
Usage of Cultured Human Fecal Microbiota for Colonization of Caenorhabditis elegans to Study Host–Microbe Interaction
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Maintenance
2.2. Human Fecal Microbiota (HFM)
2.3. Lawn Harvest for 16S Analysis
2.4. C. elegans Growth and Harvest for 16S Analysis
2.5. DNA Extraction
2.6. Library Prep and 16S Sequencing
2.7. Software and Analysis
2.8. Egg Lay and Development
2.9. GST-4 Expression
3. Results
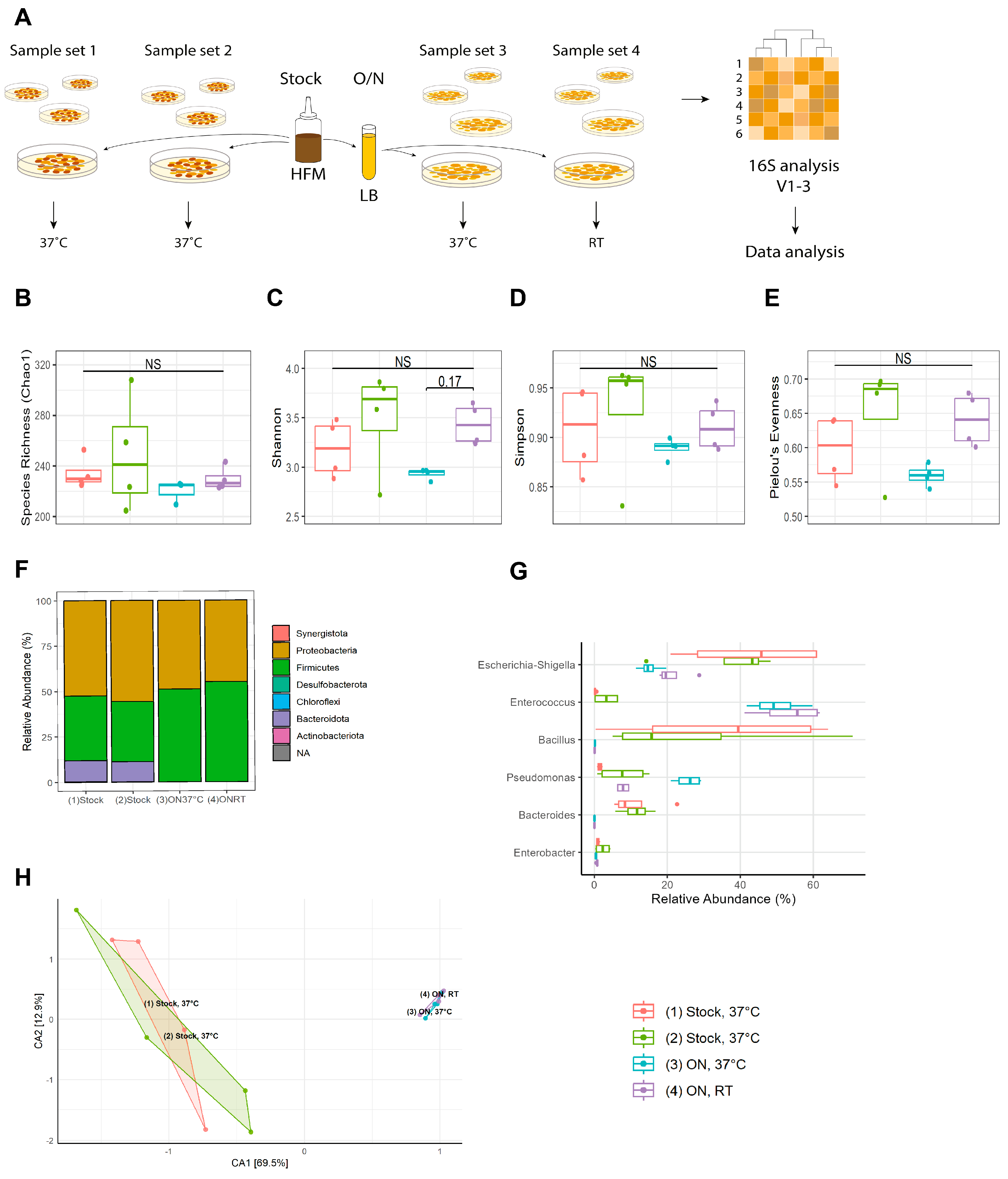
3.1. A Stable Starting Material Is Obtained through Overnight Cultivation
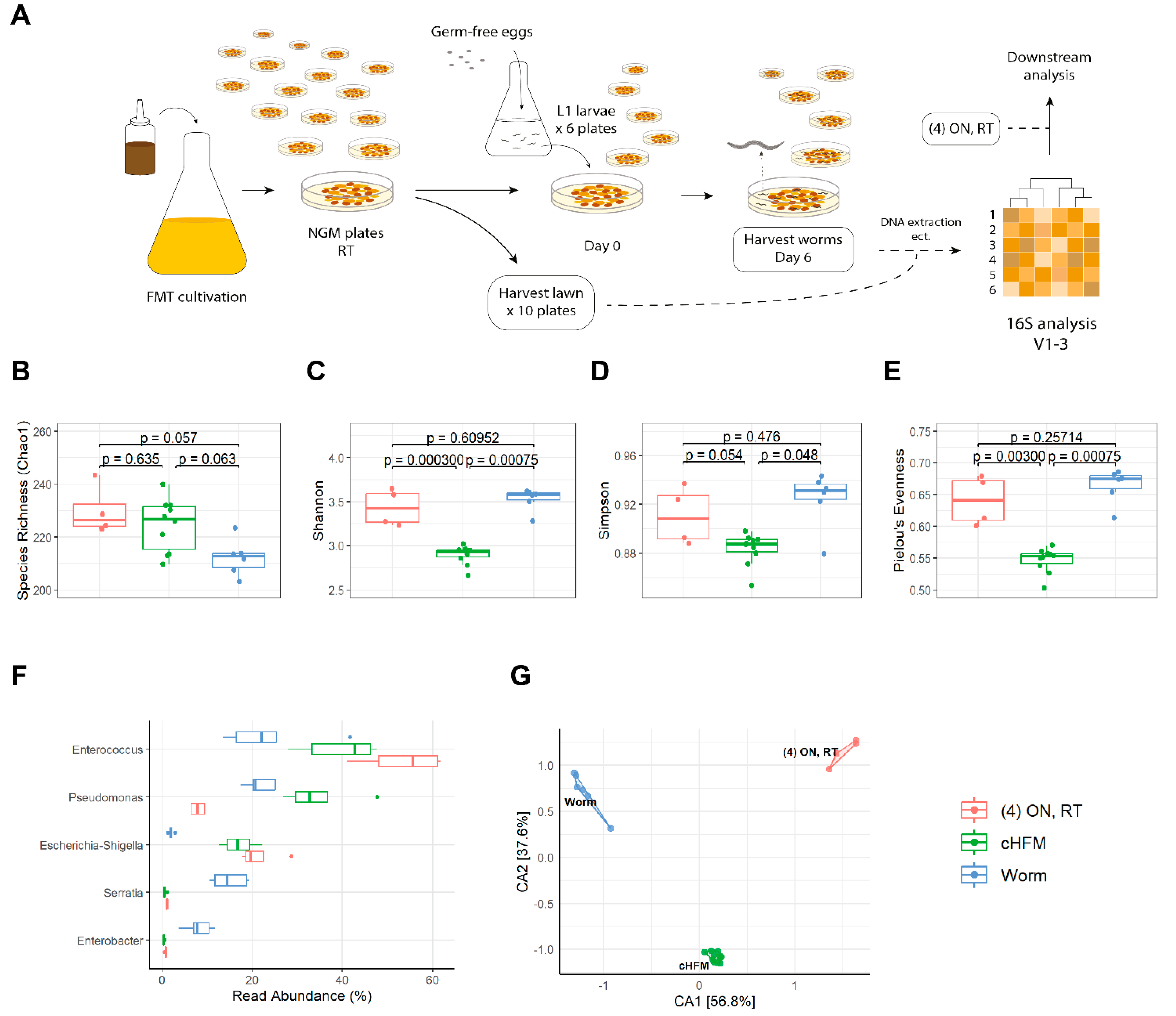
3.2. Transplantation of Cultured Human Fecal Microbiota to C. elegans
4. Discussion
4.1. The Typical E. coli Diet Can Be Replaced with Complex Sub-Set of the Human Microbiota
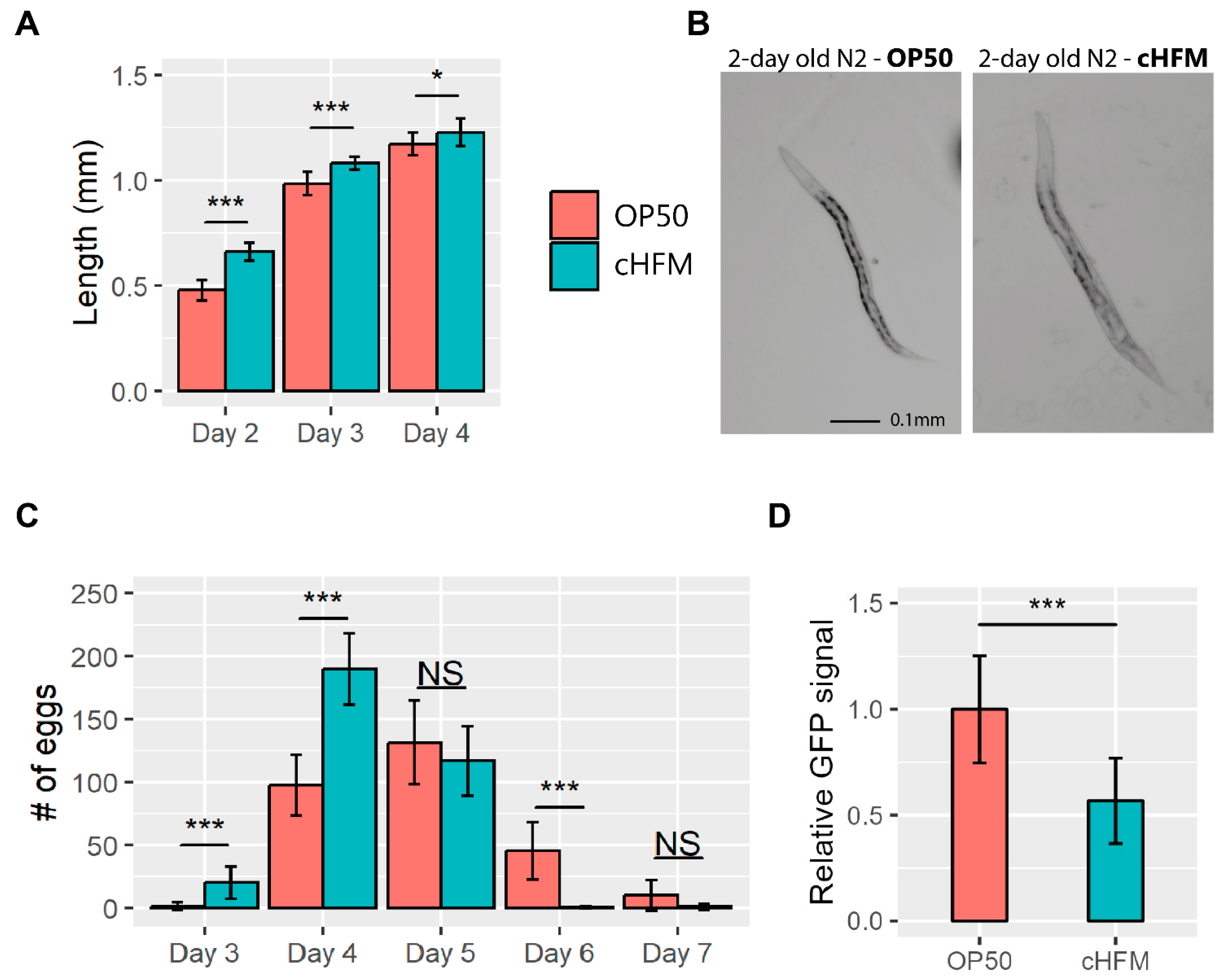
4.2. C. elegans Can Develop and Reproduce Normally When Being Fed a Sub-Microbiota of Cultivated Human Fecal Microbiota
4.3. You Are Not What You Eat—Sub-Microbiomes Colonize the Worm Intestine
4.4. Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Mitchell, A.L.; Boland, M.; Forster, S.C.; Gloor, G.B.; Tarkowska, A.; Lawley, T.D.; Finn, R.D. A New Genomic Blueprint of the Human Gut Microbiota. Nature 2019, 568, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the Gut Microbiota in Nutrition and Health. BMJ 2018, 361, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Kim, J.H. Role of Microbiome and Its Metabolite, Short Chain Fatty Acid in Prostate Cancer. Investig. Clin. Urol. 2023, 64, 3–12. [Google Scholar] [CrossRef]
- Fujisaka, S.; Watanabe, Y.; Tobe, K. The Gut Microbiome: A Core Regulator of Metabolism. J. Endocrinol. 2023, 256, e220111. [Google Scholar] [CrossRef]
- Boekhorst, J.; Venlet, N.; Procházková, N.; Hansen, M.L.; Lieberoth, C.B.; Bahl, M.I.; Lauritzen, L.; Pedersen, O.; Licht, T.R.; Kleerebezem, M.; et al. Stool Energy Density Is Positively Correlated to Intestinal Transit Time and Related to Microbial Enterotypes. Microbiome 2022, 10, 1–10. [Google Scholar] [CrossRef]
- Clemente, J.C.; Manasson, J.; Scher, J.U. The Role of the Gut Microbiome in Systemic Inflammatory Disease. BMJ 2018, 360, j5145. [Google Scholar] [CrossRef]
- Morais, L.H.; Schreiber, H.L.; Mazmanian, S.K. The Gut Microbiota–Brain Axis in Behaviour and Brain Disorders. Nat. Rev. Microbiol. 2021, 19, 241–255. [Google Scholar] [CrossRef]
- Fischbach, M.A. Microbiome: Focus on Causation and Mechanism. Cell 2018, 174, 785–790. [Google Scholar] [CrossRef]
- de Jonge, N.; Michaelsen, T.Y.; Ejbye-Ernst, R.; Jensen, A.; Nielsen, M.E.; Bahrndorff, S.; Nielsen, J.L. Housefly (Musca Domestica L.) Associated Microbiota across Different Life Stages. Sci. Rep. 2020, 10, 7842. [Google Scholar] [CrossRef]
- Xia, H.; Chen, H.; Cheng, X.; Yin, M.; Yao, X.; Ma, J.; Huang, M.; Chen, G.; Liu, H. Zebrafish: An Efficient Vertebrate Model for Understanding Role of Gut Microbiota. Mol. Med. 2022, 28, 161. [Google Scholar] [CrossRef] [PubMed]
- Poletti, M.; Arnauts, K.; Ferrante, M.; Korcsmaros, T. Organoid-Based Models to Study the Role of Host-Microbiota Interactions in IBD. J. Crohn’s Colitis 2021, 15, 1222–1235. [Google Scholar] [CrossRef]
- Cheng, A.G.; Ho, P.Y.; Aranda-Díaz, A.; Jain, S.; Yu, F.B.; Meng, X.; Wang, M.; Iakiviak, M.; Nagashima, K.; Zhao, A.; et al. Design, Construction, and in Vivo Augmentation of a Complex Gut Microbiome. Cell 2022, 185, 3617–3636. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.; Gill, M.S. (Eds.) Ageing: Lessons from C. Elegans; Springer International Publishing: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Christensen, K.; Mørch, M.; Morthorst, T.; Lykkemark, S.; Olsen, A. Microbiota, Probiotic Bacteria and Ageing. In Ageing: Lessons from C. elegans; Olsen, A., Gill, M., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; pp. 411–429. [Google Scholar]
- Cabreiro, F.; Gems, D. Worms Need Microbes Too: Microbiota, Health and Aging in Caenorhabditis elegans. EMBO Mol. Med. 2013, 5, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Mørch, M.G.M.; Møller, K.V.; Hesselager, M.O.; Harders, R.H.; Kidmose, C.L.; Buhl, T.; Fuursted, K.; Bendixen, E.; Shen, C.; Christensen, L.G.; et al. The TGF-β Ligand DBL-1 Is a Key Player in a Multifaceted Probiotic Protection against MRSA in C. elegans. Sci. Rep. 2021, 11, 10717. [Google Scholar] [CrossRef] [PubMed]
- Møller, K.V.; Nguyen, H.T.T.; Mørch, M.G.M.; Hesselager, M.O.; Mulder, F.A.A.; Fuursted, K.; Olsen, A. A Lactobacilli Diet That Confers MRSA Resistance Causes Amino Acid Depletion and Increased Antioxidant Levels in the C. elegans Host. Front. Microbiol. 2022, 13, 886206. [Google Scholar] [CrossRef]
- Brejning, J.; Nørgaard, S.; Schøler, L.; Morthorst, T.H.; Jakobsen, H.; Lithgow, G.J.; Jensen, L.T.; Olsen, A. Loss of NDG-4 Extends Lifespan and Stress Resistance in Caenorhabditis elegans. Aging Cell 2014, 13, 156–164. [Google Scholar] [CrossRef]
- Miller, B.C.; Mathai, M.; Yadav, H.; Jain, S. Geroprotective Potential of Microbiome Modulators in the Caenorhabditis elegans Model. Geroscience 2023. [Google Scholar] [CrossRef]
- Samuel, B.S.; Rowedder, H.; Braendle, C.; Félix, M.-A.; Ruvkun, G. Caenorhabditis elegans Responses to Bacteria from Its Natural Habitats. Proc. Natl. Acad. Sci. USA 2016, 113, E3941–E3949. [Google Scholar] [CrossRef]
- Dirksen, P.; Marsh, S.A.; Braker, I.; Heitland, N.; Wagner, S.; Nakad, R.; Mader, S.; Petersen, C.; Kowallik, V.; Rosenstiel, P.; et al. The Native Microbiome of the Nematode Caenorhabditis elegans: Gateway to a New Host-Microbiome Model. BMC Biol. 2016, 14, 38. [Google Scholar] [CrossRef]
- Berg, M.; Stenuit, B.; Ho, J.; Wang, A.; Parke, C.; Knight, M.; Alvarez-Cohen, L.; Shapira, M. Assembly of the Caenorhabditis elegans Gut Microbiota from Diverse Soil Microbial Environments. ISME J. 2016, 10, 1998–2009. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.; Monnin, D.; Cho, J.; Nelson, L.; Crits-Christoph, A.; Shapira, M. TGFβ/BMP Immune Signaling Affects Abundance and Function of C. elegans Gut Commensals. Nat. Commun. 2019, 10, 604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Weckhorst, J.L.; Assié, A.; Hosea, C.; Ayoub, C.A.; Khodakova, A.S.; Cabrera, M.L.; Vidal Vilchis, D.; Félix, M.-A.; Samuel, B.S. Natural Genetic Variation Drives Microbiome Selection in the Caenorhabditis elegans Gut. Curr. Biol. 2021, 31, 2603–2618.e9. [Google Scholar] [CrossRef] [PubMed]
- Dirksen, P.; Assié, A.; Zimmermann, J.; Zhang, F.; Tietje, A.M.; Marsh, S.A.; Félix, M.A.; Shapira, M.; Kaleta, C.; Schulenburg, H.; et al. CeMbio—The Caenorhabditis elegans Microbiome Resource. G3 Genes. Genomes Genet. 2020, 10, 3025–3039. [Google Scholar] [CrossRef]
- Petersen, C.; Dirksen, P.; Schulenburg, H. Why We Need More Ecology for Genetic Models Such as C. elegans. Trends Genet. 2015, 31, 120–127. [Google Scholar] [CrossRef]
- Corsi, A.K.; Wightman, B.; Chalfie, M. A Transparent Window into Biology: A Primer on Caenorhabditis Elegans. Genetics 2015, 200, 387–407. [Google Scholar] [CrossRef]
- Kumar, A.; Baruah, A.; Tomioka, M.; Iino, Y.; Kalita, M.C.; Khan, M. Caenorhabditis elegans: A Model to Understand Host–Microbe Interactions. Cell. Mol. Life Sci. 2020, 77, 1229–1249. [Google Scholar] [CrossRef]
- Walker, A.C.; Bhargava, R.; Vaziriyan-Sani, A.S.; Pourciau, C.; Donahue, E.T.; Dove, A.S.; Gebhardt, M.J.; Ellward, G.L.; Romeo, T.; Czyż, D.M. Colonization of the Caenorhabditis elegans Gut with Human Enteric Bacterial Pathogens Leads to Proteostasis Disruption That Is Rescued by Butyrate. PLoS Pathog. 2021, 17, e1009510. [Google Scholar] [CrossRef]
- Chu, Q.; Zhang, S.; Yu, X.; Wang, Y.; Zhang, M.; Zheng, X. Fecal Microbiota Transplantation Attenuates Nano-Plastics Induced Toxicity in Caenorhabditis elegans. Sci. Total Environ. 2021, 779, 146454. [Google Scholar] [CrossRef]
- Jumpstart Consortium Human Microbiome Project Data Generation Working Group. Evaluation of 16S RDNA-Based Community Profiling for Human Microbiome Research. PLoS ONE 2012, 7, e39315. [Google Scholar]
- Yashiro, E. Illumina Sequenced Amplicon Reads Processing Workflow. Available online: https://github.com/eyashiro/AmpProc (accessed on 26 September 2023).
- Edgar, R.C. Search and Clustering Orders of Magnitude Faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Wilson, J.B. A Consumer’s Guide to Evenness Indices. Oikos 1996, 76, 70–82. [Google Scholar] [CrossRef]
- Leiers, B.; Kampkötter, A.; Grevelding, C.G.; Link, C.D.; Johnson, T.E.; Henkle-Dührsen, K. A Stress-Responsive Glutathione S-Transferase Confers Resistance to Oxidative Stress in Caenorhabditis elegans. Free Radic. Biol. Med. 2003, 34, 1405–1415. [Google Scholar] [CrossRef]
- Afrizal, A.; Jennings, S.A.V.; Hitch, T.C.A.; Riedel, T.; Basic, M.; Panyot, A.; Treichel, N.; Hager, F.T.; Wong, E.O.-Y.; Wolter, B.; et al. Enhanced Cultured Diversity of the Mouse Gut Microbiota Enables Custom-Made Synthetic Communities. Cell Host Microbe 2022, 30, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Brugiroux, S.; Beutler, M.; Pfann, C.; Garzetti, D.; Ruscheweyh, H.-J.; Ring, D.; Diehl, M.; Herp, S.; Lötscher, Y.; Hussain, S.; et al. Genome-Guided Design of a Defined Mouse Microbiota That Confers Colonization Resistance against Salmonella Enterica Serovar Typhimurium. Nat. Microbiol. 2016, 2, 16215. [Google Scholar] [CrossRef]
- Wexler, H.M. Bacteroides: The Good, the Bad, and the Nitty-Gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. [Google Scholar] [CrossRef]
- Turnbull, P.C.B. Bacillus. In Medical Microbiology; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996; ISBN 0-9631172-1-1. [Google Scholar]
- Baugh, L.R.; Hu, P.J. Starvation Responses Throughout the Caenorhabditis elegans Life Cycle. Genetics 2020, 216, 837–878. [Google Scholar] [CrossRef]
- Cohen, L.B.; Troemel, E.R. Microbial Pathogenesis and Host Defense in the Nematode C. elegans. Curr. Opin. Microbiol. 2015, 23, 94–101. [Google Scholar] [CrossRef]
- Baillo, A.; Villena, J.; Albarracín, L.; Tomokiyo, M.; Elean, M.; Fukuyama, K.; Quilodrán-Vega, S.; Fadda, S.; Kitazawa, H. Lactiplantibacillus Plantarum Strains Modulate Intestinal Innate Immune Response and Increase Resistance to Enterotoxigenic Escherichia Coli Infection. Microorganisms 2023, 11, 63. [Google Scholar] [CrossRef]
- Padilla, P.A.; Nystul, T.G.; Zager, R.A.; Johnson, A.C.M.; Roth, M.B. Dephosphorylation of Cell Cycle–Regulated Proteins Correlates with Anoxia-Induced Suspended Animation in Caenorhabditis elegans. Mol. Biol. Cell 2002, 13, 1473–1483. [Google Scholar] [CrossRef]
- Harding, B.W.; Ewbank, J.J. An Integrated View of Innate Immune Mechanisms in C. elegans. Biochem. Soc. Trans. 2021, 49, 2307–2317. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Møller, K.V.; Wesseltoft, J.B.; Malazarte, R.; Kousgaard, S.J.; Nielsen, H.L.; Yashiro, E.; Olsen, A. Usage of Cultured Human Fecal Microbiota for Colonization of Caenorhabditis elegans to Study Host–Microbe Interaction. Appl. Microbiol. 2023, 3, 1130-1143. https://doi.org/10.3390/applmicrobiol3040078
Møller KV, Wesseltoft JB, Malazarte R, Kousgaard SJ, Nielsen HL, Yashiro E, Olsen A. Usage of Cultured Human Fecal Microbiota for Colonization of Caenorhabditis elegans to Study Host–Microbe Interaction. Applied Microbiology. 2023; 3(4):1130-1143. https://doi.org/10.3390/applmicrobiol3040078
Chicago/Turabian StyleMøller, Katrine V., Jonas Bruhn Wesseltoft, Richelle Malazarte, Sabrina J. Kousgaard, Hans L. Nielsen, Erika Yashiro, and Anders Olsen. 2023. "Usage of Cultured Human Fecal Microbiota for Colonization of Caenorhabditis elegans to Study Host–Microbe Interaction" Applied Microbiology 3, no. 4: 1130-1143. https://doi.org/10.3390/applmicrobiol3040078
APA StyleMøller, K. V., Wesseltoft, J. B., Malazarte, R., Kousgaard, S. J., Nielsen, H. L., Yashiro, E., & Olsen, A. (2023). Usage of Cultured Human Fecal Microbiota for Colonization of Caenorhabditis elegans to Study Host–Microbe Interaction. Applied Microbiology, 3(4), 1130-1143. https://doi.org/10.3390/applmicrobiol3040078