
A Network Analysis Approach to Detect and Differentiate Usher Syndrome Types Using miRNA Expression Profiles: A Pilot Study

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. miRNA Profiling
2.1.1. miRNA Gene Target Prediction
2.1.2. Gene Ontology Enrichment and Pathway Analysis
2.2. Preprocessing and Feature Selection
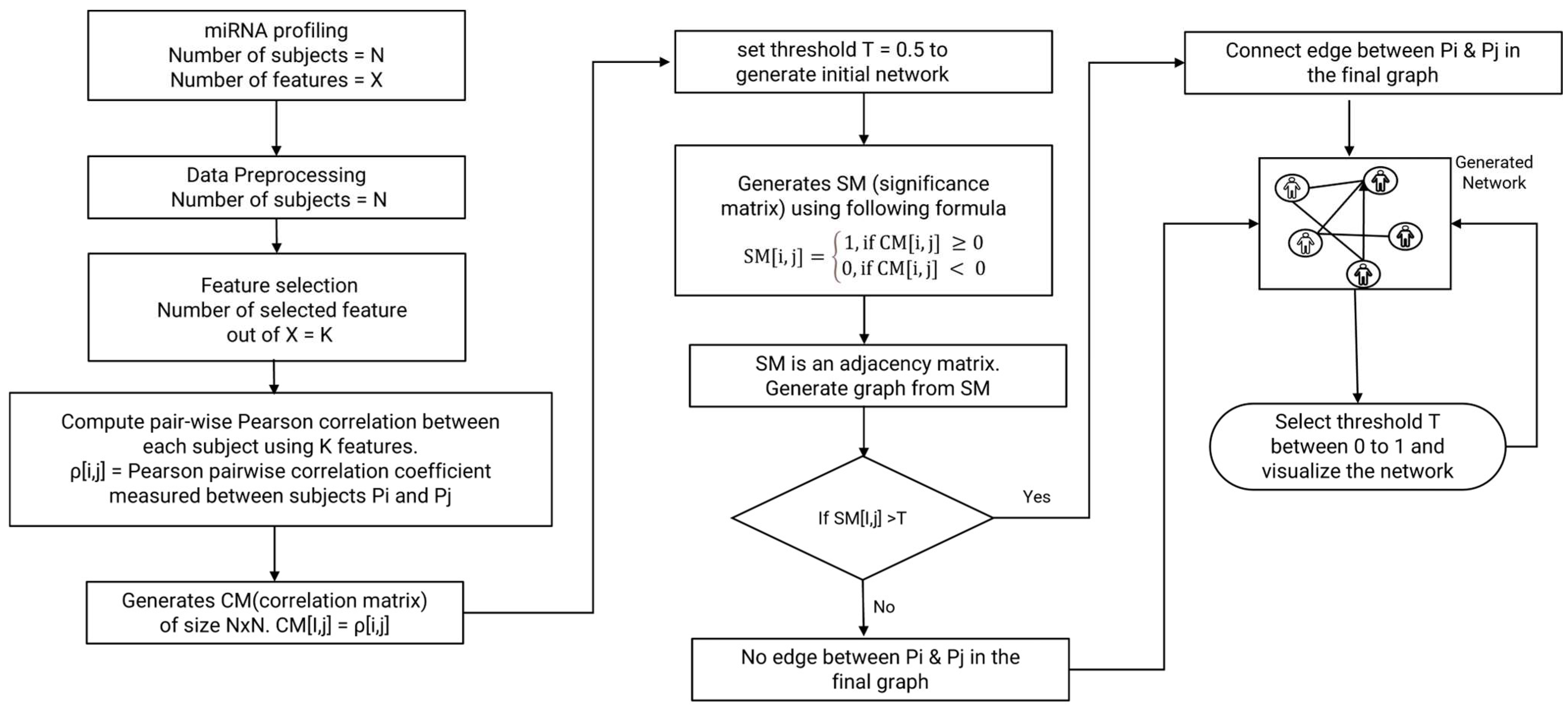
2.3. Network Modeling
2.3.1. Preliminaries
- Let N denote the total number of subjects in the study. For this case, N = 17 subjects (4 controls and 13 Usher syndrome patient cell lines).
- Let K represent the number of selected miRNA features utilized to construct the correlation network. In this analysis, we identified K = 6 key miRNA features.
- Let Pi and Pj represent two randomly selected individual subjects from the population of N. The indices i and j satisfy the condition 1 ≤ (i, j) ≤ N, ensuring valid subject pairs.
- Let ρ[i,j] correspond to the Pearson pairwise correlation coefficient between subjects Pi and Pj using their miRNA expression profiles. The Correlation Matrix (CM) stores these correlation coefficients for all subject pairs. Thus, CM[i, j] represents ρ[i, j].
- Let T represent the threshold for the minimum correlation strength required to establish an edge between two nodes.
- The Significance Matrix (SM) is derived from the Correlation Matrix by applying the threshold T. It serves as the adjacency matrix for the graph.
2.3.2. Network Construction Procedure
- Compute Pairwise Pearson Correlation
- Generate Correlation Matrix (CM)
- Set Threshold (T) for Correlation Strength
- Generate Significance Matrix (SM)
- Generate Network Graph
- Network Analysis
3. Results
3.1. Feature Selection
3.2. Network Analysis
- Network at Lower Threshold of 0.50 (Figure 2): At a lower correlation threshold, the network shows a clear separation between control subjects and Usher syndrome patient cell lines. Control subjects are grouped into a distinct cluster, while Usher patients form another interconnected cluster. This separation suggests that the control group exhibits a different miRNA expression profile compared to the Usher syndrome patient cell lines. Furthermore, at this lower threshold, Usher patient cell lines are not differentiable from one another, suggesting that while control and Usher subjects can be detected, the subtypes of Usher syndrome remain indistinguishable.
- Network at Intermediate Threshold of 0.79 (Figure 3): As the correlation threshold increases, the network begins to reveal more specific relationships within the Usher syndrome group. Notably, Usher type 1D subjects are separated into a distinct cluster, suggesting that their miRNA expression profiles are less like other Usher subtypes at this correlation strength. The control group remains isolated from the patient clusters, reinforcing the separation between healthy individuals and those with Usher syndrome.
- Network at Higher Threshold of 0.88 (Figure 4): At the highest correlation threshold, the network shows even finer granularity in the relationships between subjects. Usher type 1B samples now form a separate cluster, distinct from both the Usher type 1D cluster and other Usher subtypes. This suggests that at higher thresholds, the system can differentiate between subtypes of Usher syndrome based on subtle variations in miRNA expression. The control group continues to form its own separate cluster, reinforcing its distinct expression profile. In contrast, Usher type 2A and type 3A subjects remain strongly connected even at higher thresholds, suggesting that their similar miRNA profiles render them indistinguishable from one another.
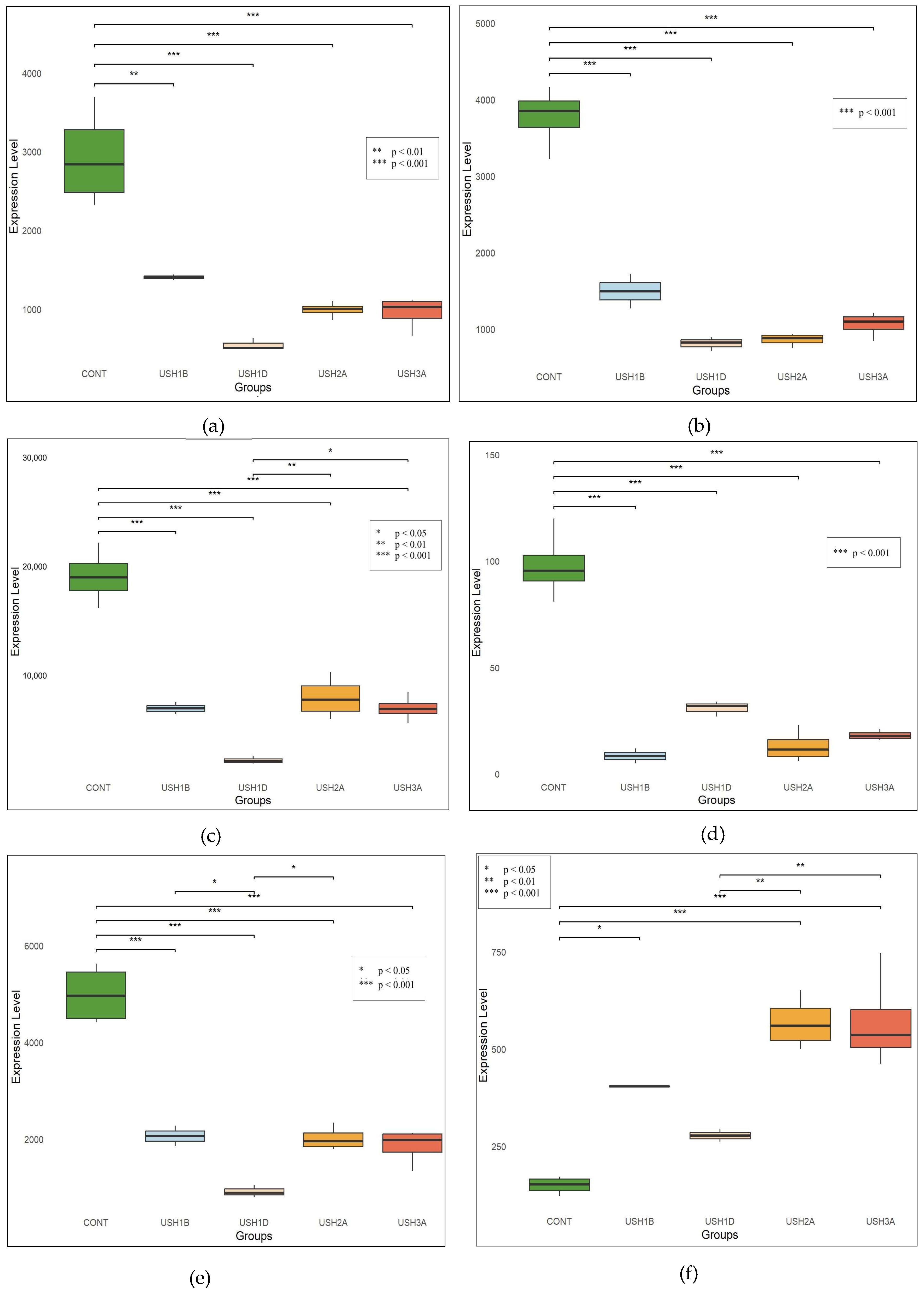
3.3. miRNA Expression Profiles
- Overall Trends
- Type-Specific Patterns
- o
- USH2A and USH3A
- o
- USH1B vs. USH1D
- Control vs. Usher Syndrome
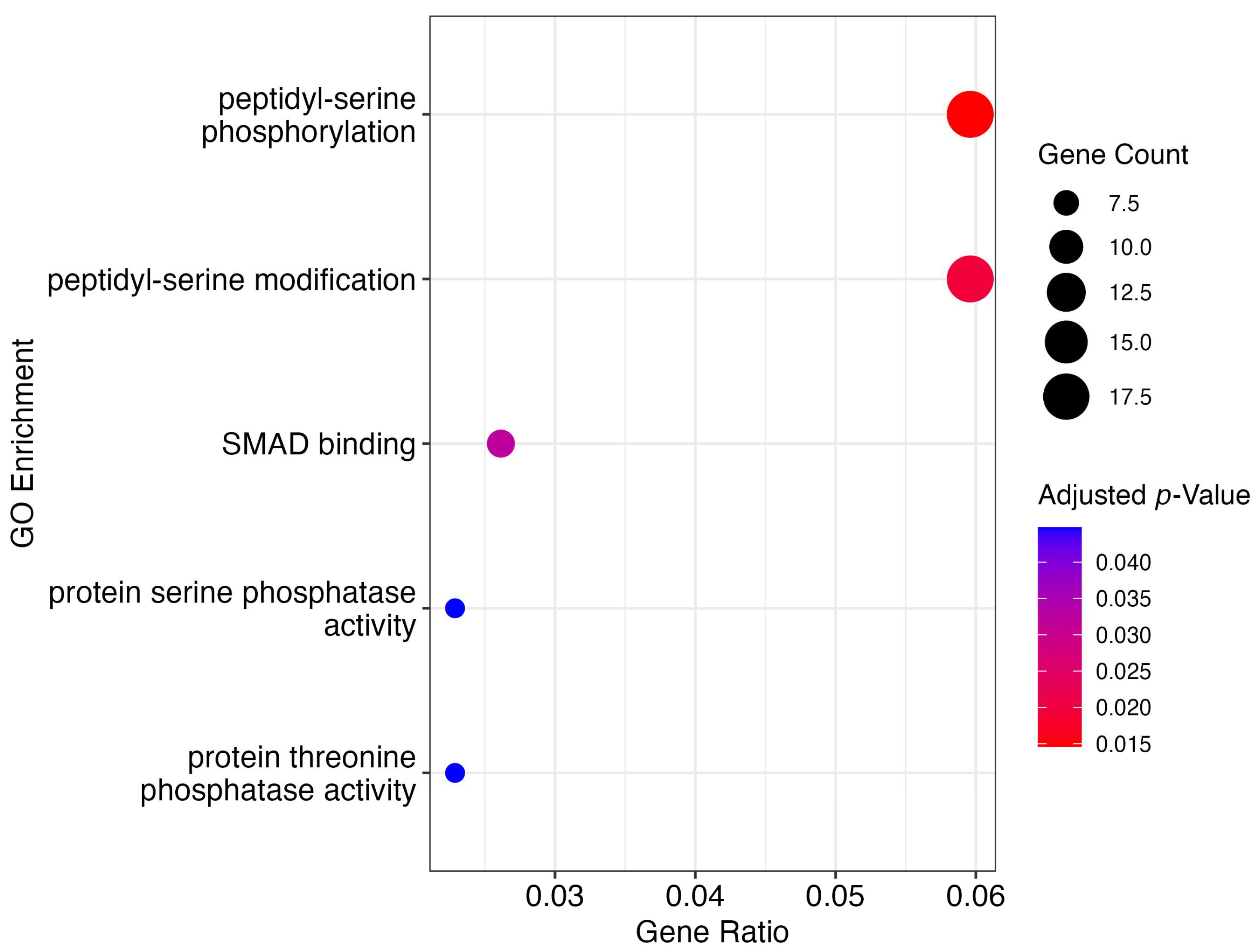
3.4. Gene Ontology Enrichment and Metabolic Pathway Analyses
4. Discussion
5. Limitations and Future Work
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boughman, J.A.; Vernon, M.; Shaver, K.A. Usher syndrome: Definition and estimate of prevalence from two high-risk populations. J. Chronic Dis. 1983, 36, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Delmaghani, S.; El-Amraoui, A. The genetic and phenotypic landscapes of Usher syndrome: From disease mechanisms to a new classification. Hum. Genet. 2022, 141, 709–735. [Google Scholar] [CrossRef] [PubMed]
- Dammeyer, J. Development and characteristics of children with Usher syndrome and CHARGE syndrome. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1292–1296. [Google Scholar] [CrossRef]
- Andersen, G.B.; Tost, J. Circulating miRNAs as biomarkers in cancer. In Tumor Liquid Biopsies; Springer: Berlin/Heidelberg, Germany, 2020; pp. 277–298. [Google Scholar]
- Seyhan, A.A. Circulating microRNAs as potential biomarkers in pancreatic cancer—Advances and challenges. Int. J. Mol. Sci. 2023, 24, 13340. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Ridzon, D.; Wong, L.; Chen, C. Characterization of microRNA expression profiles in normal human tissues. BMC Genom. 2007, 8, 166. [Google Scholar] [CrossRef]
- Tom, W.A.; Chandel, D.S.; Jiang, C.; Krzyzanowski, G.; Fernando, N.; Olou, A.; Fernando, M.R. Fernando N, Olou A, Fernando MR. Genotype characterization and miRNA expression profiling in Usher syndrome cell lines. Int. J. Mol. Sci. 2024, 25, 9993. [Google Scholar] [CrossRef]
- Lewis, M.A.; Quint, E.; Glazier, A.M.; Fuchs, H.; De Angelis, M.H.; Langford, C.; van Dongen, S.; Abreu-Goodger, C.; Piipari, M.; Redshaw, N.; et al. An ENU-induced mutation of miR-96 associated with progressive hearing loss in mice. Nat. Genet. 2009, 41, 614–618. [Google Scholar] [CrossRef]
- Weston, M.D.; Pierce, M.L.; Rocha-Sanchez, S.; Beisel, K.W.; Soukup, G.A. MicroRNA gene expression in the mouse inner ear. Brain Res. 2006, 1111, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jaber, V.; Percy, M.E.; Lukiw, W.J. A microRNA cluster (let-7c, miRNA-99a, miRNA-125b, miRNA-155, and miRNA-802) encoded at chr21q21.1-chr21q21.3 and the phenotypic diversity of Down’s syndrome (DS; trisomy 21). J. Nat. Sci. 2017, 3, e466. [Google Scholar]
- Mun, S.-K.; Chae, H.; Piao, X.-Y.; Lee, H.-J.; Kim, Y.-K.; Oh, S.-H.; Chang, M. MicroRNAs related to cognitive impairment after hearing loss. Clin. Exp. Otorhinolaryngol. 2021, 14, 76–81. [Google Scholar] [CrossRef]
- Pierce, M.L.; Weston, M.D.; Fritzsch, B.; Gabel, H.W.; Ruvkun, G.; Soukup, G.A. MicroRNA-183 family conservation and ciliated neurosensory organ expression. Evol. Dev. 2008, 10, 106–113. [Google Scholar] [CrossRef]
- Canouil, M.; Bouland, G.A.; Bonnefond, A.; Froguel, P.; ’t Hart, L.M.; Slieker, R.C. NACHO: An R package for quality control of NanoString nCounter data. Bioinformatics 2020, 36, 970–971. [Google Scholar] [CrossRef] [PubMed]
- Chance, M.R.; Chang, J.; Liu, S.; Gokulrangan, G.; Chen, D.H.-C.; Lindsay, A.; Geng, R.; Zheng, Q.Y.; Alagramam, K. Proteomics, bioinformatics and targeted gene expression analysis reveals up-regulation of cochlin and identifies other potential biomarkers in the mouse model for deafness in Usher syndrome type 1F. Hum. Mol. Genet. 2010, 19, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Linnert, J.; Knapp, B.; Güler, B.E.; Boldt, K.; Ueffing, M.; Wolfrum, U. Usher syndrome proteins ADGRV1 (USH2C) and CIB2 (USH1J) interact and share a common interactome containing TRiC/CCT-BBS chaperonins. Front. Cell Dev. Biol. 2023, 11, 1199069. [Google Scholar] [CrossRef]
- Giorgi, F.M.; Ceraolo, C.; Mercatelli, D. The R language: An engine for bioinformatics and data science. Life 2022, 12, 648. [Google Scholar] [CrossRef]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The biochemical basis of microRNA targeting efficacy. Science 2019, 366, eaav1741. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Rogers, J.; Gunn, S. Identifying feature relevance using a random forest. In International Statistical and Optimization Perspectives Workshop: Subspace, Latent Structure and Feature Selection; Springer: Berlin/Heidelberg, Germany, 2005; pp. 173–184. [Google Scholar]
- Fonti, V.; Belitser, E. Feature selection using lasso. VU Amst. Res. Pap. Bus. Anal. 2017, 30, 1–25. [Google Scholar]
- Sachdeva, R.K.; Bathla, P.; Rani, P.; Kukreja, V.; Ahuja, R. Systematic method for breast cancer classification using RFE feature selection. In Proceedings of the 2nd International Conference on Advance Computing and Innovative Technologies in Engineering (ICACITE), Greater Noida, India, 28–29 April 2022; IEEE: Piscataway, NJ, USA; pp. 1673–1676. [Google Scholar]
- Das, S. Filters, wrappers and a boosting-based hybrid for feature selection. In Proceedings of the 18th International Conference on Machine Learning (ICML), Williamstown, MA, USA, 28 June–1 July 2001; Volume 1, p. 74. [Google Scholar]
- Kunze-Schumacher, H.; Krueger, A. The Role of MicroRNAs in Development and Function of Regulatory T Cells–Lessons for a Better Understanding of MicroRNA Biology. Front. Immunol. 2020, 11, 2185. [Google Scholar] [CrossRef]
- Xia, X.; Wang, Y.; Zheng, J.C. The microRNA-17~92 family as a key regulator of neurogenesis and potential regenerative therapeutics of neurological disorders. Stem Cell Rev. Rep. 2022, 18, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Mahmoodian-Sani, M.-R.; Mehri-Ghahfarrokhi, A. The potential of miR-183 family expression in inner ear for regeneration, treatment, diagnosis and prognosis of hearing loss. J. Otol. 2017, 12, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Kong, K.Y.; Owens, K.S.; Rogers, J.H.; Mullenix, J.; Velu, C.S.; Grimes, H.L.; Dahl, R. MIR-23A microRNA cluster inhibits B-cell development. Exp. Hematol. 2010, 38, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Fu, H.; Sun, F.; Zhang, H.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res. 2008, 36, 5391–5404. [Google Scholar] [CrossRef]
- Li, L.; Shi, J.; Zhu, G.; Shi, B. MiR-17–92 cluster regulates cell proliferation and collagen synthesis by targeting TGFB pathway in mouse palatal mesenchymal cells. J. Cell. Biochem. 2012, 113, 1235–1244. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Random Forest | Lasso | RFE | SelectKBest |
|---|---|---|---|
| hsa-miR-769-5p | hsa-miR-212-3p | hsa-miR-106a-5p + hsa-miR-17-5p | hsa-miR-106a-5p +hsa-miR-17-5p |
| hsa-miR-23a-3p | hsa-miR-107 | hsa-miR-142-3p | hsa-miR-129-2-3p |
| hsa-miR-183-5p | hsa-miR-15a-5p | hsa-miR-146a-5p | hsa-miR-16-5p |
| hsa-miR-20a-5p+hsa-miR-20b-5p | hsa-miR-132-3p | hsa-miR-155-5p | hsa-miR-183-5p |
| hsa-let-7e-5p | hsa-let-7a-5p | hsa-miR-16-5p | hsa-miR-194-5p |
| hsa-miR-299-3p | hsa-let-7d-5p | hsa-miR-19b-3p | hsa-miR-20a-5p+hsa-miR-20b-5p |
| hsa-miR-28-3p | hsa-miR-146b-5p | hsa-miR-29a-3p | hsa-miR-296-5p |
| hsa-miR-1244 | hsa-miR-23a-3p | hsa-miR-29b-3p | hsa-miR-484 |
| hsa-miR-3934-5p | hsa-miR-18a-5p | hsa-miR-4454 + hsa-miR-7975 | hsa-miR-92a-3p |
| hsa-miR-363-3p | hsa-let-7b-5p | hsa-miR-92a-3p | hsa-miR-96-5p |
| Pathway | Overlap | p-Value | Adjusted p-Value | Odds Ratio | Combined Score | Genes |
|---|---|---|---|---|---|---|
| Signal Transduction R-HSA-162582 | 72/2465 | 3.13 × 10−6 | 0.00237826 | 1.92394484 | 24.3828992 | CHRM2; ITGB1; TFRC; HTR2A; HTR4; CRKL; AKAP12; FGF7; SPRED1; DUSP10; CCND1; MYB; AKT3; TAGAP; PDK4; PDE4B; PROK2; RSPO3; ZNF367; SH3GL2; TGIF1; DUSP5; PRKCI; DUSP2; CNOT6L; TGIF2; FBXW7; OMG; AXIN2; RHOC; VRK3; TGFBR2; DYNC1LI2; CCNE1; PKP4; PFN2; AMER1; GNAI3; PSEN2; DERL2; ARL2; ADRB1; PTHLH; MYL12B; ARHGAP12; PPP2CA; PPP2CB; ARHGAP20; CCL7; GNG5; MKNK1; SHOC2; CCL1; RHPN2; E2F5; USP25; TCF7L2; CBX4; WNT3A; WNT7A; GNG12; VEGFA; SMAD7; APLN; BAMBI; VAPB; PDE7A; CENPQ; RAB9B; CDC42SE2; CDK5R1; PTPN3 |
| MASTL Facilitates Mitotic Progression R-HSA-2465910 | 4/10 | 1.63 × 10−5 | 0.00617227 | 38.7613412 | 427.403609 | PPP2CA; PPP2CB; MASTL; ARPP19 |
| Negative Regulation Of MAPK Pathway R-HSA-5675221 | 6/41 | 6.49 × 10−5 | 0.01642841 | 10.0117347 | 96.5344737 | PPP2CA; PPP2CB; DUSP5; DUSP2; DUSP10; PTPN3 |
| Cyclin D Associated Events In G1 R-HSA-69231 | 6/47 | 0.00014227 | 0.02699549 | 8.54398955 | 75.6808883 | PPP2CA; PPP2CB; CCND2; CCND1; CCNE1; E2F5 |
| Signaling By WNT In Cancer R-HSA-4791275 | 5/33 | 0.00022733 | 0.03450933 | 10.4016532 | 87.2604006 | AMER1; PPP2CA; PPP2CB; TCF7L2; WNT3A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thelagathoti, R.K.; Tom, W.A.; Jiang, C.; Chandel, D.S.; Krzyzanowski, G.; Olou, A.; Fernando, R.M. A Network Analysis Approach to Detect and Differentiate Usher Syndrome Types Using miRNA Expression Profiles: A Pilot Study. BioMedInformatics 2024, 4, 2271-2286. https://doi.org/10.3390/biomedinformatics4040122
Thelagathoti RK, Tom WA, Jiang C, Chandel DS, Krzyzanowski G, Olou A, Fernando RM. A Network Analysis Approach to Detect and Differentiate Usher Syndrome Types Using miRNA Expression Profiles: A Pilot Study. BioMedInformatics. 2024; 4(4):2271-2286. https://doi.org/10.3390/biomedinformatics4040122
Chicago/Turabian StyleThelagathoti, Rama Krishna, Wesley A. Tom, Chao Jiang, Dinesh S. Chandel, Gary Krzyzanowski, Appolinaire Olou, and Rohan M. Fernando. 2024. "A Network Analysis Approach to Detect and Differentiate Usher Syndrome Types Using miRNA Expression Profiles: A Pilot Study" BioMedInformatics 4, no. 4: 2271-2286. https://doi.org/10.3390/biomedinformatics4040122
APA StyleThelagathoti, R. K., Tom, W. A., Jiang, C., Chandel, D. S., Krzyzanowski, G., Olou, A., & Fernando, R. M. (2024). A Network Analysis Approach to Detect and Differentiate Usher Syndrome Types Using miRNA Expression Profiles: A Pilot Study. BioMedInformatics, 4(4), 2271-2286. https://doi.org/10.3390/biomedinformatics4040122