Abstract
Group contribution (GC) methods to predict thermochemical properties are eminently important to process design. Following earlier work which presented a GC model in which, for the first time, chemical accuracy (1 kcal/mol or 4 kJ/mol) was accomplished, we here discuss classes of molecules for which the traditional GC approach does not hold, i.e., many results are beyond chemical accuracy. We report new ring-strain-related parameters which enable us to evaluate the heat of formation of alkyl-substituted cycloalkanes. In addition, the definition of the appropriate group size is important to obtain reliable and accurate data for systems in which the electron density varies continuously but slowly between related species. For this and in the case of ring strain, G4 quantum calculations are shown to be able to provide reliable heats of formation which provide the quantitative data which we can use, in the case of absence of experimental data, to establish group and nearest-neighbour interaction parameters to extend the range of applicability of the GC method whilst retaining chemical accuracy. We also found that the strong van der Waals that overlap in highly congested branched alkanes can be qualitatively investigated by applying DFT quantum calculations, which can provide an indication of the GC approach being inappropriate.
1. Introduction
Starting from the fact that the heat of formation is a crucial parameter with respect to the stability of molecules and chemical transformation, one can approach the availability of data by experimental or theoretical methods. The aim of this work is to have a method available which allows one to obtain accurate (chemical accuracy = difference between model and experiment less than, as originally known, 1 kcal/mol, i.e., 4 kJ/mol) and reliable (very few, if any, and certainly not with large deviation, outliers) values for this property at one’s fingertips. For the process developer or the experimental chemist, it is more than useful to have a method with such qualities, especially when multiple process routes are to be compared.
For the purpose of the evaluation of the enthalpy of formation ∆Hf of organic molecules from their molecular structure, the group contribution (GC) approach is one of the most important and widely applied methods. In this work, the ∆Hf (in this paper also indicated as dHf) is the enthalpy of formation for the ideal gas species at the reference temperature of 298.15 K. The original GC method [1] is based on the assumption that a molecule can be decomposed into molecular fragments which are in essence mutually independent, and the molecular property of interest is the sum of the individual properties of the molecular fragments. In the course of time, a larger number of further studies employing the GC methodology to evaluate the heat of formation of organic molecules have been reported, including the works by Benson and co-workers [2], Joback and Reid [3] and Gani et al. [4,5]. Recently, a study relevant in this context and employing neural networks was also reported [6].
In three earlier papers, the group contribution (GC) method was reevaluated for which, despite the method being old, a substantial and necessary increase in performance was still achieved [7,8,9]. It was demonstrated that it is possible to establish a GC parametrization for organic molecules, achieving chemical accuracy. This result could be obtained because some distinct aspects/approaches were taken into account: (i) using accurate and reliable experimental data (as the GC method is a so-called data-driven model), with experimental data being used to parametrize the model, our self-imposed requirement of chemical accuracy means that the quality of the experimental data is preeminent), (ii) optimizing parameters group by group and introducing an absolute minimum number of additional group-specific parameters related to nearest- or next-nearest-neighbour interactions, (iii) recognition of the limitations of the GC approach, i.e., the breakdown related to the conditions of linearity and additivity that can arise as a result of steric hindrance, ring strain, geminal effects or electronic conjugation effects and (iv) the judicious definition of chemical groups, e.g., a phenyl group rather than six individual aromatic carbon atoms. These recent results outperform previous parametrizations and show both a low absolute average deviation between experimental and model values as well as exceptionally few outliers.
To achieve wide applicability to a larger variety of organic molecules, further groups should be parametrized. However, for this to be possible under the constraint of chemical accuracy, we need many more experimental data, which, however, are scarce. Alternatively, ab initio quantum chemical calculations can be used to obtain heats of formation. The high-level calculations required cannot be applied to larger molecules due to the excessive computational expense, but calculations on smaller entities can be used to determine additional group contribution parameters. Whereas we will comment on the ab initio-type calculations in the present paper, the determination of additional group parameters for molecules that can be treated using the linear additive GC approach is not the main subject of the present paper.
This paper will be concerned with the issue of molecules for which the linear additive GC method breaks down, i.e., is not directly applicable without the consequence of large deviations. This can be the consequence of effects including steric hindrance, ring strain, geminal effects or electronic conjugation effects. We will foster workable solutions for how to cope with these, as previous attempts to handle this with higher-order contributions in the GC method have led to both overfitting as well as very substantial differences between experimental and GC model values for multiple species [4,5,10,11].
2. Experimental Data and Computational Methods
2.1. Experimental Data
The heat of formation is a property which is not measured directly, but a variety of approaches exist to obtain such data. It is common to report the gas-phase heat of formation at ambient pressure and room temperature, i.e., 298 K, and as many compounds are not gaseous at these conditions, this requires an additional action to obtain the gas phase equivalent heat of formation. The way in which the original data are measured and subsequently processed largely determines the quality of the heat of formation values. It should also to be mentioned that all of this by no means implies more recent data are more reliable than old data (more recent versus old publications). For details, we refer to the original experimental works quoted in the present paper. The experimental errors are commonly around 1–1.5 kJ/mol (see, e.g., [12]), though for some species, the error is indicated as being larger.
The GC method is a so-called data-driven model, with experimental data being used to parametrize the model, and the molecular property of interest is the sum of the individual properties of the molecular fragments j
and is thus based on the assumption that a molecule can be decomposed in molecular fragments which are in essence mutually independent. We did the utmost to exclusively use literature experimental data from a few sources and which are considered reliable by experimental thermodynamic experts. This also implies preferably larger data sets from experimental groups using the same measuring equipment, measuring protocol and data processing therewith having data which are at least internally consistent. Moreover, we have seen [7,8,9], as we will see in the present paper, that specific experimental data assigned as less reliable by experimental thermodynamic experts usually lead to a less good model (larger difference between model and experimental values). The limited availability of high-quality verified experimental data has made us accept other sources of experimental data as well, where critical evaluation was required to judge to what extent we are developing a proper model. Various papers in the field have used available databases as the source of experimental data, which is unlikely to lead to a good predictive model because of a larger number of inaccurate or unreliable data. In this context, a very interesting study was recently published by Chan [13] in which he assesses NIST database values with results from high-level quantum calculations. According to the results reported by Chan, the heats of formation of only less than 40% of all species included were found within chemical accuracy. Moreover, even a pretty normal organic molecule such as 2-methyl-4-methylene-1,3-dioxolane was found to be off by more than 100 kJ/mol.
ΔHf = ∑ Nj.ΔHf (j)
j = 1,N
j = 1,N
2.2. Computational Methods
There are various ways to calculate the heat of formation using ab initio methods, and we briefly discuss these as, in the end, we want to suggest a procedure describing how strained molecules can be handled best and most quickly while achieving (near) chemical accuracy. The main generic method is evaluating the heat of formation using the atomization energy method in which the following formula is used, as illustrated for hydrocarbons:
CmHn = mC + nH
This is thus simply a calculation based on the standard definition of the heat of formation of a compound being the sum of the enthalpy change of the reaction by which the compound is formed from the elements.
The Weizmann-n ab initio methods (Wn, n = 1–4) [14,15] are highly accurate composite theories devoid of empirical parameters. However, the high accuracy of Wn theories comes with the price of a significant computational cost, and only pretty small molecules can be handled. It was reported that the alternative G4 method (to be discussed below) is 28 times faster than the W1BD method [16], not considering memory requirements. Larger molecules (still small in an absolute sense) treated with high-level ab initio methods are usually those with high symmetry, which can be calculated much more efficiently by taking into account all symmetry elements, which very strongly reduces the computational demand. Whereas, e.g., the highly symmetrical benzene can be easily calculated at a very high level, a mono-substituted benzene might involve a much higher computational cost.
To alleviate the issue of computational expense, the computationally more feasible and for this purpose popular Gn methods [17,18,19] have been devised to calculate, in particular, also the heat of formation of a molecule. As the Wn methods, the Gn methods are in reality composite methods comprising a series of calculations, including specific calculated corrections selected and adjusted in a way so that a set of reference experimental data is well accounted for. Starting with the theoretical atomic parameters, the program parameters were adjusted to give the lowest error for a large set of compounds, leading to an error of only 0.8 kcal/mol, i.e., 3.4 kJ/mol. This is a seemingly good result, but it should be realized that this composite method, in fact semi-empirical in nature, was devised such as to minimize this error. In addition, this was accomplished using a test set comprising a total number of entries in the G3/05 test set of 454 energies divided into subgroups and containing only 270 experimentally obtained enthalpies of formation (and, furthermore, 105 ionization potentials, 63 electron affinities, 10 proton affinities and 6 hydrogen-bonded complexes). Some are not organic molecules, and the molecule set consists of really small molecules, including species such as H2O, NH3, etcetera, so the number of species which is really useful to assess the quality regarding the heat of formation of realistic molecules is rather limited. Of course, molecules with non-standard geometries and thus non-standard bonding behaviour, such as heavily strained, are not included. Consequently, there is no ‘ab initio’ guarantee that G4 delivers quantitatively good results for an arbitrary molecule. On the other hand, neither does it mean that results are a priori not good, and for some indoles, very good results (within a few kJ/mol from experimental values) have been reported [20]. In this context, another very relevant paper by van der Spoel et al. [21] reports a larger number of G4 heats of formation.
Furthermore, smaller corrections ought to be applied when a bond is of the restricted rotor type; the C-C bond is a typical example of this type of bond to which this applies. You et al. have studied this explicitly for branched alkanes [22], see in particular Table S2 in the Supplementary Material in [22]. These corrections are small: for the linear alkanes, the effect of the hindered rotor, depending on the method of calculating, this effect (MS-T, PO-N, PI-N or PI-A) is about 0.4 kJ/mol for butane and 1.5 kJ/mol for heptane, where it is logical that more flexible bonds in longer alkanes will make the effect larger. The direction of this change is that dHf becomes less negative. For the singly branched alkanes, the effect was calculated as up to 1.4 kJ/mol for 3-ethylpentane. For the dimethylalkanes, the effect is very similar to that for the unbranched alkanes when comparing structures with the same main chain length (pentanes in You et al. [22]), though the effect is a little smaller with up to 1.2 kJ/mol. Thus, in summary, the effect is small but not totally negligible, and we will not take it into further consideration in the present paper.
Whereas we mentioned before that the heat of formation can be calculated using atomization energies, alternatively, one could adopt an approach which resembles a group contribution method but now based on ab initio calculations. Ring strain values were reported [23] for a series of 66 molecules using state-of-the-art ab initio methods, including W1BD, G4, CBS-APNO and CBS-QB3. In addition to the ring strain values, the G4-calculated heats of formation were obtained, both at 0 K and 298 K, with the latter being the values we need for comparison to the GC method and results. Whereas the different ab initio methods W1BD, G4, CBS-APNO and CBS-QB3 gave somewhat different numbers, which vary by as much as 4 kJ/mol with some exceptions exhibiting larger variation (8–12 kJ/mol for [2.2.2]propellane, cubane and [3.4.4.4]fenestrane), the mutual energy differences are sufficiently acceptable to fit with a model requiring chemical accuracy, but this assumes that these ab initio calculated values reflect true experimental data, something which needs to be substantiated and which will be pursued in the present work.
In summary, the problem for all ab initio methods is what was recently underpinned by Wiberg and Rablen [24]: despite some publications suggesting the contrary, it was noted that ‘Complete ab initio calculations of the heats of formation are very difficult and only have been carried out for some small molecules’. Consequently, even though for some species the G4 level has revealed good results, we should validate the accuracy and reliability for new classes of molecules, in particular when they are much larger than those in the test set or have specific characteristics, such as ring strain. Considering the way how the G4 method was developed and its performance according to various sources, the most promising way forward seems to be to validate, using reliable experimental data, the results for the particular class of interest before relying on the results.
In the present study, ab initio-type calculations were performed based on the G4 method [17,18,19], employing either Gaussian 09 (G09) [25] or Gaussian 16 (G16) [26]. For geometry optimization, force constants were calculated analytically, and tight convergence criteria were used (fopt = (calcfc, tight)). Structures were verified as minima on the potential energy surface via calculation of second derivatives (frequency calculation). For structures with multiple low-energy conformations, the conformational searching was performed manually, and subsequently, Boltzmann-averaged enthalpies were obtained. In Table S1 (Supplementary Material), we provide the optimized G4 structures (XZY coordinates) of the lowest energy conformations of all the cycloalkane and cycloalkene species studied in one list and the structures of all the other higher energy conformations in a separate list. Along with the structures, we have listed the G4 energies, ZPEs, enthalpy corrections, Gibbs free energy corrections and internal energy corrections. In two separate tables, we summarize the G4 enthalpies at 298 K, which are the quantities used in our analysis (Table S2).
The G4 data were converted into the enthalpies of formation in the way described by Wiberg and Rablen [24], which, in essence, follows the correction scheme proposed previously by Saeys et al. [27]. This procedure aims to correct for systematic deviation in the calculation of the atomization energies of the elements. Wiberg and Rablen accomplished this by applying
dHf(CmHnNpOw; 298 K) = dHG4(CmHn; 298 K) − mX − nY − pZ − qW
The term on the left is the computed enthalpy of formation, whereas the first term on the right-hand side is the direct G4 result; for further details see the original publication. X, Y, Z and W are the empirically corrected per-atom G4 enthalpies of C, H, N and O in their standard states. The numerical values of these pre-factors are determined by taking a series of equations, such as Equation (3), for which we have experimental values, and the left-hand term, available. For convenience, Equation (3) can be extended to include other atoms. This overcomes the problem of the systematic increase in the deviation in ab initio- and DFT-based methods in terms of the number of atoms because the atomic energies, which are the reference for the evaluation of the heat of formation from the elements, are not perfect. In other works [28], a similar approach was practised, but the correction was calibrated individually per class of molecules. Yet, other studies [21] have used the G4 results as they come directly from the calculations with the Gaussian program suite. The important corollary is that G4 data from different publications but for the same molecules might be different. The differences are in the kJ/mol range and are, therefore, relevant to the accuracy we want to achieve. Therefore, we have applied Equation (3) because it is the more generic and more reliable approach.
For comparison, even though the method would not be applicable to larger still molecules, we also provide the W1BD dHf values obtained by the Wiberg and Rablen procedure. In general, we found that the differences in the results obtained by the W1BD and G4 approaches were small: 1.6 kJ/mol average absolute deviation and 2.2 kJ/mol root mean square (rms) deviation between the two methods. Since neither set of values gave a noticeably and consistently better agreement with the experiment, we use the more easily calculated G4 values for analysis. The fact that the two different methods yield very similar results nonetheless lends further confidence to these results. The comparison also suggests that ~2 kJ/mol is a lower limit of the magnitude of the inherent error.
Density Functional Type (DFT) quantum calculations were performed using the Spartan program [29] and by invoking the B3LYP functional and the 6-311 + G** basis set.
3. Breakdown of the GC Approach When Achieving Chemical Accuracy
3.1. Ring Strain
Ring strain results from a combination of angle strain, conformational strain or Pitzer strain (torsional eclipsing interactions) and trans-annular strain, also known as van der Waals strain (or Prelog) strain. Ring strain is a typical non-linear effect and can therefore not be handled by a linear additive method. Whereas there is no direct way to measure the ring strain, it can be determined indirectly using combustion experiments. This would, however, require experimental heats of formation for all species of interest, which is unrealistic as well as expensive, whereas the aim is to have a well-performing GC method to predict the heat of formation without having all individual experimental data.
The strain energy is commonly evaluated as the difference between the energy of the strained molecule and the sum of the energies of the (unconstrained) constituting chemical groups, so in essence similar to the working of a group contribution method. For example, cyclohexane consists of six CH2 groups, the energy of a single CH2 being derived from the increment for a CH2 group as obtained from a larger series of linear alkanes. The strain energy can be obtained from ab initio calculations on the relevant chemical entities [23,24] or from a group contribution analysis based on the experimental data of the strained molecule and the group parameter values derived from the linear alkane series, viz. Refs. [1,7].
3.1.1. Ring Strain Evaluated from Group Contribution Involving Experimental Heats of Formation
Previously [7], excellent agreement between experimental and GC model values was obtained when adopting the values for the ring strain presented by Anslyn and Dougherty [30]. The averaged absolute deviation was 2.32 kJ/mol, and only methylcyclohexane had a value (5.77 kJ/mol) somewhat beyond chemical accuracy. Despite the good agreement we reported formerly, we need to have a closer look for several reasons. First of all, the text by Anslyn and Dogherty does not provide us with information on how these values were obtained, although this is not really surprising for a text book. Secondly, there are a lot more strained molecules, and to widen the applicability of the GC model, we would need to establish a good and unique procedure to evaluate the strain energy.
In Table 1, we have collected experimental data on cyclic molecules, preferably and many from reliable sources of experimental data. These include data on the cycloalkanes, alkylcyclopentanes and alkylcyclohexanes [31,32,33,34,35,36]. The complete set of literature references to all species considered is given in the Supplementary Material, Table S3. The Supplementary Material Table S3 comprises all G4-calculated data, as well as the W1BD results we will not discuss any further (see Section 2.2). We note that the NIST data base [12] provides a value for methylcyclobutane, namely −44.8 kJ/mol, which must be in error, firstly, when comparing to ethylcyclobutane and, secondly, because of the very large error when comparing to our model GC value adjusted by the G4-calculated ring strain value. This was reported to NIST, and we subsequently adopted an experimental value from another source in Table 1.
Next, regarding the group contribution approach, from the high-quality experimental data on the alkanes (paraffins), i.e., data from Rossini et al. [37], it was possible to obtain the CH2 group contribution parameter with a variation of less than 0.1 kJ/mol for the range from pentane up to and including eicosane (C5–C20) [7]. This group contribution parametrisation developed previously [1] can now be applied to the cyclic structures in Table 1. The calculated heat of formation values dHf = Σ ‘Group Contribution of constituting Groups’ are shown in column 4 in Table 1 (model dHf). As an example, within the context of the GC model, each cycloalkane is solely constituted of CH2 groups. Now, the dHf of the strained molecule which should correspond to the experimental value
dHf experimental (strained molecule) =
Σ Group Contribution of constituting Groups + strain energy
Σ Group Contribution of constituting Groups + strain energy
The strain energy can be corroborated by G4 quantum calculations. The sixth column in Table 1 comprises the G4-calculated strain energies for the various cycloalkanes and cycloalkenes, as obtained by the procedure outlined in [23].
The numbers in column 5, entitled ‘model-exp + G4 ring strain’, reveal that the accuracy with which the GC model enhanced with G4 strain energies resembles the experimental values. For the unsubstituted cycloalkanes (first four entries), we observe agreement within chemical accuracy. It is often stated that cyclohexane would have no ring strain, but whether this is genuinely true may require verification. Our current G4 calculations revealed a small but non-zero ring strain of 1.2 kJ/mol.
Whereas the older data from the Rossini group are considered very reliable, which was also underpinned by recent GC parametrisations [7,8,9], from experimental thermodynamics expertise one may argue whether the Wiberg and Finoglio data [38] are sufficiently reliable. Regarding the entries in Table 1, this concerns methylenecyclopropane, cyclopropane, cyclobutene and 1-methylcyclopropene (values in red in Table 1). Interestingly, however, we observe that when we use the G4-calculated heats of formation (column 7 in Table 1) for these four species in the calculations in column 5 of Table 1 (so the experiment is now replaced by the G4 value), we obtain much better and in essence good agreement with chemical accuracy for three of the four species and one slightly beyond (1-methylcyclopropene with −5.6 kJ/mol). Subsequently, one may now argue that we could, as we value the G4 results, replace all experimental values by corresponding the calculated G4 data. However, both G4 and experimental values have a typical error of a few kJ/mol, which is also the typical average difference between the G4 and experimental values in Table 1.
For the three methylenecycloalkanes, we have invoked the terminal C=C- bond GC parameter, next to the parameter for the CH2 group. We also observe results within chemical accuracy in column 5. It needs to be mentioned at this point that we have replaced the experimental value for methylenecyclopropane (201 kJ/mol, column 3) with the G4-calculated dHf (190.4 kJ/mol, last column) in the evaluation of the performance criterion ‘model-exp + G4 ring strain’ (column 5). As discussed above, the reason for this is that the experimental data reported by Wiberg et al. [38] (methylene cyclopropane, cyclopropene, cyclobutene, 1-methylcyclopropene) are to be considered less reliable than, e.g., the Rossini data.

Table 1.
Experimental and model heats of formation and ring strain. All values in kJ/mol. The model dHf values are the pure GC model values adopted from [1]. The ring strain (column 6) was evaluated from G4 quantum calculations following the procedure outlined previously [23]. The values in red in the last column are the G4-calculated heats of formation which replaced the experimental values from Wiberg and Fenoglio [38] in column 3 (see text for explanation). The values in column 5 ‘model-exp + G4 ring strain’ are the differences between the experimental values and, when not available, the G4-calculated dHf (these are all species for which the G4 dHf in column 7 are printed in italics), and our model corrected for the G4-calculated ring strain. The values in the last column, Final model GC − xp (or G4), are the differences between the experimental (and if not available, the G4-calculated) heat of formation and the GC value with the addition of the proposed fixed strain energy values per class (e.g., 102.5 kJ/mol for the substituted cyclobutanes), as discussed in Section 3.1.2. These numbers (last column) thus reveal the quality of the GC prediction as enhanced with the class typical strain energy values or, in other words, the accuracy with which the model can predict the heat of formation whilst adopting typical ring strain for each class.
Table 1.
Experimental and model heats of formation and ring strain. All values in kJ/mol. The model dHf values are the pure GC model values adopted from [1]. The ring strain (column 6) was evaluated from G4 quantum calculations following the procedure outlined previously [23]. The values in red in the last column are the G4-calculated heats of formation which replaced the experimental values from Wiberg and Fenoglio [38] in column 3 (see text for explanation). The values in column 5 ‘model-exp + G4 ring strain’ are the differences between the experimental values and, when not available, the G4-calculated dHf (these are all species for which the G4 dHf in column 7 are printed in italics), and our model corrected for the G4-calculated ring strain. The values in the last column, Final model GC − xp (or G4), are the differences between the experimental (and if not available, the G4-calculated) heat of formation and the GC value with the addition of the proposed fixed strain energy values per class (e.g., 102.5 kJ/mol for the substituted cyclobutanes), as discussed in Section 3.1.2. These numbers (last column) thus reveal the quality of the GC prediction as enhanced with the class typical strain energy values or, in other words, the accuracy with which the model can predict the heat of formation whilst adopting typical ring strain for each class.
| Cycloalkanes/Cycloalkenes | Exp. Rossini et al. [31,32,33,34,35,36] | Exp. Origin see Table S3 | Model dHf | Model-Exp + G4 Ring Strain | G4 Ring Strain | G4 dHf | Boltzmann-Averaged G4 dHf | Final Model-Exp. (or G4) |
|---|---|---|---|---|---|---|---|---|
| cyclopropane | 53.3 ± 0.6 | −61.89 | 0.2 | 115.4 | 53.7 | −0.6 | ||
| cyclobutane | 28.4 ± 0.6 | −82.52 | −0.9 | 110.0 | 27.8 | −0.3 | ||
| cyclopentane | −77.3 ± 0.8 | −103.15 | 3.6 | 29.4 | −73.4 | 0.7 | ||
| cyclohexane | −123.2 ± 0.8 | −123.78 | 0.6 | 1.2 | −121.1 | −4.2 | ||
| methylcyclopropane | 25.0 | −91.62 | −0.6 | 116 | 24.3 | −0.9 | ||
| ethylcyclopropane | 3.2 | −112.25 | −1.1 | 114.4 | 2.1 | 2.3 | 3.8 | |
| propylcyclopropane | −132.88 | 0.0 | 113.5 | −19.4 | −18.4 | 1.5 | ||
| isopropylcyclopropane | −137.98 | 4.3 | 113.2 | −29.1 | −28.4 | 6.1 | ||
| methylcyclobutane | −6.8 | −112.25 | 1.1 | 106.5 | −5.8 | −5.0 | −4.2 | |
| ethylcyclobutane | −26.3 | −132.88 | −1.8 | 104.8 | −28.1 | −27.1 | −2.5 | |
| propylcyclobutane | −153.51 | −0.1 | 103.7 | −49.7 | −47.9 | −1.5 | ||
| isopropylcyclobutane | −158.61 | 4.3 | 102.9 | −60.0 | −59.0 | 3.7 | ||
| methylcyclopentane | −106.7 ± 0.8 | −132.88 | 1.5 | 27.7 | −102.4 | −102.7 | −4.0 | |
| ethylcyclopentane | −127.1 ± 1.0 | −153.51 | 0.8 | 27.2 | −126.2 | −125.7 | −0.8 | |
| propylcyclopentane | −148.2 ± 1.3 | −174.14 | −0.1 | 25.8 | −148.1 | −147.4 | 0.5 | |
| isopropylcyclopentane | −179.24 | 4.2 | 27.5 | −155.9 | −153.6 | 3.2 | ||
| methylcyclohexane | −154.9 ± 1.0 | −153.51 | 0.0 | −1.4 | −154.8 | −2.2 | ||
| propylcyclohexane | −193.4 ± 1.3 | −194.77 | −2.4 | −0.9 | −195.4 | −194.3 | −3.0 | |
| isopropylcyclohexane | −199.87 | 4.1 | 6.4 | −197.6 | −197.5 | −5.9 | ||
| 1,1-dimethylcyclopropane | −8.2 ± 1.2 | −123.98 | 1.6 | 114.1 | −11.5 | 2.5 | ||
| cis-1,2-dimethylcyclopropane | 0.7 | −117.55 | 3.8 | 122.1 | 0.3 | −2.8 | ||
| trans-1,2-dimethylcyclopropane | −3.2 | −117.55 | 2.0 | 116.3 | −5.4 | 2.9 | ||
| 1,1-dimethylcyclobutane | −143.61 | 2.6 | 104.1 | −42.1 | 0.8 | |||
| cis-1,2-dimethylcyclobutane | −138.18 | 4.2 | 109.1 | −33.3 | −2.9 | |||
| trans-1,2-dimethylcyclobutane | −138.18 | 4.1 | 102.2 | −40.1 | 3.7 | |||
| cis-1,3-dimethylcyclobutane | −138.18 | 4.1 | 103.4 | −38.9 | 3.0 | |||
| trans-1,3-dimethylcyclobutane | −138.18 | 4.2 | 106.9 | −35.5 | −0.4 | |||
| 1,1-dimethylcyclopentane | −138.3 ± 1.2 | −165.24 | −0.2 | 26.7 | −140.1 | −139.4 | −0.4 | |
| 1,2-dimethylcyclopentane cis | −129.5 ± 1.3 | −158.81 | 2.2 | 31.5 | −131.4 | −130.2 | −2.8 | |
| 1,3-dimethylcyclopentane trans | −135.9 ± 1.2 | −158.81 | 5.1 | 28 | −134.8 | 3.6 | ||
| 1,2-dimethylcyclopentane trans | −136.7 ± 1.3 | −158.81 | 3.3 | 25.4 | −137.5 | 4.4 | ||
| 1,3-dimethylcyclopentane cis | −133.7 ± 1.5 | −158.81 | 1.8 | 26.9 | −136 | 1.4 | ||
| cis-1,3-dimethylcyclohexane | −179.44 | 4.0 | −4 | −187.4 | 4.4 | |||
| trans-1,4-dimethylcyclohexane | −179.44 | 4.1 | −3.6 | −187.1 | 4.1 | |||
| cis-1,3,5-trimethylcyclohexane | −212.97 | 0.5 | −6.5 | −220 | 3.4 | |||
| methylenecyclopropane | 201 | 28.74 | −4.0 | 157.7 | 190.4 | |||
| methylenecyclobutane | 121.6 | 8.11 | 3.3 | 116.8 | 120.3 | |||
| methylenecyclopentane | 12 ± 1.1 | −12.52 | 2.6 | 27.1 | 10.0 | |||
| cyclopropene | 277 | 50.87 | −0.9 | 230.4 | 282.2 | |||
| cyclobutene | 157 | 30.24 | −0.1 | 130.1 | 160.4 | |||
| cyclopentene | 33 | 9.61 | 1.9 | 25.3 | 35.1 | |||
| cyclohexene | −5.3 | −11.02 | 0.0 | 5.7 | −5 | |||
| 4-methylcyclopentene | −16.12 | 4.2 | 25.6 | 5.3 | 5.1 | |||
| 3-methylcyclopentene | 8 ± 2 | −16.12 | 0.4 | 24.5 | 5.1 | 5.7 | ||
| 1-methylcyclopentene | −4 ± 2 | −3.80 | −32.75 | −5.3 | 23.5 | −4.4 | ||
| 1-methylcyclopropene | 243.6 | 8.51 | −5.6 | 223.9 | 238.0 |
For the mono-substituted alkylcycloalkanes (methylcyclopropane up till isopropylcyclohexane), we observe agreement within chemical accuracy with the exception of isopropylcycloalkanes. However, when we introduce an additional nearest-neighbour interaction parameter of −4 kJ/mol species for all these mono-substituted alkylcycloalkanes, which we relate to an alkyl group attached to a cycloalkane, we observe agreement within chemical accuracy for all named species. Only the two isopropylcycloalkanes are at the edge of chemical accuracy. However, recognizing that the experimental values are averaged values over the possible conformations at a given temperature, Boltzmann averaging of various conformations (the detailed figures are collected in Table S3 of the Supplementary Material), which we evaluated using the G4 ab initio approach, showed the agreement for isopropylcyclopentane improves (column 5 in Table 1) by a further 2.3 kJ/mol (Boltzmann-averaged values in the one but final column of Table 1, −155.9 versus −153.6 kJ/mol. In addition, for isopropylcyclopropane, a small improvement (0.7 kJ/mol) is observed, bringing the model result within chemical accuracy, and a similar result is observed for isopropylcyclobutane.
One may wonder now why 1,1-dimethylcyclopropane reveals good agreement (column 5) without the –4 kJ/mol nearest-neighbour correction. However, when we take this correction twice (2 methyl substituents) and then add the correction of +11 kJ/mol for two methyl groups on the same carbon atoms (see Scheme 1, which was taken from Ref. [8]), we see practically a cancellation (+2*(-4) + 11), and the same applies to 1,1-dimethylcyclopentane. These neighbour interactions were taken into account in the results shown in Table 1.
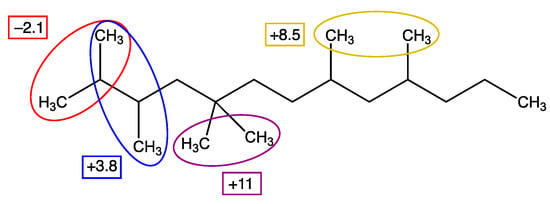
Scheme 1.
Illustration of the corrections accounting for the methyl–methyl interactions we have introduced (taken from [8]).
For the 1,2-dimethylcycloalkanes, we should add the −4 kJ/mol correction twice but in addition to the +3.8 kJ/mol correction set earlier (Scheme 1), leading to an overall contribution of −4.2 kJ/mol. For cis-1,3,5-trimethylcyclohexane, along the same arguments and quantitative corrections, we find +3 × (−4) = −12 kJ/mol. The Me-Me-1,5 interactions we considered previously in methyl-substituted linear alkanes [2] are different from those in the cyclic alkanes as, due to the cyclic structure, the Me groups are much further apart, and consequently, the −8.5 kJ/mol correction (see Scheme 1) is not expected to apply. This procedure leads to very satisfactory and consistent results, viz. Table 1.
Originally, we obtained larger deviations between the experimental and the model values (including the G4 strain energy) for the cycloalkenes, with an average difference of around 9 kJ/mol. In our model, we had taken one of the C=C GC parameters we had set in our previous studies, but all of these did not lead to good agreement for the cycloalkenes. However, there is no independent reason why one of these C=C GC parameters is also the appropriate one for the C=C in a (strained) ring, and therefore, we may need to introduce an additional GC parameter. With the value GCC=C ring of +71.5 kJ/mol, we obtain very good agreement between experimental and model values. On the other hand, methods involving higher-order group contribution, such as one of the more recent approaches due to Marrero and Gani [4,14], only include the C-C and C=C group contribution and thus neglect the ring strain, and consequently, the difference with the experimental values is enormous for the high ring strain species cyclopropene and cyclobutene. The Constantinou and Gani approach [11,39] does a better job as it has a three-membered ring as the group representing ring strain. However, this does not account for the significant differences in ring strain upon substitution of cyclopropane, viz. the G4 strain energies in Table 1 with which in conjunction with our previous GC model we obtain good agreement with the experiment.
3.1.2. Discussion and Conclusions on How to Tackle Ring Strain
When looking at the data collected in Table 1, two conclusions can be drawn straightforwardly: (i) our G4-calculated heats of formation are in good agreement with the reliable experimental data (when available) and (ii) our GC model in combination with the G4-calculated strain energies also agrees very well with the available experimental values, and this is generally within chemical accuracy.
These are highly satisfactory results, but we now recall that the goal of this work is to obtain a GC model with chemical accuracy and, at the same time, a method which delivers instantaneous answers to the process developer, as it is common for GC models and does not require expertise to perform quantum calculations. So, what we are now looking for are more generic trends based on the previous analysis including G4-calculated data, which could help us predict ring strain without having to resort to quantum calculations.
When we take all data in Table 1 into account, in particularly also taking into account the G4-calculated data, we can observe the following:
- -
- The isopropylcycloalkanes seem systematically off the GC prediction by 4.2 kJ/mol (Table 1, column 5). The Boltzmann-averaged G4 enthalpies did show some improvement but not for all. One could introduce an additional parameter correcting all isopropylalkanes by 4.2 kJ/mol and obtain very good agreement between the model and the experiment. However, as the deviation is not much beyond chemical accuracy and we do not have more than four experimental values, it seems more appropriate to suggest further studies including other substituted cycloalkanes before introducing an additional parameter. Therefore, at present, the isopropylalkanes are excluded from the conclusions on the other cycloalkanes discussed below.
- -
- For cyclopropane, a single alkyl substitution has a small influence on the ring strain, which is around 115 kJ/mol independent of the alkyl chain length. When we consider all cyclopropanes in Table 1, we do not see very large deviations. It should be mentioned here that, in part, we cannot compare with experimental data and need to rely on a mix of experimental and G4-calculated data. When we take the GC model value and add a ring strain of 115 kJ/mol for all substituted cyclopropanes, we obtain heats of formation within chemical accuracy from the G4-computed result. This even applies for cis-1,2-dimethylcyclopropane for which the G4 ring strain as such was calculated as 122 kJ/mol and therewith is clearly larger than for all other cyclopropanes.
- -
- For cyclobutane, the ring strain slowly drops with the lengthening of the alkyl chain, 6 kJ/mol from cyclobutane up till propylcyclobutane, but levels off with longer alkyl chain length. For the mono- and di-methyl-substituted cyclobutanes, the ring strain is roughly constant and around 103 kJ/mol. Again, the exception is cis-1,2-dimethylcyclobutane, with a G4-calculated ring strain of 109.1 kJ/mol being somewhat higher than those for other substituted cyclobutanes, but the overall result is still, albeit just, within chemical accuracy. The GC model does not (yet) discriminate between cis and trans in the current context. Still, when we take the GC model value and add a ring strain of 102.5 kJ/mol for the substituted cyclobutanes, we obtain a heat of formation within chemical accuracy from the G4-computed result. Only cyclobutane itself needs to be considered separately, but this is not a problem because for the isolated species, we can adopt the experimental value anyway.
- -
- For cyclopentane, the ring strain is almost constant with the lengthening of the alkyl chain.
- -
- For both the mono- as well as the dimethylcyclopentanes, we see pretty good agreement (column 4 in Table 1), but the G4 strain energies vary. Here, we observed a somewhat higher value for the G4-calculated ring strain for cis-1,2-dimethylcyclopentane. When we add to the pure GC approach, a strain energy of 26.5 kJ/mol throughout, we obtain chemical accuracy for all named species. Note that for most species, we rely on available experimental heat of formation data.
- -
- For the alkyl-substituted cyclohexanes, we observe similar trends as for the cyclopentanes: the results presented in Table 6 in [7] reveal, upon considering our GC model, that we find a very constant ring strain of −2 kJ/mol (note that it is indeed a minus sign!) for the series methylcyclohexane up till tetradecylcyclohexane, compared to 0.4 kJ/mol for the parent cyclohexane. Interestingly, the G4 results also suggest a small but negative ring strain for n-alkylcyclohexanes (see Table 1).
When looking at the dimethylcyclohexanes in Table 1, the ring strain energies as derived from the G4 calculations lie in the range −3.6 till −6.5 kJ/mol, compared to +1.3 kJ/mol for the parent cyclohexane. More important is the observation, in view of our main goals of these studies, that when adopting −3.6 kJ/mol for the ring strain for the di- and trimethylcyclohexanes, we obtain agreement within chemical accuracy between the experimental and our GC values (viz. Table 1), except for isopropylcyclohexane (−5.9 kJ/mol).
In summary, by combining our GC model enhanced with the addition of fixed values for the strain energy for each cycloalkanes class (cyclopropanes, cyclobutanes, cyclopentanes, cyclohexanes) as just proposed, it was shown that we can obtain model heats of formation largely within chemical accuracy. A refinement could be possible, e.g., because for the mono-substituted cyclohexanes, the GC + ring strain value is more negative than either the available experimental or alternatively the G4 value, whereas it is more positive for the three di- and tri-substituted cyclohexanes. However, due to the scarcity of data, we do not consider this appropriate at this stage. However, having seen the good performance of the G4 method for these cycloalkanes, extension with possible definition of additional strain energy parameters is to be considered possible by performing further G4 calculations on other molecules in these families.
3.2. Selection of the Group Size: Problems with Systems Not Obeying ’Simple’ GC
This section is perhaps, formally speaking, not exactly about systems definitely not obeying a GC approach, but most GC approaches reported in the past have adopted small chemical groups, e.g., CH3, CH, etcetera, and applying a linear additive method does not lead to a generic GC method with chemical accuracy. We recall, being particularly relevant as an introduction to this section, that the group contribution method assumes the chemical groups to be independent, i.e., there are virtually no interactions between them and the molecular property value is the sum of the group values. For many of the classes of molecules we have reviewed in the previous three papers, the group contribution concept worked very well, and we also know this from the literature. Comparatively small additional effects could be treated as a correction, namely nearest- or next-nearest-neighbour interactions or a 1,5-methyl-methyl-interaction, as in methyl-substituted alkanes [8], 1,1- or 1,2-difluoro interactions, mono- up till tri-alkyl-substituted phenyl rings and naphthalenes.
One of the reasons we previously obtained good results, i.e., within chemical accuracy, was because we had chosen the appropriate size of certain groups. The phenyl ring was adopted as a group, whereas other approaches handled the phenyl ring as a collection of six aCH (aromatic CH) groups [4,10,39] or as =CH)- groups [3]. Obviously, the latter choice has fewer groups and thus fewer parameters are involved but comes at the cost of insufficient accounting for effects between the groups and therewith not achieving chemical accuracy. The introduction of higher-order group contribution, as in [4] and [10], leads to a very large number of additional parameters, as there are many triple-group combinations, and consequently, the problem of overfitting arises (in essence such approaches are group-interaction models for which it is generally known that many more parameters are involved). For example, Kadda et al. [10], employing the MG approach [4], considered a data base of 750 molecules and employed just over 300 group parameters (115 first-order, 77 s-order, 36 third-order and 83 new groups) at a ratio of almost 2. Our previous studies [7,8,9] involved a total of 44 parameters, 35 groups and 9 neighbour-type interaction parameters on a total of 458 molecules, which gives a ratio of around 10. It is also important that the selection of these parameters was also based on chemical understanding of the parameters, e.g., the observation of germinal effects or the introduction of methyl–methyl interaction parameters, when we know these are both qualitatively expected as well as (semi-)quantitatively supported by quantum calculations. Thus, there is an explicit rationale behind each individual parameter, and we cannot obtain results within chemical accuracy with fewer parameters.
When we take into account chemical knowledge (the practical knowledge about molecules chemists have collected over many decades), we know, for instance, that there is a redistribution of the electron density in heterocycles, which is different between a phenyl ring which has a single N substitution (pyridine), two N substitutions (pyrimidine) or three N substitutions (triazine). The fact, and this is very important, that we have a non-discrete scale for electron distribution, therewith for bond strength and therewith for the heat of formation, inevitably implies that the additive character whilst adopting the smallest of groups, i.e., CH3, CH2, aromatic CH, etcetera, cannot hold.
The arguments put forward in favour of a less standard definition of the groups become even more relevant when we require chemical accuracy. In retrospect, what we introduced in our previous papers on the topic is in essence what Verevkin et al. [40] introduced as what they have called the ‘centerpiece’ approach: ‘The idea of this approach is to select a “centerpiece” molecule (e.g., benzene or methoxybenzene, or toluene or etc.) with the well-established thermodynamic properties. Various substituents (mostly relevant for the lignin are alkoxy, hydroxy and carbonyl substituents) can be attached to these “centerpieces” in different positions on the benzene ring’.
In our previous work, we presented the example of 1,3-dioxolane acetals [9], which revealed that adopting the appropriate size of a group representing the acetal leads to a model revealing chemical or close to chemical accuracy. Another very clear-cut example illustrating the need for well-chosen groups is the melamine molecule. Whereas molecules including pyridine, pyrimidine and triazine can be well represented by some GC methods [4,11] (though the GC parameters might have been determined very specifically on the basis of the experimental values for these compounds and then biased), for melamine, a GC prediction was 255 kJ/mol [4,11]. This value is the logical consequence of the value for triazine (226 kJ/mol [41,42]) complemented with three amino groups. Our current G4 result reads 42 kJ/mol, and we obtained a value of 43 kJ/mol from W1BD calculations. In addition, an older Quantum Monte Carlo result [43] revealed a much lower value (56.5 kJ/mol). Thus, whereas melamine seems to be a challenging molecule for GC methods, the true value is likely to be around 43 kJ/mol, based on ab initio data. Chemically, this is understandable due to the interaction of the amino nitrogen lone pairs with the heterocycle. Consequently, to be able to employ a GC approach successfully here we need to adopt melamine as a group and use a quantum mechanically calculated heat of formation as the numerical value associated with this group.
Quinoline is naphthalene but with one aromatic carbon replaced by a nitrogen, similar to the pair benzene–pyridine. In Table 2, we have collected experimental [28] and model heat of formation data. We have adopted both pyridine and quinoline themselves as new groups. The values for these two groups in the column model values were set at values which resulted in the best agreement for all other entries in the table. All other model values were calculated using the group values for pyridine and quinoline, enhanced with additional group contributions determined in our earlier work [7]. Except for 3,4-dimethylpyridine, all values are within chemical accuracy from the experimental value, and the absolute averaged deviation is 2.76 kJ/mol. We emphasize that we only needed two additional parameters, namely the group values for pyridine and quinoline, whereas other methods have introduced pyridine-specific substitution parameters, actually a separate parameter for each substitution pattern [5]. We would also like to note that the value of 197 kJ/mol which we fixed for the quinolone group compares very favourably with the G4 result of 198.1 kJ/mol reported by Verevkin et al. [28], similarly for our model value of 142 kJ/mol for pyridine compared to 141.3 kJ/mol from Verevkin et al. [28]. These results also indicate that we could have taken the G4 results for pyridine and quinoline as ‘Centerpiece’-related dHf values and subsequently obtained very good, mostly within chemical accuracy, predictions for the dHf for the substituted species presented in Table 2.
Table 2.
Experimental [28] and GC model heats of formation for selected pyridines and quinolones. All values in kJ/mol. Both pyridine and quinoline have been adopted as a group themselves. Therefore, the model values for these two, indicated with an *, are calculated GC values but set to a value such that the differences between experimental and GC model values (last column) for all substituted species are, whenever possible, within chemical accuracy. The pyridine data in [28] were obtained from [44]. The ethylpyridines are G4-calculated values were obtained from [28].
The overall conclusion of the present paper and the former three papers [7,8,9] is that, wherever a linear additive group contribution method is applicable, it is feasible to achieve chemical accuracy when using high-quality experimental data and the judicious definition of the groups. This includes taking phenyl as a group, as well as the examples provided above.
3.3. Steric Hindrance
In previous work [2], we considered many alkyl-substituted alkanes for which accurate and reliable experimental data are available. Most of these were not too very heavily branched but lightly branched and could be accounted for well by our GC model, which included mono-, di- and tri-alkyl-substituted alkanes, within chemical accuracy. In addition, tetra-substituted alkanes could still be described using the GC model when the substituents were not bonded to neighbouring carbon atoms, e.g., 2,2,5,5-tetramethylhexane was reproduced with a deviation from the experiment of 1.69 kJ/mol. In addition, 2,2,3-trimethyl-3-ethylpentane could be well described with a deviation of 3.4 kJ/mol and, thus, within chemical accuracy. However, for 2,2,3,3,4-pentamethylpentane and 3,3,4,4-tetraethylhexane, the deviations were significantly beyond chemical accuracy, 17 and 30 kJ/mol, respectively, despite the neighbour interaction parameters we had already defined based on alkyl-substituted alkanes, viz. Scheme 1, and including a specific ethyl substituent related parameter [2]. The same, a strong deviation from our GC model, was found for several alcohols, including 2,2,4,4-tetramethyl-3-iPr-3-pentanol.
As the range of potential steric interactions, including those exhibiting larger deviations when comparing the GC result with the experimental value, is so large, it is unrealistic to look for generic trends, e.g., as for the cycloalkanes and cases with ring strain. Thus, the GC approach cannot be applied here. Still, there is a need to predict properties, such as the heat of formation for molecules, for which this plays a non-negligible role. The direct evaluation of the heat of formation using ab initio calculations, e.g., the G4 or the Wn methods, is computationally prohibitive for larger molecules. Therefore, it is adequate to look for an alternative procedure to obtain information on the effect of high steric congestion on the heat of formation, i.e., look for a comparatively simple and straightforward procedure to capture the main effects. It would, however, have been an illusion that a simple ready-to-use procedure would be found by which we could obtain reliable and accurate heat of formation prediction for any congested system. Still, the minimum to be achieved, hopefully, is to have a strategy for how to assess whether the GC approach should be considered with utmost care, something which is not foreseen in the GC methods up till now.
We have, therefore, investigated whether Density Functional Theory (DFT) quantum calculations (B3LYP-type involving the 6-311 + G** basis set) can provide us with the insights we are looking for. This is an older density functional, being a mix of density functional character and the traditional Hartree–Fock character, which turned out to give good relative energies for organic molecules [45]. The idea is that one can compare differences in experimental heats of formation with differences in B3LYP-calculated total energies for the same pair of molecules. This can be illustrated in the case of Scheme 2. Whereas steric congestion is present in 3,3,4,4-tetraethylhexane, comparison with 2,2,5,5-tetraethylhexane (formally correct chemical name 2,6-di methyl-2,6-diethyldecane) will provide quantitative information on the effect of the congestion. When including all neighbour interaction parameters previously developed, the difference between the experimental and model values for 3,3,4,4-tetraethylhexane is still 69.6 kJ/mol and, thus, much off chemical accuracy. Just for comparison, the Marrero–Gani method [4], as implemented in ICAS23 [11], leads to 337 kJ/mol, which is 73 kJ/mol off the experimental value, a similar deviation. As reported earlier [2], we found a B3LYP-calculated difference between the two structures shown in Scheme 2 of 110.3 kJ/mol, compared to the 115.6 kJ/mol discrepancy between the experimental heat of formation [46] and the value according to the GC model with CH, CH2 and CH3 parameters only. This comparison is fair as both structures exhibit the same GC groups and the same parameters, including the additional neighbour-related parameters, as established in [2].
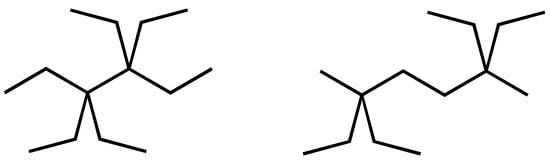
Scheme 2.
Chemical Structures for 3,3,4,4-tetraethylhexane (left) and ‘2,2,5,5-tetraethylhexane’, (formally correct chemical name 2,6-di methyl-2,6-diethyldecane (right).
This was, however, only a single example with a positive quantitative result. Before we apply these to the more heavily crowded system, we first need to validate the approach for less crowded molecules. Table 3 provides a series of results. The first entry actually corresponds to the 2.1 kJ/mol for 2-methyl substitution, i.e., interaction with terminal methyl group, as illustrated in Scheme 1. The difference between 2,3-dimethylhexane and 2,4-dimethylhexane corresponds to the 1,5-interaction with the value of 8.5 kJ/mol determined previously ([2], also see Scheme 1). These interaction parameter values were, however, determined from a larger range of branched alkanes and therefore deviate from the specific values for the hexanes in Table 2. Moreover, the DFT energies are bare energies; they do not include conformational averages, ZPE (zero-point energy) or finite temperature enthalpy corrections. In summary, it should be taken into account that the experimental heats of formation, G4-calculated heats of formation and B3LYP-evaluated energy differences will all have an uncertainty of a few kJ/mol. Despite this, the differences between values in the two columns in Table 3 are all within chemical accuracy and apply to both small energy differences as well as the larger values for the 2,2-dimethylhexane/2,3-dimethylhexane and the 2,2,5-trimethylhexane/2,3,4-trimethylhexane duos where we observe small differences, but this might be a coincidence. It thus seems that the B3LYP level of calculation may be well capable of capturing differences in energies for more congested and multiple methyl-substituted molecules which may be compared to differences in heat of formation. This means that the additional interaction parameters we defined previously [2] are well accounted for, if you wish supported, by the B3LYP calculations. Note that the heats of formation of all named molecules were also well accounted for by the GC model we had developed previously, within chemical accuracy.
Table 3.
Molecule pairs and the differences in heat of formation (experimental) and DFT total energies. All experimental values were referenced in [2].
A further interesting and relevant class exhibiting steric hindrance are phenyl-containing molecules including a t-butyl group. The simplest one is t-butylbenzene. The difference between the experimental heat of formation and our GC-model-based value is almost 15 kJ/mol. To investigate the effect of steric hindrance on the difference, we compared the two species depicted in Scheme 3. They have the same groups, and thus the GC-based heat of formation is initially the same. The B3LYP/6-311 + G** energy difference was calculated as 11.3 kJ/mol in favour of the p-ethyl-t-butylbenzene as, expectedly, the more stable species. When we add this value to the bare GC values for t-butylbenzene and 3- and 4-t-butyltoluene, we obtain good, within chemical accuracy, agreement between the experimental and model values. However, as the B3LYP is a good indication but not a good absolute value, by varying the interaction parameter, we found that 13.5 kJ/mol yielded the best agreement, viz. Table 4.
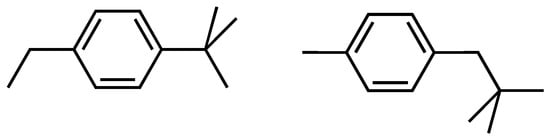
Scheme 3.
Structure of p-ethyl-t-butylbenzene and 4-tert-butyltoluene.
Table 4.
Experimental and model heats of formation for t-butylbenzenes. All values in kJ/mol. The model dHf values are based on the GC model we established previously with a steric contribution of 13.5 kJ/mol added (for explanation see text). For 2-tert-butyl-toluene, the correction due to steric interaction was initially calculated (B3LYP) as 26 kJ/mol in addition to the 13.5 kJ/mol for the other t-butylbenzenes (for explanation see text).
Not unexpectedly, the proximity of the methyl and the t-butyl group make 2-t-butyl-toluene behave differently. B3LYP calculations revealed a difference of 26 kJ/mol with 4-t-butyltoluene. By adding this value to the GC model result for 4-t-butyl-toluene (Table 4), we obtain a difference of just over 5 kJ/mol between the experimental and model value for 2-t-butyltoluene, just beyond chemical accuracy. In a similar type of analysis (not explicitly reported here), we were able to account for several mono- and di-t-butyl-phenols.
Before we discuss the interesting case of 2,2,4,4-tetramethyl-3-iPr-3-pentanol, a pretty congested molecule, we consider the related molecule 2,2,4,4-tetramethyl-3-pentanol for which we also have an experimental heat of formation [49]. The experimental value of −397 kJ/mol can be compared with the value of −430.1 kJ/mol according to our GC model [1,3], a difference of 33.1 kJ/mol. The fact that the GC value is more negative is in accordance with an anticipated van der Waals overlap (steric effects). What we have not accounted for in the GC approach is the possible influence of congestion or van der Waals overlap between the 2,3,4-substituted groups and the potential interaction between the hydroxyl group and the methyl groups, and therefore, it is not surprising that we see a difference between the experimental and the GC values in such a congested molecule. B3LYP quantum calculations revealed an energy difference of 29.6 kJ/mol between the two structures shown in Scheme 4, a value astonishingly close to the 33.1 kJ/mol difference we quoted above. Interactions between the OH and the methyl groups cannot be cancelled out, and B3LYP also has a limited accuracy in describing these systems. Still, we believe the good news is that in case we suspect steric hindrance of this kind, performing these quantum calculations and finding a clear energy difference will warn us against the straightforward use of a GC method.
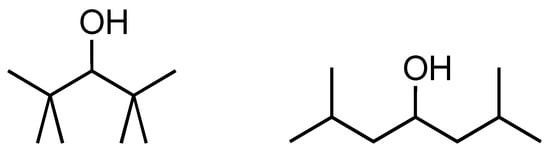
Scheme 4.
Chemical structures for 2,2,4,4-tetramethyl-3-pentanol (left) and ‘1,1,5,5-tetramethyl-3-pentanol’ (right); the latter name is formally incorrect but is used to indicate it is the structure with the methyl groups shifted by one carbon atom on each end.
The next case study is the very congested molecule 2,2,4,4-tetramethyl-3-iPr-3-pentanol, viz. Scheme 5, so the same molecule as just discussed but with the addition of an isopropyl group at the 3-position, making the molecule clearly more congested. This molecule was selected because it is one of the few of this kind of system for which we have a reliable experimental heat of formation [49].
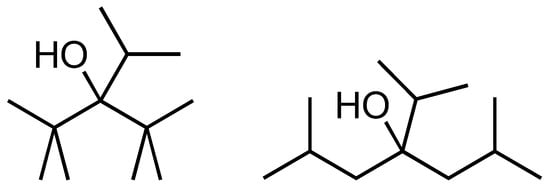
Scheme 5.
Chemical structures for 2,2,4,4-tetramethyl-iPr-3-pentanol (left) and ‘1,1,5,5-tetramethyl-iPr-3-pentanol’ (right), the latter name is formally incorrect but to indicate it is the structure with the methyl groups shifted by one carbon atom on each end.
The difference between the experimental value −418.1 kJ/mol and the GC value −509.1 kJ/mol (according to our previously developed model) is 91.7 kJ/mol. When we now compare the B3LYP calculations for 2,2,4,4-tetramethyl-3-iPr-3-pentanol and, as in the previous case, ‘1,1,5,5-tetramethyl-3-iPr-3-pentanol’, we find an energy difference of 51.5 kJ/mol. This value is much smaller than the 91.7 kJ/mol quoted before, but with the methyl groups at the 1 and 5 positions, they still have a clear interaction with the bulky isopropyl group and an underestimation of the steric effect is not surprising. When we increase the main chain length, e.g., compare 4,4,6,6-tetramethyl-5-iPr-5-nonanol (central part is 2,2,4,4-tetramethyl-3-iPr-3-pentanol) with 2,2,8,8-tetramethyl-5-iPr-5-nonanol, we find an energy difference of 136 kJ/mol. The large number of possible conformations does not allow us to determine the proper energy difference. Whereas we cannot use the B3LYP calculations to predict a reliable energy difference, the calculated difference provides us with a clear indication that we cannot trust the GC model value for the heat of formation.
Whereas B3LYP is an accepted good tool for calculations on organics in general, the van der Waals overlap and resulting dispersion interactions are not well accounted for. There are a variety of density functionals which have been designed to better account for these, and we have tested the ωB97X-D functional [50]. Interestingly, the energy difference for 2,2,4,4-tetramethyl-3-iPr-3-pentanol and 2,2,8,8-tetramethyl-5-iPr-5-nonanol decreased (compared to the B3LYP results) to 110 kJ/mol, which is much closer to the 91.7 kJ/mol difference between the experimental value and the GC result for 2,2,4,4-tetramethyl-3-iPr-3-pentanol. However, when we consider the previously considered pair, 2,2,4,4-tetramethyl-3-propanol (left) and ‘1,1,5,5-tetramethyl-3-propanol’, the ωB97X-D result compares less favourably compared to the B3LYP result.
In summary, we have seen that the named B3LYP quantum calculations are capable of reproducing energy differences between not very congested species within chemical accuracy, viz. Table 3. Whereas this, however, had already been achieved using the additional nearest-neighbour interaction parameters we had assigned earlier, viz. Scheme 1, these B3LYP results served to validate the B3LYP method for obtaining semi-quantitative estimates for steric effects. For truly congested systems, this level of quantum calculations can, when one selects appropriately similar but not identical molecules in which the steric hindrance is clearly much less (space filling model), provide numerical values which indicate that the GC approach cannot be applied. In some cases, e.g., the 2,2,4.4-tetramethyl-3-pentanol, the B3LYP value was in good quantitative agreement, but such a result cannot be used effectively to obtain the heat of formation if there is no way to check this independently if no experimental data are available. For t-butyl-substituted benzenes, we could assign an additional parameter, magnitude +13.5 kJ/mol, which is justified based on B3LYP calculations and which indeed brings the GC-calculated values to within chemical accuracy. For the more congested 2-t-butyltoluene with its additional t-Bu–methyl interaction, we could, likewise, calculate a steric energy contribution based on B3LYP calculations, and after adding this to the GC value, we obtained a heat of formation slightly beyond chemical accuracy. Altogether, these results are highly relevant, as other GC methods have never been reported in conjunction with measures to evaluate the validity in case of congestion, and heats of formation are often simply generated even when highly off the correct value. It is possible that other density functionals, in particular those better designed to account for van der Waals interactions, could do a better job, which would require a quantum chemical study involving various methods and more molecules to arrive at a possible quantitative procedure which is beyond the scope of the present paper.
4. Conclusions
In the previous three papers in this series, we developed a GC parametrization for various classes of organic molecules whilst achieving chemical accuracy, while in the present paper we have looked at three different groups of molecules which lead or may lead to problems when applying the GC method, which in essence is a linear and additive method.
The first class we considered in the present work were the alkyl-substituted cycloalkanes/enes for which we, based on G4 quantum calculations verified with respect to available experimental data, established strain energy contributions for each ring size to be added to the ‘pure’ GC expression in order to obtain accurate heats of formation. Next, we provided further examples (quinoline, pyridine, melamine) to show the need to adopt what Verevkin c.s. have called the ‘centerpiece approach,’ namely to define the group sufficiently large enough to capture electronic effects which would be otherwise impossible when the aim is to achieve a sufficiently high accuracy of the final heats of formation. Such a result opens the way to account for the enthalpy of formation for many more classes of molecules whilst exhibiting chemical accuracy. G4 calculations, preferably along with one molecule from the class for which an experimental value is available for validation, combined with the already established group parameter values [7,8,9], can provide the required dHf values.
The most difficult class of molecules is that with the most diverse, stronger steric interactions, i.e., strong van der Waals interactions. We reported on some highly branched alkanes and substituted benzenes and were able to assess, based on B3LYP calculations (which can be executed on a laptop), when a species has such strong interactions and therefore the GC method should be used with utmost care. As a by-product of this, we were able to establish an additional parameter for t-butyl phenyl type molecules.
Regarding further extension of the range of applicability, we have seen that G4-level quantum calculations can play a key role in obtaining further parameters for other cycloalkane/ene species as well as in determining the heat of formation for other ‘centerpiece’ fragments (when no reliable experimental value is available), e.g., establishing a GC-based approach with chemical accuracy for all heterocycles. It might also be investigated whether G4 calculations are capable of quantitatively capturing the correct heat of formation of fragments involving high steric congestion.
Finally, and this was also a recommendation by one of the referees, we have compiled all the GC parameter values we have collected thus far ([7,8,9] and the current manuscript) into a single document, which can be found in the Supplementary Material S4.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/thermo3020018/s1. Table S1: G4 calculated data on cycloalkanes and cycloalkenes. (a) Data related to the lowest energy structures. (b) Data related to all other conformations. Table S2: G4 and W1BD calculated data. Table S3: references to original papers quoting the heats of formation of various cycloalkanes. Table S4: overview of the GC parameter values developed in the present and the previous [7,8,9] three papers.
Author Contributions
Conceptualization, R.J.M.; methodology, R.J.M. and P.R.R.; software, P.R.R.; validation, R.J.M. and P.R.R.; formal analysis, R.J.M. and P.R.R. investigation, R.J.M. and P.R.R.; resources, P.R.R. and R.J.M.; data curation, R.J.M.; writing—original draft preparation, R.J.M.; writing—review and editing, P.R.R. and R.J.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article or the Supplementary Materials.
Acknowledgments
The authors gratefully acknowledge Sergey Verevkin for truly interesting and relevant discussions. The authors also sincerely thank Georgios Kontogeorgis, Gürkan Sin and Guoliang Wang (all Danish Technical University, DTU) for allowing the use and providing a copy of the ICAS23 software suite, particularly the ProPred module, which was, in part, used in this study.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The authors declare no conflict of interest.
References
- Van Krevelen, D.W.; Chermin, H.A.G. Estimation of the free enthalpy (Gibbs free energy) of formation of organic compounds from group contributions. Chem. Eng. Sci. 1951, 1, 66–80, Erratum in 1952, 1, 238. [Google Scholar] [CrossRef]
- Cohen, N.; Benson, S. Estimation of the heats of formation of organic compounds. Chem. Rev. 1993, 93, 2419–2438. [Google Scholar] [CrossRef]
- Joback, K.G.; Reid, R.C. Estimation of Pure-Component Properties from Group-Contributions. Chem. Eng. Commun. 1987, 57, 233–243. [Google Scholar] [CrossRef]
- Marrero, J.; Gani, R. Group-contribution based estimation of pure component properties. Fluid Phase Equilibria 2001, 183–184, 183–208. [Google Scholar] [CrossRef]
- Hukkerikar, A.S.; Meier, R.J.; Sin, G.; Gani, R. A method to estimate the enthalpy of formation of organic compounds with chemical accuracy. Fluid Phase Equilibria 2013, 348, 23–32. [Google Scholar] [CrossRef]
- Aouichaoui, A.R.N.; Fan, F.; Mansouri, S.S.; Abildskov, J.; Sin, G. Combining Group-Contribution Concept and Graph Neural Networks Toward Interpretable Molecular Property Models. J. Chem. Inf. Model. 2023, 63, 725–744. [Google Scholar] [CrossRef]
- Meier, R.J. Group contribution revisited: The enthalpy of formation of organic compounds with “chemical accuracy”. ChemEngineering 2021, 5, 24. [Google Scholar] [CrossRef]
- Meier, R.J. Group contribution revisited: The enthalpy of formation of organic compounds with “chemical accuracy” Part II. AppliedChem 2021, 1, 111–129. [Google Scholar] [CrossRef]
- Meier, R.J. Group contribution revisited: The enthalpy of formation of organic compounds with “chemical accuracy” Part III. AppliedChem 2022, 2, 213–228. [Google Scholar] [CrossRef]
- Kadda, A.; Mustapha, B.A.; Yahiaoui, A.; Khaled, T.; Hadji, D. Enthalpy of Formation Modeling Using Third Order Group Contribution Technics and Calculation by DFT Method. Int. J. Thermodyn. (IJoT) 2020, 23, 34–41. [Google Scholar] [CrossRef]
- ICS23 Software Package Which Includes the Marrero-Gani Method as Well as the Constantinou-Gani Approach and Also Provides Results Based on the Joback and Reid GC Method. Available online: https://www.kt.dtu.dk/english/research/kt-consortium/software (accessed on 21 February 2023).
- NIST Data Base. Available online: https://webbook.nist.gov/ (accessed on 16 March 2023).
- Chan, B. High-Level Quantum Chemistry Reference Heats of Formation for a Large Set of C, H, N, and O Species in the NIST Chemistry Webbook and the Identification and Validation of Reliable Protocols for Their Rapid Computation. J. Phys. Chem. A 2022, 126, 4981–4990. [Google Scholar] [CrossRef]
- Parthiban, S.; Martin, J.M.L. Assessment of W1 and W2 theories for the computation of electron affinities, ionization potentials, heats of formation, and proton affinities. J. Chem. Phys. 2001, 114, 6014. [Google Scholar] [CrossRef]
- Karton, A.; Daon, S.; Martin, J.M.L. W4-11: A high-confidence benchmark dataset for computational thermochemistry derived from first-principles W4 data. Chem. Phys. Lett. 2011, 510, 165–178. [Google Scholar] [CrossRef]
- Simmie, J.M.; Somers, K.P. Benchmarking compound methods (CBSQB3, CBS-APNO, G3, G4, W1BD) against the active thermochemical tables: A litmus test for cost-effective molecular formation enthalpies. J. Phys. Chem. A 2015, 119, 7235–7246. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 theory. J. Chem. Phys. 2007, 126, 084108. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian 4 theory using reduced order perturbation theory. J. Chem. Phys. 2007, 127, 1241. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, B.; Redfern, P.C.; Assary, R.S.; Curtiss, L.A. Accurate quantum chemical energies for 133,000 organic molecules. Chem. Sci. 2019, 10, 7449. [Google Scholar] [CrossRef] [PubMed]
- Konnova, M.E.; Li, S.; Bösmann, A.; Müller, K.; Peter Wasserscheid, P.; Andreeva, I.V.; Turovtzev, V.V.; Zaitsau, D.H.; Pimerzin, A.A.; Verevkin, S.P. Thermochemical Properties and Dehydrogenation Thermodynamics of Indole Derivates. Ind. Eng. Chem. Res. 2020, 59, 20539–20550. [Google Scholar] [CrossRef]
- Ghahremanpour, M.M.; van Maaren, P.J.; Ditz, J.C.; Lindh, R.; van der Spoel, D. Large-scale calculations of gas phase thermochemistry: Enthalpy of formation, standard entropy, and heat capacity. J. Chem. Phys. 2016, 145, 114305. [Google Scholar] [CrossRef]
- Gao, Y.; He, T.; Li, X.; You, X. Effect of hindered internal rotation treatments on predicting the thermodynamic properties of alkanes. Phys. Chem. Chem. Phys. 2019, 21, 1928–1936. [Google Scholar] [CrossRef]
- Rablen, P. A Procedure for Computing Hydrocarbon Strain Energies Using Computational Group Equivalents, with Application to 66 Molecules. Chemistry 2020, 2, 22. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Rablen, P.R. Increase in Strain Energy during Conversion of [4.4.4.5]Fenestrane to [4.4.4.4]Fenestrane: A Method for Estimating the Heats of Formation of Hydrocarbons and Their Derivatives from Ab Initio Energies. J. Org. Chem. 2020, 85, 4981–4987. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Peterson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Peterson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Saeys, M.; Reyniers, M.-F.; Marin, G.B.; VanSpeybroeck, V.; Waroquier, M. Ab Initio Calculations for Hydrocarbons: Enthalpy of Formation, Transition State Geometry, and Activation Energy for Radical Reactions. J. Phys. Chem. 2003, 107, 9147–9159. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Safronov, S.P.; Samarov, A.A.; Vostrikov, S.V. Hydrogen Storage: Thermodynamic Analysis of Alkyl-Quinolines and Alkyl-Pyridines as Potential Liquid Organic Hydrogen Carriers (LOHC). Appl. Sci. 2021, 11, 11758. [Google Scholar] [CrossRef]
- Wavefunction Inc. Spartan’10; Wavefunction Inc.: Irvine, CA, USA, 2010; Available online: https://www.wavefun.com/ (accessed on 21 February 2023).
- Anslyn, E.V.; Dougherty, D.A. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, USA, 2006; ISBN 1-891389-31-9. [Google Scholar]
- Knowlton, J.W.; Rossini, F.D. Heats of Combustion and Formation of Cyclopropane. J. Res. Natl. Bur. Stand. 1949, 43, 113–115. [Google Scholar] [CrossRef]
- Prosen, E.J.; Johnson, W.H.; Rossini, F.D. Heats of formation and combustion of the normal alkylcyclopentanes and cyclohexanes and the increment per CH2 group for several homologous series of hydrocarbons. J. Res. NBS. 1946, 37, 51–56. [Google Scholar] [CrossRef]
- Kilpatrick, J.E.; Werner, H.G.; Beckett, C.W.; Pitzer, K.S.; Rossini, F.D. Heats, Equilibrium Constants, and Free Energies of Formation of the Alkylcyclopentanes and Alkylcyclohexanes. J. Res. Natl. Bur. Stand. 1947, 39, 523–543. [Google Scholar] [CrossRef]
- Labbauf, A.; Rossini, F.D. Heats of combustion, formation, and hydrogenation of 14 selected cyclomonoolefin hydrocarbons. J. Phys. Chem. 1961, 65, 476–480. [Google Scholar] [CrossRef]
- Johnson, W.H.; Prosen, E.J.; Rossini, F.D. Heats of Combustion and Isomerization of the Six C7H14 Alkylcyclopentanes. J. Res. NBS 1949, 42, 251–255. [Google Scholar] [CrossRef]
- Good, W.D.; Moore, R.T.; Osborn, A.G.; Douslin, D.R. The enthalpies of formation of ethylcyclobutane, methylenecyclobutane, and 1,1-dimethylcyclopropane. J. Chem. Thermodyn. 1974, 6, 303–310. [Google Scholar] [CrossRef]
- Prosen, E.J.; Rossini, F.D. Heats of combustion and formation of the paraffin hydrocarbons at 25 °C. J. Res. Natl. Bur. Stand. 1945, 34, 263–269. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Fenoglio, R.A. Heats of formation of C4H6 hydrocarbons. J. Am. Chem. Soc. 1968, 90, 3395–3397. [Google Scholar] [CrossRef]
- Constantinou, L.; Gani, R. New group contribution method for estimating properties of pure compounds. AIChE J. 1994, 40, 1697–1710. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Vladimir, V.; Turovtsev, V.V.; Andreeva, I.V.; Orlovd, Y.D.; Pimerzin, A.P. Webbing a network of reliable thermochemistry around lignin building blocks: Tri-methoxybenzenes. RSC Adv. 2021, 11, 10727–10737. [Google Scholar] [CrossRef]
- Bystrom, K. The stabilization energy of 1,3,5-triazine derived from measurements of the enthalpies of combustion and sublimation. J. Chem. Thermodyn. 1982, 14, 865–870. [Google Scholar] [CrossRef]
- Iniguez, J.C.; Lopez, M.E.; McEachern, D.M. Vapor pressure and sublimation enthalpy of s-triazine. Rev. Soc. Quim. Mex. 1982, 26, 122–124. [Google Scholar]
- VanSpeybroeck, V.; Gani, R.; Meier, R.J. The calculation of thermodynamic properties of molecules. Chem. Soc. Rev. 2010, 39, 1764–1779. [Google Scholar] [CrossRef]
- Pedley, J.B.; Naylor, R.D.; Kirby, S.P. Thermochemical Data of Organic Compounds, 2nd ed.; Chapman & Hall: New York, NY, USA, 1986; pp. 1–792. ISBN 0412271001. [Google Scholar]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Emel’yanenko, V.N.; Diky, V.; Muzny, C.D.; Chirico, R.D.; Frenkel, M. New Group-Contribution Approach to Thermochemical Properties of Organic Compounds: Hydrocarbons and Oxygen-Containing Compounds. J. Phys. Chem. Ref. Data 2013, 42, 033102. [Google Scholar] [CrossRef]
- Prosen, E.J.; Johnson, W.H.; Rossini, F.D. Heats of combustion and formation at 25 °C of the alkylbenzenes through C10H14, and of the higher normal monoalkylbenzenes. J. Res. NBS 1946, 36, 455–461. [Google Scholar] [CrossRef]
- Brown, H.C.; Domash, L. Steric effects in displacement reactions. XI. The heats of reaction of diborane with pyridine bases. Steric strains in homomorphs of o-t-butyltoluene and hemimellitene. J. Am. Chem. Soc. 1956, 78, 5384–5386. [Google Scholar] [CrossRef]
- Roganov, G.N.; Pisarev, P.N.; Emel’yanenko, V.N.; Verevkin, S.P. Measurement and Prediction of Thermochemical Properties. Improved Benson-Type Increments for the Estimation of Enthalpies of Vaporization and Standard Enthalpies of Formation of Aliphatic Alcohols. J. Chem. Eng. Data 2005, 50, 1114–1124. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).