
Metabolically Active Microbial Communities in Oilfields: A Systematic Review and Synthesis of RNA Preservation, Extraction, and Sequencing Methods

 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
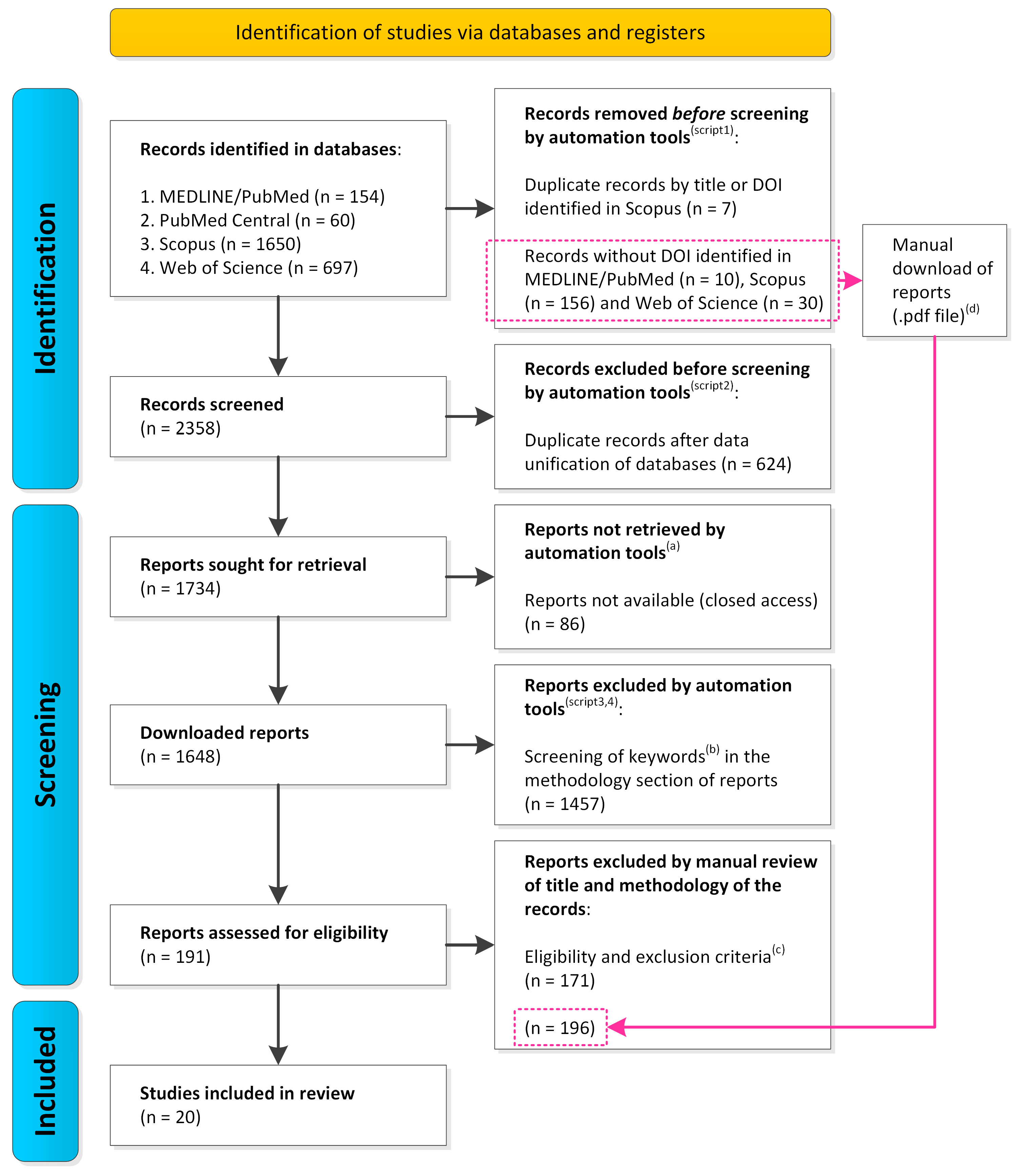
2.1. Identification of Records in Databases
2.2. Automated and Manual Screening of Records
2.3. Assessment of Eligibility and Inclusion of Studies
2.4. Synthesis and Analysis of Data of Included Studies
Analysis of Data from 16S rRNA Gene Transcripts and Metatranscriptome
3. Results
3.1. Identification and Selection of Studies
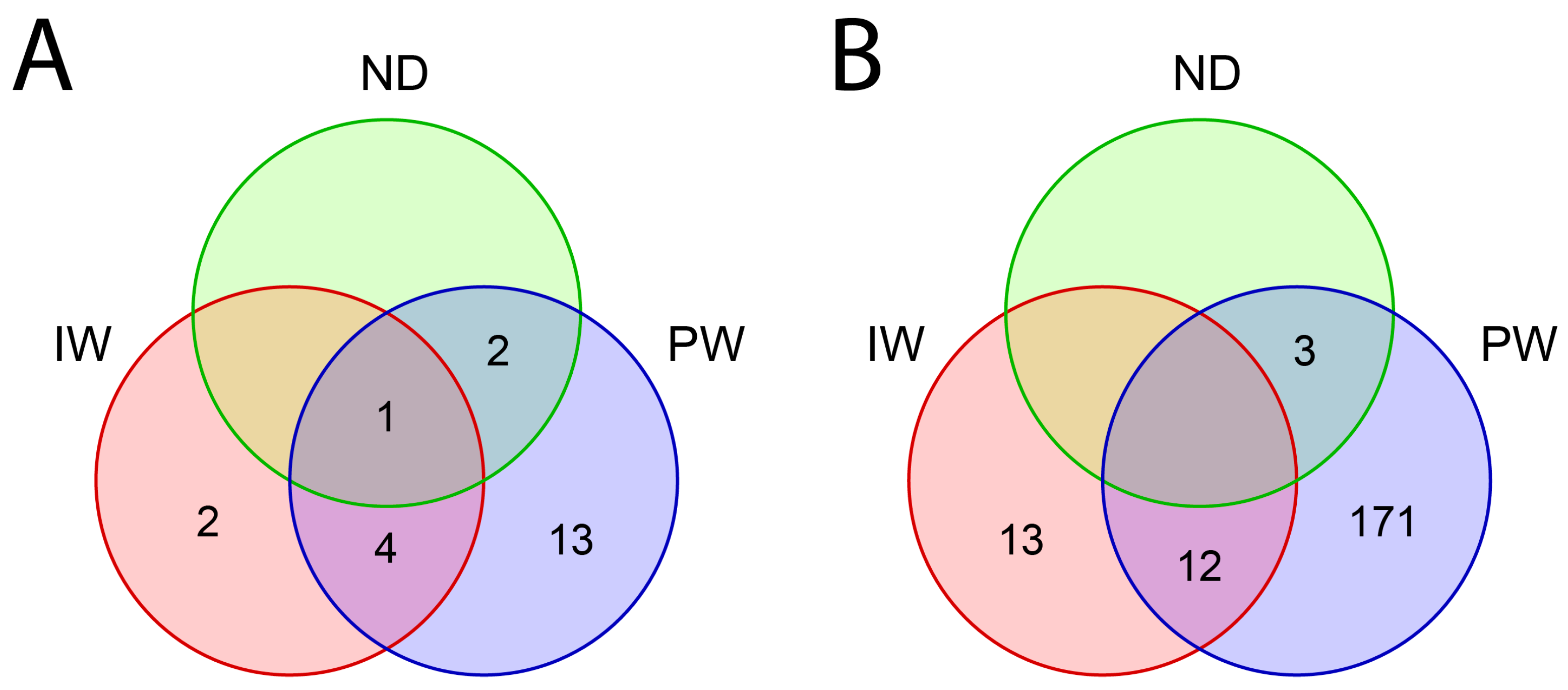
3.2. Included Studies
3.3. Methods of Preprocessing, Preservation, and Extraction of RNA
3.4. Methods for Amplification and Sequencing RNA
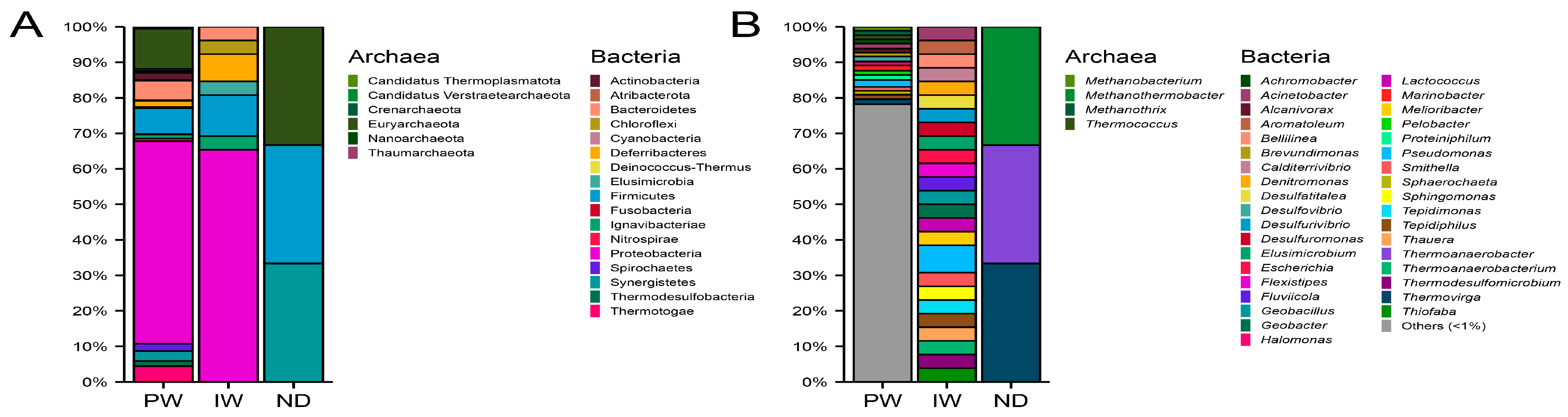
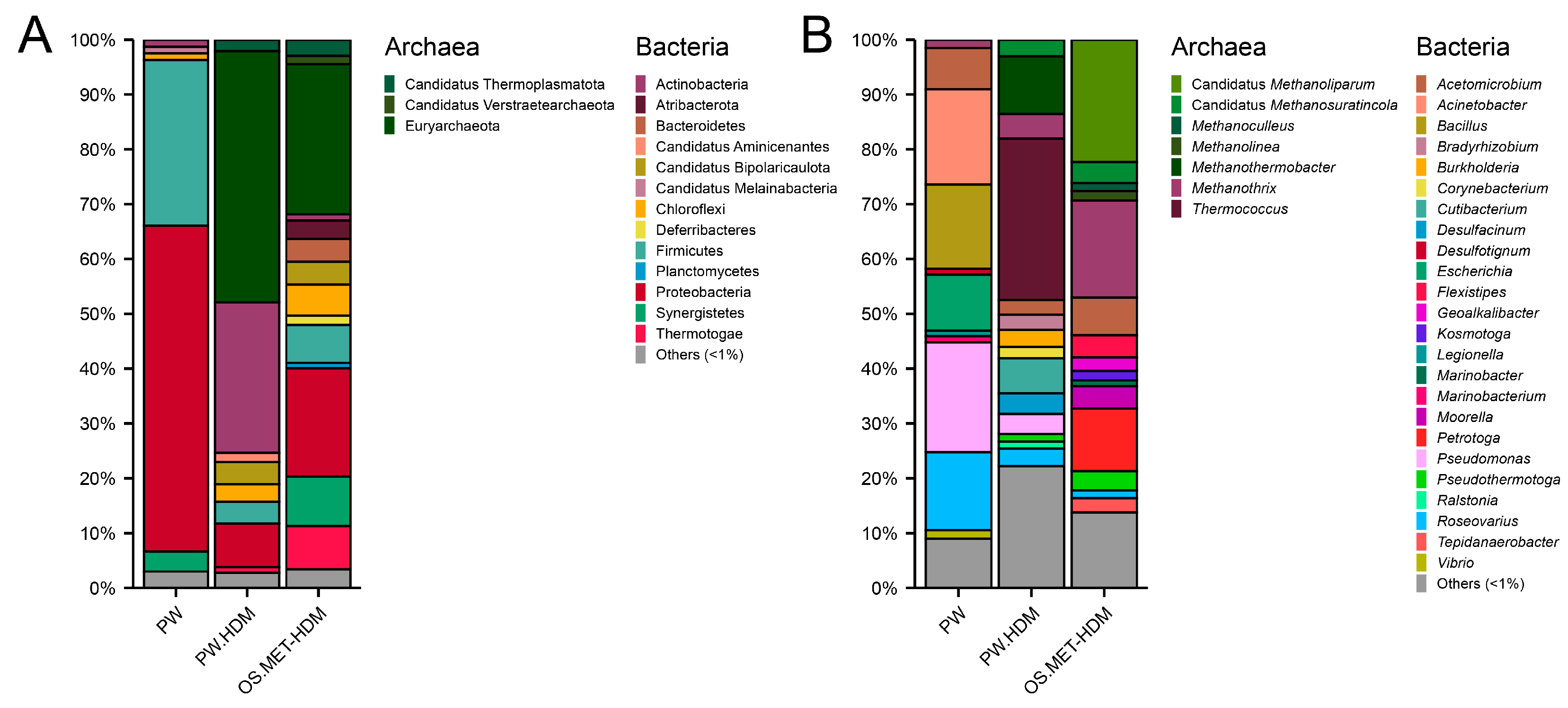
3.5. Composition of the Metabolically Active Microbiota
4. Discussion
4.1. Identification and Selection of Studies
4.2. Studies Included
4.3. RNA Preprocessing, Preservation, and Extraction Method
4.4. RNA Amplification and Sequencing Method
4.5. Composition of the Metabolically Active Microbiota
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salgar-Chaparro, S.J.; Machuca, L.L. Complementary DNA/RNA-based profiling: Characterization of corrosive microbial communities and their functional profiles in an oil production facility. Front. Microbiol. 2019, 10, 2587. [Google Scholar] [CrossRef] [PubMed]
- Neff, J.; Lee, K.; DeBlois, E.M. Produced water: Overview of composition, fates, and effects. In Produced Water: Environmental Risks and Advances in Mitigation Technologies; Springer Science Business Media: New York, NY, USA, 2011; pp. 3–54. [Google Scholar]
- Nazina, T.N.; Shestakova, N.M.; Semenova, E.M.; Korshunova, A.V.; Kostrukova, N.K.; Tourova, T.P.; Liu, M.; Feng, Q.; Poltaraus, A.B. Diversity of metabolically active bacteria in water-flooded high-temperature heavy oil reservoir. Front. Microbiol. 2017, 8, 707. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Peñasco, I.; Salazar-Coria, L.; Saucedo-García, M.; Villa-Tanaca, L.; Hernández-Rodríguez, C. Bisulfite reductase gene expression of thermophilic sulphate-reducing bacteria from saline connate water of oil reservoirs with high temperature. Int. Biodeterior. Biodegrad. 2016, 108, 198–206. [Google Scholar] [CrossRef]
- Liang, Y.; Ning, Y.; Liao, L.; Yuan, B. Special focus on produced water in oil and gas fields: Origin, management, and reinjection practice. In Formation Damage during Improved Oil Recovery: Fundamentals and Applications; Yuan, B., Wood, D.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 515–586. [Google Scholar]
- Sanders, P.F.; Sturman, P.J. Biofouling in the oil industry. In Petroleum Microbiology; Ollivier, B., Magot, M., Eds.; ASM Press: Washington, DC, USA, 2005; pp. 171–198. [Google Scholar]
- Gieg, L.M.; Jack, T.R.; Foght, J.M. Biological souring and mitigation in oil reservoirs. Appl. Microbiol. Biotechnol. 2011, 92, 263–282. [Google Scholar] [CrossRef]
- Johnson, R.J.; Folwell, B.D.; Wirekoh, A.; Frenzel, M.; Skovhus, T.L. Reservoir Souring–Latest developments for application and mitigation. J. Biotechnol. 2017, 256, 57–67. [Google Scholar] [CrossRef]
- Xu, D.; Gu, T.; Lovley, D.R. Microbially mediated metal corrosion. Nat. Rev. Microbiol. 2023, 1–14. [Google Scholar] [CrossRef]
- Dutra, J.; García, G.; Gomes, R.; Cardoso, M.; Côrtes, Á.; Silva, T.; de Jesus, L.; Rodrigues, L.; Freitas, A.; Góes-Neto, A.; et al. Effective biocorrosive control in oil industry facilities: 16S rRNA gene metabarcoding for monitoring microbial communities in produced water. Microorganisms 2023, 11, 846. [Google Scholar] [CrossRef]
- Eckert, R.B. Emphasis on biofilms can improve mitigation of microbiologically influenced corrosion in oil and gas industry. Corros. Eng. Sci. Technol. 2015, 50, 163–168. [Google Scholar] [CrossRef]
- Vigneron, A.; Head, I.M.; Tsesmetzis, N. Damage to offshore production facilities by corrosive microbial biofilms. Appl. Microbiol. Biotechnol. 2018, 102, 2525–2533. [Google Scholar] [CrossRef]
- Alhefeiti, M.A.; Athamneh, K.; Vijayan, R.; Ashraf, S.S. Bioremediation of various aromatic and emerging pollutants by Bacillus cereus sp. isolated from petroleum sludge. Water Sci. Technol. 2021, 83, 1535–1547. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, C.J.; Liu, P.F.; Fu, L.; Laso-Pérez, R.; Yang, L.; Bai, L.-P.; Li, J.; Yang, M.; Cheng, L.; et al. Non-syntrophic methanogenic hydrocarbon degradation by an archaeal species. Nature 2022, 601, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Shestakova, N.M.; Korshunova, A.V.; Mikhailova, E.M.; Sokolova, D.S.; Tourova, T.P.; Belyaev, S.S.; Poltaraus, A.B.; Nazina, T.N. Characterization of the aerobic hydrocarbon-oxidizing enrichments from a high-temperature petroleum reservoir by comparative analysis of DNA-and RNA-derived clone libraries. Microbiology 2011, 80, 60–69. [Google Scholar] [CrossRef]
- Liu, Y.F.; Galzerani, D.D.; Mbadinga, S.M.; Zaramela, L.S.; Gu, J.D.; Mu, B.Z.; Zengler, K. Metabolic capability and in situ activity of microorganisms in an oil reservoir. Microbiome 2018, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Li, X.X.; Liu, J.F.; Zhou, L.; Mbadinga, S.M.; Yang, S.Z.; Gu, J.D.; Mu, B.Z. Diversity and composition of sulfate-reducing microbial communities based on genomic DNA and RNA transcription in production water of high temperature and corrosive oil reservoir. Front. Microbiol. 2017, 8, 1011. [Google Scholar] [CrossRef] [PubMed]
- Albahri, M.B.; Barifcani, A.; Iglauer, S.; Lebedev, M.; O’Neil, C.; Salgar-Chaparro, S.J.; Machuca, L.L. Investigating the mechanism of microbiologically influenced corrosion of carbon steel using X-ray micro-computed tomography. J. Mater. Sci. 2021, 56, 13337–13371. [Google Scholar] [CrossRef]
- Zheng, X.; Lin, X.; Yan, Y.; Wen, Y.; Shi, Y.; Xie, X.; Hou, A.; Lai, N. Effect of reservoir salinity between bioacid and carbonate rock based on biometabolic analysis. Energy Fuels 2019, 33, 8135–8144. [Google Scholar] [CrossRef]
- Su, Z.; Wang, S.; Yang, S.; Yin, Y.; Cao, Y.; Li, G.; Ma, T. Genetic and comparative genome analysis of Exiguobacterium aurantiacum SW-20, a petroleum-degrading bacteria with salt tolerance and heavy metal-tolerance isolated from produced water of changqing oilfield, China. Microorganisms 2022, 10, 66. [Google Scholar] [CrossRef]
- Lomans, B.P.; de Paula, R.; Geissler, B.; Kuijvenhoven, C.A.; Tsesmetzis, N. Proposal of improved biomonitoring standard for purpose of microbiologically influenced corrosion risk assessment. In Proceedings of the SPE International Oilfield Corrosion Conference and Exhibition, Aberdeen, UK, 9–10 May 2016; OnePetro: Richardson, TX, USA, 2016. [Google Scholar]
- Zhou, L.; Lu, Y.W.; Wang, D.W.; Zhang, S.L.; Tang, E.G.; Qi, Z.Z.; Xie, S.-N.; Wu, J.; Liang, B.; Mu, B.Z.; et al. Microbial community composition and diversity in production water of a high-temperature offshore oil reservoir assessed by DNA-and RNA-based analyses. Int. Biodeterior. Biodegrad. 2020, 151, 104970. [Google Scholar] [CrossRef]
- Neria-González, I.; Wang, E.T.; Ramírez, F.; Romero, J.M.; Hernández-Rodríguez, C. Characterization of bacterial community associated to biofilms of corroded oil pipelines from the southeast of Mexico. Anaerobe 2006, 12, 122–133. [Google Scholar] [CrossRef]
- Gittel, A.; Sørensen, K.B.; Skovhus, T.L.; Ingvorsen, K.; Schramm, A. Prokaryotic community structure and sulfate reducer activity in water from high-temperature oil reservoirs with and without nitrate treatment. Appl. Environ. Microbiol. 2009, 75, 7086–7096. [Google Scholar] [CrossRef]
- Li, H.; Chen, S.; Mu, B.Z.; Gu, J.D. Molecular detection of anaerobic ammonium-oxidizing (anammox) bacteria in high-temperature petroleum reservoirs. Microb. Ecol. 2010, 60, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Lenchi, N.; İnceoğlu, Ö.; Kebbouche-Gana, S.; Gana, M.L.; Llirós, M.; Servais, P.; García-Armisen, T. Diversity of microbial communities in production and injection waters of Algerian oilfields revealed by 16S rRNA gene amplicon 454 pyrosequencing. PLoS ONE 2013, 8, 66588. [Google Scholar] [CrossRef] [PubMed]
- Machuca Suarez, L.; Salgar-Chaparro, S. Effect of sample storage conditions on the molecular assessment of MIC. In Proceedings of the Corrosion & Prevention Conference, Adelaide, Australia, 11–14 November 2018. [Google Scholar]
- Nasser, B.; Saito, Y.; Alarawi, M.; Al-Humam, A.A.; Mineta, K.; Gojobori, T. Characterization of microbiologically influenced corrosion by comprehensive metagenomic analysis of an inland oil field. Gene 2021, 774, 145425. [Google Scholar] [CrossRef] [PubMed]
- Hampton-Marcell, J.T.; Frazier, A.; Moormann, S.M.; Owens, S.M.; Gilbert, J.A. Preparation and analysis of metatranscriptomic libraries in petroleum hydrocarbon microbe systems. In Hydrocarbon and Lipid Microbiology Protocols: Genetic, Genomic and System Analyses of Communities; McGenity, T., Timmis, K., Nogales, B., Eds.; Springer Protocols Handbooks; Springer: Berlin/Heidelberg, Germany, 2017; pp. 51–67. [Google Scholar]
- Simister, R.L.; Schmitt, S.; Taylor, M.W. Evaluating methods for the preservation and extraction of DNA and RNA for analysis of microbial communities in marine sponges. J. Exp. Mar. Biol. Ecol. 2011, 397, 38–43. [Google Scholar] [CrossRef]
- Rachel, N.M.; Gieg, L.M. Preserving microbial community integrity in oilfield produced water. Front. Microbiol. 2020, 11, 581387. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.C.F.; Lopes, D.R.G.; Lima, H.S.; Quartaroli, L.; de Sousa, M.P.; de Abreu Waldow, V.; Akamine, R.N.; de Paula, S.O.; da Silva, C.C. Comparison of methods for preservation of activated sludge samples for high-throughput nucleic acid sequencing and bacterial diversity analysis. Int. Biodeterior. Biodegrad. 2021, 157, 105139. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Moher, D.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Int. J. Surg. 2021, 88, 105906. [Google Scholar] [CrossRef]
- Rethlefsen, M.L.; Kirtley, S.; Waffenschmidt, S.; Ayala, A.P.; Moher, D.; Page, M.J.; Koffel, J.B. PRISMA-S: An extension to the PRISMA statement for reporting literature searches in systematic reviews. Syst. Rev. 2021, 10, 39. [Google Scholar] [CrossRef]
- Liu, Y.F.; Chen, J.; Liu, Z.L.; Shou, L.B.; Lin, D.D.; Zhou, L.; Yang, S.Z.; Liu, J.F.; Li, W.; Mu, B.Z.; et al. Anaerobic degradation of paraffins by thermophilic Actinobacteria under methanogenic conditions. Environ. Sci. Technol. 2020, 54, 10610–10620. [Google Scholar] [CrossRef]
- Tamames, J.; Puente-Sánchez, F. SqueezeMeta, a highly portable, fully automatic metagenomic analysis pipeline. Front. Microbiol. 2019, 9, 3349. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Puente-Sánchez, F.; García-García, N.; Tamames, J. SQMtools: Automated processing and visual analysis of’omics data with R and anvi’o. BMC Bioinform. 2020, 21, 358. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2. Wiley interdisciplinary reviews. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Salgar-Chaparro, S.J.; Lepkova, K.; Pojtanabuntoeng, T.; Darwin, A.; Machuca, L.L. Microbiologically influenced corrosion as a function of environmental conditions: A laboratory study using oilfield multispecies biofilms. Corros. Sci. 2020, 169, 108595. [Google Scholar] [CrossRef]
- Salgar-Chaparro, S.J.; Lepkova, K.; Pojtanabuntoeng, T.; Darwin, A.; Machuca, L.L. Nutrient level determines biofilm characteristics and subsequent impact on microbial corrosion and biocide effectiveness. Appl. Environ. Microbiol. 2020, 86, e02885-19. [Google Scholar] [CrossRef]
- Zhou, L.; Hu, Q.Q.; Lu, Y.W.; Mbadinga, S.M.; Liu, Y.F.; Li, X.X.; Wang, B.; Lv, H.; Liu, J.F.; Mu, B.Z.; et al. Dominant and active methanogens in the production waters from a high-temperature petroleum reservoir by DNA-and RNA-based analysis. Geomicrobiol. J. 2021, 38, 191–198. [Google Scholar] [CrossRef]
- Prajapat, G.; Jain, S.; Lal, B.; Lavania, M.; Agrawal, A. Control of reservoir souring by incomplete nitrate reduction in Indian oil fields. Bioresour. Technol. Rep. 2023, 21, 101302. [Google Scholar] [CrossRef]
- Liu, Y.F.; Chen, J.; Zaramela, L.S.; Wang, L.Y.; Mbadinga, S.M.; Hou, Z.W.; Wu, X.L.; Gu, J.D.; Zengler, K.; Mu, B.Z. Genomic and transcriptomic evidence supports methane metabolism in Archaeoglobi. mSystems 2020, 5, e00651-19. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Lu, Y.W.; Liu, X.B.; Li, B.G.; Sun, Y.F.; Zhou, L.; Liu, Y.F.; Yang, S.Z.; Gu, J.D.; Mu, B.Z. Dominance of Pseudomonas in bacterial community and inhibition of fumarate addition pathway by injection of nutrients in oil reservoir revealed by functional gene and their transcript analyses. Int. Biodeterior. Biodegrad. 2020, 153, 105039. [Google Scholar] [CrossRef]
- Liu, J.F.; Lu, Y.W.; Zhou, L.; Li, W.; Hou, Z.W.; Yang, S.Z.; Wu, X.L.; Gu, J.D.; Mu, B.Z. Simultaneous detection of transcribed functional assA gene and the corresponding metabolites of linear alkanes (C4, C5, and C7) in production water of a low-temperature oil reservoir. Sci. Total Environ. 2020, 746, 141290. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Tomo, S.; Modi, A.; Purohit, P.; Sharma, P. Optimising total RNA quality and quantity by phenol-chloroform extraction method from human visceral adipose tissue: A standardisation study. MethodsX 2020, 7, 101113. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Zhao, Y.; Wei, Q.; Shi, S.; Jiang, Z. Isolation of high-quality RNA from Platycladus orientalis and other Cupressaceae plants. Electron. J. Biotechnol. 2016, 23, 21–27. [Google Scholar] [CrossRef]
- Amirouche, A.; Ait-Ali, D.; Nouri, H.; Boudrahme-Hannou, L.; Tliba, S.; Ghidouche, A.; Bitam, I. TRIzol-based RNA extraction for detection protocol for SARS-CoV-2 of coronavirus disease 2019. New Microbes New Infect. 2021, 41, 100874. [Google Scholar] [CrossRef]
- Nóbrega, B.B.; Soares, D.M.; Zamuner, C.K.; Stevani, C.V. Optimized methodology for obtention of high-yield and-quality RNA from the mycelium of the bioluminescent fungus Neonothopanus gardneri. J. Microbiol. Methods 2021, 191, 106348. [Google Scholar] [CrossRef]
- Oldham, A.L.; Sandifer, V.; Duncan, K.E. Effects of sample preservation on marine microbial diversity analysis. J. Microbiol. Methods 2019, 158, 6–13. [Google Scholar] [CrossRef]
- Klappenbach, J.A.; Saxman, P.R.; Cole, J.R.; Schmidt, T.M. rrndb: The ribosomal RNA operon copy number database. Nucleic Acids Res. 2001, 29, 181–184. [Google Scholar] [CrossRef]
- Moeseneder, M.M.; Arrieta, J.M.; Herndl, G.J. A comparison of DNA-and RNA-based clone libraries from the same marine bacterioplankton community. FEMS Microbiol. Ecol. 2005, 51, 341–352. [Google Scholar] [CrossRef]
- Blazewicz, S.J.; Barnard, R.L.; Daly, R.A.; Firestone, M.K. Evaluating rRNA as an indicator of microbial activity in environmental communities: Limitations and uses. ISME J. 2013, 7, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Atlas, R.M.; Atlas, M.C. Biodegradation of oil and bioremediation of oil spills. Curr. Opin. Biotechnol. 1991, 2, 440–443. [Google Scholar] [CrossRef]
- Salama, A.M.; Rashad, E.; Elgarahy, A.M.; Elwakeel, K. Effect of green synthesized iron oxide nanoparticles on bacterial microbiome for clean up the crude oil. Aswan Univ. J. Environ. Stud. 2023, 4, 49–81. [Google Scholar] [CrossRef]
- Yang, Z.; Li, M.; Cheng, D.; Xiao, H.; Lai, H.; Chen, Q. Geochemistry and possible origins of biodegraded oils in the Cretaceous reservoir of the Muglad Basin and their application in hydrocarbon exploration. J. Pet. Sci. Eng. 2019, 173, 889–898. [Google Scholar] [CrossRef]
- Cruz, G.F.D.; Marsaioli, A.J. Processos naturais de biodegradação do petróleo em reservatórios. Química Nova 2012, 35, 1628–1634. [Google Scholar] [CrossRef]
- Head, I.M.; Jones, D.M.; Larter, S.R. Biological activity in the deep subsurface and the origin of heavy oil. Nature 2003, 426, 344–352. [Google Scholar] [CrossRef]
- Wolodko, J.; Haile, T.; Khan, F.; Taylor, C.; Eckert, R.; Hashemi, S.J.; Ramirez, A.M.; Skovhus, T.L. Modeling of microbiologically influenced corrosion (MIC) in the oil and gas industry-past, present and future. In Proceedings of the NACE Corrosion, Phoenix, AZ, USA, 15–19 April 2018. [Google Scholar]
- Hampton-Marcell, J.T.; Frazier, A.; Moormann, S.M.; Owens, S.M.; Gilbert, J.A. Preparation and Analysis of Metatranscriptomic Libraries in Petroleum Hydrocarbon Microbe Systems. In Laboratory Methods in Food Microbiology; McGenity, T., Timmis, K., Nogales, B., Eds.; Gulf Professional Publishing: Houston, TX, USA, 2014. [Google Scholar]
- Gupta, R.S. The phylogeny of proteobacteria: Relationships to other eubacterial phyla and eukaryotes. FEMS Microbiol. Rev. 2000, 24, 367–402. [Google Scholar] [CrossRef]
- Goveas, L.C.; Selvaraj, R.; Vinayagam, R.; Alsaiari, A.A.; Alharthi, N.S.; Sajankila, S.P. Nitrogen dependence of rhamnolipid mediated degradation of petroleum crude oil by indigenous Pseudomonas sp. WD23 in seawater. Chemosphere 2022, 304, 135235. [Google Scholar] [CrossRef]
- Muthukumar, B.; Al Salhi, M.S.; Narenkumar, J.; Devanesan, S.; Rao, T.N.; Kim, W.; Rajasekar, A. Characterization of two novel strains of Pseudomonas aeruginosa on biodegradation of crude oil and its enzyme activities. Environ. Pollut. 2022, 304, 119223. [Google Scholar] [CrossRef]
- de Oliveira, H.L.; Dias, G.M.; Neves, B.C. Genome sequence of Pseudomonas aeruginosa PA1-Petro—A role model of environmental adaptation and a potential biotechnological tool. Heliyon 2022, 8, e11566. [Google Scholar] [CrossRef]
- Sengupta, K.; Pal, S. A review on microbial diversity and genetic markers involved in methanogenic degradation of hydrocarbons: Futuristic prospects of biofuel recovery from contaminated regions. Environ. Sci. Pollut. Res. 2021, 28, 40288–40307. [Google Scholar] [CrossRef] [PubMed]
- Dolfing, J.; Larter, S.R.; Head, I.M. Thermodynamic constraints on methanogenic crude oil biodegradation. ISME J. 2008, 2, 442–452. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criteria | Description |
|---|---|
| Sampling location | Reservoir; pipeline; tank |
| Sample type | environmental (oil, injection water, produced water, biofilm, pig residue, and oil sludge); laboratory (cultures inoculated with environmental samples) |
| Analysis | RNA sequencing |
| Study | Original |
| Reference | Sampling Country | Sample Type | N | Sample Studied | ID |
|---|---|---|---|---|---|
| Shestakova et al. (2011) [15] | Hebei, China | PW | 1 | Culture | PW.HDM (a) |
| IW | 1 | Culture | IW.HDM (a) | ||
| Zapata-Peñasco et al. (2016) [4] | Mexico | PW | 2 | Culture | PW.SRB (a) |
| Nazina et al. (2017) [3] | Hebei, China | IW | 1 | Environmental | IW (a) |
| Li et al. (2017) [17] | Jiangsu, China | PW | 6 | Environmental | PW (a) |
| Liu et al. (2018) [16] | Jiangsu, China | PW | 3 | Environmental | PW (a) |
| Salgar-Chaparro e Machuca (2019) [1] | Australia | PW | 6 | Environmental | PW (a) |
| IW | 1 | Environmental | IW (a) | ||
| SD | 4 | Environmental | SD (a) | ||
| Zheng et al. (2019) [19] | China | OIL | 1 | Culture | OIL.HDM (a) |
| Liu et al. (2020) [35] | Shandong, China | PW | 1 | Environmental | PW (a) |
| Culture | PW.HDM (a) | ||||
| Liu et al. (2020) [47] | China | PW | 2 | Environmental | PW (a) |
| Liu et al. (2020) [48] | Jiangsu, China | PW | 1 | Environmental | PW (a) |
| Zhou et al. (2020) [22] | China | PW | 2 | Environmental | PW (a) |
| Liu et al. (2020) [49] | China | PW | 1 | Environmental | PW (a) |
| Salgar-Chaparro et al. (2020) [43] | Australia | PW | 1 | Consortium | PW.CIM (a),(b) |
| Salgar-Chaparro et al. (2020) [44] | Australia | PW | 2 | Consortium | PW.CIM (a),(b) |
| Alhefeiti et al. (2021) [13] | United Arab Emirates | OS | 1 | Culture | OS.OCDM (a) |
| Zhou et al. (2021) [45] | Jiangsu, China | PW | 6 | Environmental | PW (a) |
| Albahri et al. (2021) [18] | Australia | OIL | 1 | Consortium | ND.CIM (a),(b),(c) |
| Su et al. (2022) [20] | China | PW | 1 | Culture | PW.HDM (a) |
| Zhou et al. (2022) [14] | Shengli, China | OS | 1 | Culture | OS.MET-HDM (a) |
| Prajapat et al. (2023) [46] | Rajasthan, India | PW | 1 | Environmental | PW (a) |
| IW | 3 | Environmental | IW (a) |
| Reference | Sample | Preprocessing | RNA Preserving Agent | RNA Extraction Method |
|---|---|---|---|---|
| Shestakova et al. (2011) [15] | PW.HDM | Centrifugation (b) | Not declared | TRIzol reagent (d) |
| IW.HDM | Centrifugation (b) | Not declared | TRIzol reagent (d) | |
| Zapata-Peñasco et al. (2016) [4] | PW.SRB | Centrifugation (b) | RNAProtect Bacteria Reagent (e) | RNeasy Protect Bacteria kit (e) |
| Nazina et al. (2017) [3] | IW | Filtration (b) | Ethanol reagent (h) | TRIzol reagent (d) |
| Li et al. (2017) [17] | PW | Filtration (a) | 95:5 v/v ethanol/trizol (h) | High Pure RNA Isolation Kit (f) |
| Liu et al. (2018) [16] | PW | Centrifugation (b) | 95:5 v/v ethanol/trizol (h) | PowerMicrobiome RNA Isolation kit (g) |
| Salgar-Chaparro e Machuca (2019) [1] | PW | Filtration (a) | RNAProtect Bacteria Reagent (e) | RNeasy PowerWater kit (e) |
| IW | Filtration (a) | RNAProtect Bacteria Reagent (e) | RNeasy PowerWater kit (e) | |
| SD | Not declared | RNAProtect Bacteria Reagent (e) | RNeasy PowerSoil kit (e) | |
| Zheng et al. (2019) [19] | OIL.HDM | Not declared | Not declared | TRIzol reagent (d) |
| Liu et al. (2020) [35] | PW | Centrifugation (b) | 95:5 v/v ethanol/trizol (h) | PowerMicrobiome RNA Isolation kit (g) |
| PW.HDM | Centrifugation (b) | Not declared | PowerMicrobiome RNA Isolation kit (g) | |
| Liu et al. (2020) [48] | PW | Centrifugation (b) | 95:5 v/v ethanol/trizol (h) | TRIzol reagent (d) |
| Liu et al. (2020) [47] | PW | Centrifugation (b) | 95:5 v/v ethanol/trizol (h) | PowerMicrobiome RNA Isolation kit (g) |
| Zhou et al. (2020) [22] | PW | Centrifugation (b) | 95:5 v/v ethanol/trizol (h) | TRIzol reagent (d) |
| Liu et al. (2020) [49] | PW | Centrifugation (b) | 95:5 v/v ethanol/trizol (h) | TRIzol reagent (d) |
| Salgar-Chaparro et al. (2020) [43] | PW.CIM (c) | Centrifugation (b) | Not declared | RNeasy PowerBiofilm kit (e) |
| Salgar-Chaparro et al. (2020) [44] | PW.CIM (c) | Centrifugation (b) | Not declared | RNeasy PowerBiofilm kit (e) |
| Alhefeiti et al. (2021) [13] | OS.OCDM | Centrifugation (b) | 20% glycerol reagent (h) | TRIzol reagent (i) |
| Zhou et al. (2021) [45] | PW | Filtration (a) | 95:5 v/v ethanol/trizol (h) | High Pure RNA Isolation kit (f) |
| Albahri et al. (2021) [18] | ND.CIM (c),(i) | Centrifugation (b) | Not declared | RNeasy PowerBiofilm kit (e) |
| Su et al. (2022) [20] | PW.HDM | Centrifugation (b) | Not declared | TRIzol reagent (d) |
| Zhou et al. (2022) [14] | OS.MET-HDM | Centrifugation (b) | Liquid nitrogen (n2) | acid phenol chloroform/ isoamyl alcohol reagent (h) |
| Prajapat et al. (2023) [46] | PW | Not declared | Not declared | RNeasy plant mini kit (e) |
| IW | Not declared | Not declared | RNeasy plant mini kit (e) |
| Reference | Sample | Gene | Primer | Platform (Sequencer Model) |
|---|---|---|---|---|
| Shestakova et al. (2011) [15] | PW.HDM and IW.HDM | alkB | AlkBFB/AlkBRB | ABI (3730) |
| 16S rRNA | Bact-827F/519R | |||
| 16S rRNA | Arch-A109F/A1041R | |||
| Zapata-Penãsco et al. (2016) [4] | PW.SRB | dsrA | DSRAVibF/DSRAVIbR | ABI (310) |
| Nazina et al. (2017) [3] | IW | 16S rRNA | Bact-827F/519R | ABI (3730) |
| Li et al. (2017) [17] | PW | 16S rRNA | Bact-515F/907R | NGS (Miseq) |
| 16S rRNA | Arch-344F/915R | NGS (Miseq) | ||
| aprA | aprA-1-FW/aprA-5-RV’ | ABI (377) | ||
| dsrA | DSR-1Fdeg/PJdsr853Rdeg | ABI (377) | ||
| Liu et al. (2018) [16] | PW | Metatranscriptomic | NGS (Miseq) | |
| Salgar-Chaparro and Machuca (2019) [1] | PW, IW (a) and DS (b) | 16S rRNA | Bact-341F/806R | NGS (Miseq) |
| Zheng et al. (2019) [19] | OIL.HDM | Transcriptomic (Bacillus licheniformis) | NGS (NextSeq) | |
| Liu et al. (2020) [35] | PW and PW.HDM | Metatranscriptomic | NGS (HiSeq X ten) | |
| Liu et al. (2020) [48] | PW | assA | assA2F/assA2R | Sanger (not specified) |
| mcrA | mlas-mod-F/mcrA-rev-R | |||
| Liu et al. (2020) [47] | PW | Metatranscriptomic (c) | NGS (MiSeq) | |
| Zhou et al. (2020) [22] | PW | 16S rRNA | Bact-515F/907R | NGS (MiSeq) |
| 16S rRNA | Arch-524F10extF/Arch958RmodR | |||
| Liu et al. (2020) [49] | PW | assA | assA2F/assA2R | ABI (377) |
| mcrA | mlas-mod-F/mcrA-rev-R | |||
| Salgar-Chaparro et al. (2020) [43] | PW.CIM (f) | 16S rRNA | Bact-341F/806R | NGS (MiSeq) |
| Salgar-Chaparro et al. (2020) [44] | PW.CIM (f) | 16S rRNA | Bact-341F/806R | NGS (MiSeq) |
| Alhefeiti et al. (2021) [13] | OS.OCDM | Transcriptomic (Bacillus cereus) | NGS (not specified) | |
| Zhou et al. (2021) [45] | PW | mcrA | mlas-mod-F/mcrA-rev-R | ABI (377) |
| Albahri et al. (2021) [18] | ND.CIM (e),(f) | 16S rRNA | Bact-341F/806R | NGS (MiSeq) |
| Su et al. (2022) [20] | PW.HDM | Transcriptomic (Exiguobacterium aurantiacum SW-20) | NGS (HiSeq X ten) | |
| Zhou et al. (2022) [14] | OS.MET-HDM | Metatranscriptomic | NGS (NovaSeq 6000) | |
| Prajapat et al. (2023) [46] | PW and IW | 16S rRNA | EUB341F/EUB534R | Unrealized (d) |
| narG | narG 1575F/narG 1748R | |||
| nirS | nirS 1189F/nirS 1376R | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, R.F.; García, G.J.Y.; Dutra, J.d.C.F.; Cardoso, M.S.; Costa, E.A.; de Abreu Waldow, V.; Groposo, C.J.; Akamine, R.N.; de Sousa, M.P.; Figueiredo, H.; et al. Metabolically Active Microbial Communities in Oilfields: A Systematic Review and Synthesis of RNA Preservation, Extraction, and Sequencing Methods. Appl. Microbiol. 2023, 3, 1144-1163. https://doi.org/10.3390/applmicrobiol3040079
Gomes RF, García GJY, Dutra JdCF, Cardoso MS, Costa EA, de Abreu Waldow V, Groposo CJ, Akamine RN, de Sousa MP, Figueiredo H, et al. Metabolically Active Microbial Communities in Oilfields: A Systematic Review and Synthesis of RNA Preservation, Extraction, and Sequencing Methods. Applied Microbiology. 2023; 3(4):1144-1163. https://doi.org/10.3390/applmicrobiol3040079
Chicago/Turabian StyleGomes, Rosimeire Floripes, Glen Jasper Yupanqui García, Joyce da Cruz Ferraz Dutra, Mariana Santos Cardoso, Eduardo Almeida Costa, Vinicius de Abreu Waldow, Claudia Julia Groposo, Rubens Nobumoto Akamine, Maira Paula de Sousa, Henrique Figueiredo, and et al. 2023. "Metabolically Active Microbial Communities in Oilfields: A Systematic Review and Synthesis of RNA Preservation, Extraction, and Sequencing Methods" Applied Microbiology 3, no. 4: 1144-1163. https://doi.org/10.3390/applmicrobiol3040079
APA StyleGomes, R. F., García, G. J. Y., Dutra, J. d. C. F., Cardoso, M. S., Costa, E. A., de Abreu Waldow, V., Groposo, C. J., Akamine, R. N., de Sousa, M. P., Figueiredo, H., Azevedo, V. A. d. C., & Góes-Neto, A. (2023). Metabolically Active Microbial Communities in Oilfields: A Systematic Review and Synthesis of RNA Preservation, Extraction, and Sequencing Methods. Applied Microbiology, 3(4), 1144-1163. https://doi.org/10.3390/applmicrobiol3040079