C.; Krishna, A.; Wampach, L.; Schneider, J.G.; Hogan, A.; de Beaufort, C.; et al. Integrated Multi-Omics of the Human Gut Microbiome in a Case Study of Familial Type 1 Diabetes. Nat. Microbiol. 2016, 2, 16180. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum Information about a Single Amplified Genome (MISAG) and a Metagenome-Assembled Genome (MIMAG) of Bacteria and Archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef]
- Busi, S.B.; Bourquin, M.; Fodelianakis, S.; Michoud, G.; Kohler, T.J.; Peter, H.; Pramateftaki, P.; Styllas, M.; Tolosano, M.; Staercke, V.D.; et al. Genomic and Metabolic Adaptations of Biofilms to Ecological Windows of Opportunity in Glacier-Fed Streams. Nat. Commun. 2022, 13, 2168. [Google Scholar] [CrossRef] [PubMed]
- Antunes, T.C.; Marconatto, L.; Borges, L.G.d.A.; Giongo, A.; Sand, S.T.V.D. Analysis of Microbial Community Biodiversity in Activated Sludge from a Petrochemical Plant. Rev. Ambient. Água—Interdiscip. J. Appl. Sci. 2021, 16, 1–22. [Google Scholar] [CrossRef]
- Lima, C.P.V.; Grisi, D.C.; Guimarães, M.D.C.M.; Salles, L.P.; Kruly, P.d.C.; Do, T.; Borges, L.G.D.A.; Dame-Teixeira, N. Enrichment of Sulphate-Reducers and Depletion of Butyrate-Producers May Be Hyperglycaemia Signatures in the Diabetic Oral Microbiome. J. Oral Microbiol. 2022, 14, 2082727. [Google Scholar] [CrossRef]
- Saito, D.; Lemos, L.N.; Ferreira, A.T.R.N.; Saito, C.P.B.; de Oliveira, R.F.; Cannavan, F.d.S.; Tsai, S.M. Draft Genome Sequences of Five Putatively Novel Saccharibacteria Species Assembled from the Human Oral Metagenome. Microbiol. Resour. Announc. 2022, 11, e00246-22. [Google Scholar] [CrossRef]
- Mousavi, S.H.; Motahar, S.F.S.; Salami, M.; Kavousi, K.; Mamaghani, A.S.A.; Ariaeenejad, S.; Salekdeh, G.H. In Vitro Bioprocessing of Corn as Poultry Feed Additive by the Influence of Carbohydrate Hydrolyzing Metagenome Derived Enzyme Cocktail. Sci. Rep. 2022, 12, 405. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Hsieh, A.-H.; Wang, L.-C.; Huang, Y.-J.; Tsai, Y.-C.; Tseng, W.-Y.; Kuo, Y.-L.; Luo, S.-F.; Yu, K.-H.; Kuo, C.-F. Fecal Microbiota Changes in NZB/W F1 Mice after Induction of Lupus Disease. Sci. Rep. 2021, 11, 22953. [Google Scholar] [CrossRef]
- Lodowska, J.; Wolny, D.; Węglarz, L. The Sugar 3-Deoxy-d-Manno-Oct-2-Ulosonic Acid (Kdo) as a Characteristic Component of Bacterial Endotoxin—A Review of Its Biosynthesis, Function, and Placement in the Lipopolysaccharide Core. Can. J. Microbiol. 2013, 59, 645–655. [Google Scholar] [CrossRef]
- Cartmell, A.; Lowe, E.C.; Baslé, A.; Firbank, S.J.; Ndeh, D.A.; Murray, H.; Terrapon, N.; Lombard, V.; Henrissat, B.; Turnbull, J.E.; et al. How Members of the Human Gut Microbiota Overcome the Sulfation Problem Posed by Glycosaminoglycans. Proc. Natl. Acad. Sci. USA 2017, 114, 7037–7042. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Sarkar, A.; Lehto, S.M.; Harty, S.; Dinan, T.G.; Cryan, J.F.; Burnet, P.W.J. Psychobiotics and the Manipulation of Bacteria-Gut-Brain Signals. Trends Neurosci. 2016, 39, 763–781. [Google Scholar] [CrossRef]
- Wang, S.; Ishima, T.; Zhang, J.; Qu, Y.; Chang, L.; Pu, Y.; Fujita, Y.; Tan, Y.; Wang, X.; Hashimoto, K. Ingestion of Lactobacillus Intestinalis and Lactobacillus Reuteri Causes Depression- and Anhedonia-like Phenotypes in Antibiotic-Treated Mice via the Vagus Nerve. J. Neuroinflamm. 2020, 17, 241. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus Strain Regulates Emotional Behavior and Central GABA Receptor Expression in a Mouse via the Vagus Nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Wang, T.; Hu, X.; Luo, J.; Li, W.; Wu, X.; Duan, Y.; Jin, F. Administration of Lactobacillus Helveticus NS8 Improves Behavioral, Cognitive, and Biochemical Aberrations Caused by Chronic Restraint Stress. Neuroscience 2015, 310, 561–577. [Google Scholar] [CrossRef] [PubMed]
- Chatelier, E.L.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of Human Gut Microbiome Correlates with Metabolic Markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Vila, A.V.; Imhann, F.; Collij, V.; Jankipersadsing, S.A.; Gurry, T.; Mujagic, Z.; Kurilshikov, A.; Bonder, M.J.; Jiang, X.; Tigchelaar, E.F.; et al. Gut Microbiota Composition and Functional Changes in Inflammatory Bowel Disease and Irritable Bowel Syndrome. Sci. Transl. Med. 2018, 10, eaap8914. [Google Scholar] [CrossRef]
- Schirmer, M.; Garner, A.; Vlamakis, H.; Xavier, R.J. Microbial Genes and Pathways in Inflammatory Bowel Disease. Nat. Rev. Microbiol. 2019, 17, 497–511. [Google Scholar] [CrossRef]
- Moskowitz, J.E.; Andreatta, F.; Amos-Landgraf, J. The Gut Microbiota Modulates Differential Adenoma Suppression by B6/J and B6/N Genetic Backgrounds in Apc(Min) Mice. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2019, 30, 237–244. [Google Scholar] [CrossRef]
- Blachier, F.; Andriamihaja, M.; Larraufie, P.; Ahn, E.; Lan, A.; Kim, E. Production of Hydrogen Sulfide by the Intestinal Microbiota and Epithelial Cells and Consequences for the Colonic and Rectal Mucosa. Am. J. Physiol.-Gastrointest. Liver Physiol. 2021, 320, G125–G135. [Google Scholar] [CrossRef]
- Attene-Ramos, M.S.; Wagner, E.D.; Plewa, M.J.; Gaskins, H.R. Evidence That Hydrogen Sulfide Is a Genotoxic Agent. Mol. Cancer Res. MCR 2006, 4, 9–14. [Google Scholar] [CrossRef]
- Guo, F.F.; Yu, T.C.; Hong, J.; Fang, J.Y. Emerging Roles of Hydrogen Sulfide in Inflammatory and Neoplastic Colonic Diseases. Front. Physiol. 2016, 7, 156. [Google Scholar] [CrossRef]
- Flannigan, K.L.; Agbor, T.A.; Motta, J.P.; Ferraz, J.G.; Wang, R.; Buret, A.G.; Wallace, J.L. Proresolution Effects of Hydrogen Sulfide during Colitis Are Mediated through Hypoxia-Inducible Factor-1alpha. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 1591–1602. [Google Scholar] [CrossRef]
- Motta, J.P.; Flannigan, K.L.; Agbor, T.A.; Beatty, J.K.; Blackler, R.W.; Workentine, M.L.; Silva, G.J.D.; Wang, R.; Buret, A.G.; Wallace, J.L. Hydrogen Sulfide Protects from Colitis and Restores Intestinal Microbiota Biofilm and Mucus Production. Inflamm. Bowel Dis. 2015, 21, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Blackler, R.W.; Chan, M.V.; Silva, G.J.D.; Elsheikh, W.; Flannigan, K.L.; Gamaniek, I.; Manko, A.; Wang, L.; Motta, J.P.; et al. Anti-Inflammatory and Cytoprotective Actions of Hydrogen Sulfide: Translation to Therapeutics. Antioxid. Redox Signal. 2015, 22, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Tain, Y.L. Maternal N-Acetylcysteine Therapy Prevents Hypertension in Spontaneously Hypertensive Rat Offspring: Implications of Hydrogen Sulfide-Generating Pathway and Gut Microbiota. Antioxidants 2020, 9, 856. [Google Scholar] [CrossRef]
- Feil, E.J.; Holmes, E.C.; Bessen, D.E.; Chan, M.S.; Day, N.P.; Enright, M.C.; Goldstein, R.; Hood, D.W.; Kalia, A.; Moore, C.E.; et al. Recombination within Natural Populations of Pathogenic Bacteria: Short-Term Empirical Estimates and Long-Term Phylogenetic Consequences. Proc. Natl. Acad. Sci. USA 2001, 98, 182–187. [Google Scholar] [CrossRef]
- Duchene, S.; Holt, K.E.; Weill, F.X.; Hello, S.L.; Hawkey, J.; Edwards, D.J.; Fourment, M.; Holmes, E.C. Genome-Scale Rates of Evolutionary Change in Bacteria. Microb. Genom. 2016, 2, e000094. [Google Scholar] [CrossRef]
- Didelot, X.; Walker, A.S.; Peto, T.E.; Crook, D.W.; Wilson, D.J. Within-Host Evolution of Bacterial Pathogens. Nat. Rev. Microbiol. 2016, 14, 150–162. [Google Scholar] [CrossRef]
- Garud, N.R.; Good, B.H.; Hallatschek, O.; Pollard, K.S. Evolutionary Dynamics of Bacteria in the Gut Microbiome within and across Hosts. PLoS Biol. 2019, 17, e3000102. [Google Scholar] [CrossRef]
- Young, B.C.; Wu, C.H.; Gordon, N.C.; Cole, K.; Price, J.R.; Liu, E.; Sheppard, A.E.; Perera, S.; Charlesworth, J.; Golubchik, T.; et al. Severe Infections Emerge from Commensal Bacteria by Adaptive Evolution. eLife 2017, 6, e30637. [Google Scholar] [CrossRef]
- Yilmaz, B.; Mooser, C.; Keller, I.; Li, H.; Zimmermann, J.; Bosshard, L.; Fuhrer, T.; de Aguero, M.G.; Trigo, N.F.; Tschanz-Lischer, H.; et al. Long-Term Evolution and Short-Term Adaptation of Microbiota Strains and Sub-Strains in Mice. Cell Host Microbe 2021, 29, 650–663.e9. [Google Scholar] [CrossRef]
- Bor, B.; Poweleit, N.; Bois, J.S.; Cen, L.; Bedree, J.K.; Zhou, Z.H.; Gunsalus, R.P.; Lux, R.; McLean, J.S.; He, X.; et al. Phenotypic and Physiological Characterization of the Epibiotic Interaction Between TM7x and Its Basibiont Actinomyces. Microb. Ecol. 2016, 71, 243–255. [Google Scholar] [CrossRef] [PubMed]
- He, X.; McLean, J.S.; Edlund, A.; Yooseph, S.; Hall, A.P.; Liu, S.Y.; Dorrestein, P.C.; Esquenazi, E.; Hunter, R.C.; Cheng, G.; et al. Cultivation of a Human-Associated TM7 Phylotype Reveals a Reduced Genome and Epibiotic Parasitic Lifestyle. Proc. Natl. Acad. Sci. USA 2015, 112, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Soro, V.; Dutton, L.C.; Sprague, S.V.; Nobbs, A.H.; Ireland, A.J.; Sandy, J.R.; Jepson, M.A.; Micaroni, M.; Splatt, P.R.; Dymock, D.; et al. Axenic Culture of a Candidate Division TM7 Bacterium from the Human Oral Cavity and Biofilm Interactions with Other Oral Bacteria. Appl. Environ. Microbiol. 2014, 80, 6480–6489. [Google Scholar] [CrossRef] [PubMed]
- Dinis, J.M.; Barton, D.E.; Ghadiri, J.; Surendar, D.; Reddy, K.; Velasquez, F.; Chaffee, C.L.; Lee, M.C.; Gavrilova, H.; Ozuna, H.; et al. In Search of an Uncultured Human-Associated TM7 Bacterium in the Environment. PLoS ONE 2011, 6, e21280. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McAdams, Z.L.; Busi, S.B.; Gustafson, K.L.; Bivens, N.; Franklin, C.L.; Wilmes, P.; Ericsson, A.C. Multi-Omics Analysis of Mouse Fecal Microbiome Reveals Supplier-Dependent Functional Differences and Novel Metagenome-Assembled Genomes. Appl. Microbiol. 2024, 4, 1600-1615. https://doi.org/10.3390/applmicrobiol4040109
McAdams ZL, Busi SB, Gustafson KL, Bivens N, Franklin CL, Wilmes P, Ericsson AC. Multi-Omics Analysis of Mouse Fecal Microbiome Reveals Supplier-Dependent Functional Differences and Novel Metagenome-Assembled Genomes. Applied Microbiology. 2024; 4(4):1600-1615. https://doi.org/10.3390/applmicrobiol4040109
Chicago/Turabian StyleMcAdams, Zachary L., Susheel Bhanu Busi, Kevin L. Gustafson, Nathan Bivens, Craig L. Franklin, Paul Wilmes, and Aaron C. Ericsson. 2024. "Multi-Omics Analysis of Mouse Fecal Microbiome Reveals Supplier-Dependent Functional Differences and Novel Metagenome-Assembled Genomes" Applied Microbiology 4, no. 4: 1600-1615. https://doi.org/10.3390/applmicrobiol4040109
APA StyleMcAdams, Z. L., Busi, S. B., Gustafson, K. L., Bivens, N., Franklin, C. L., Wilmes, P., & Ericsson, A. C. (2024). Multi-Omics Analysis of Mouse Fecal Microbiome Reveals Supplier-Dependent Functional Differences and Novel Metagenome-Assembled Genomes. Applied Microbiology, 4(4), 1600-1615. https://doi.org/10.3390/applmicrobiol4040109