
Vascular NADPH Oxidases and Atherothrombotic Stroke



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. NADPH Oxidases in the Vessel Wall
3. NADPH Oxidases in Atherosclerosis
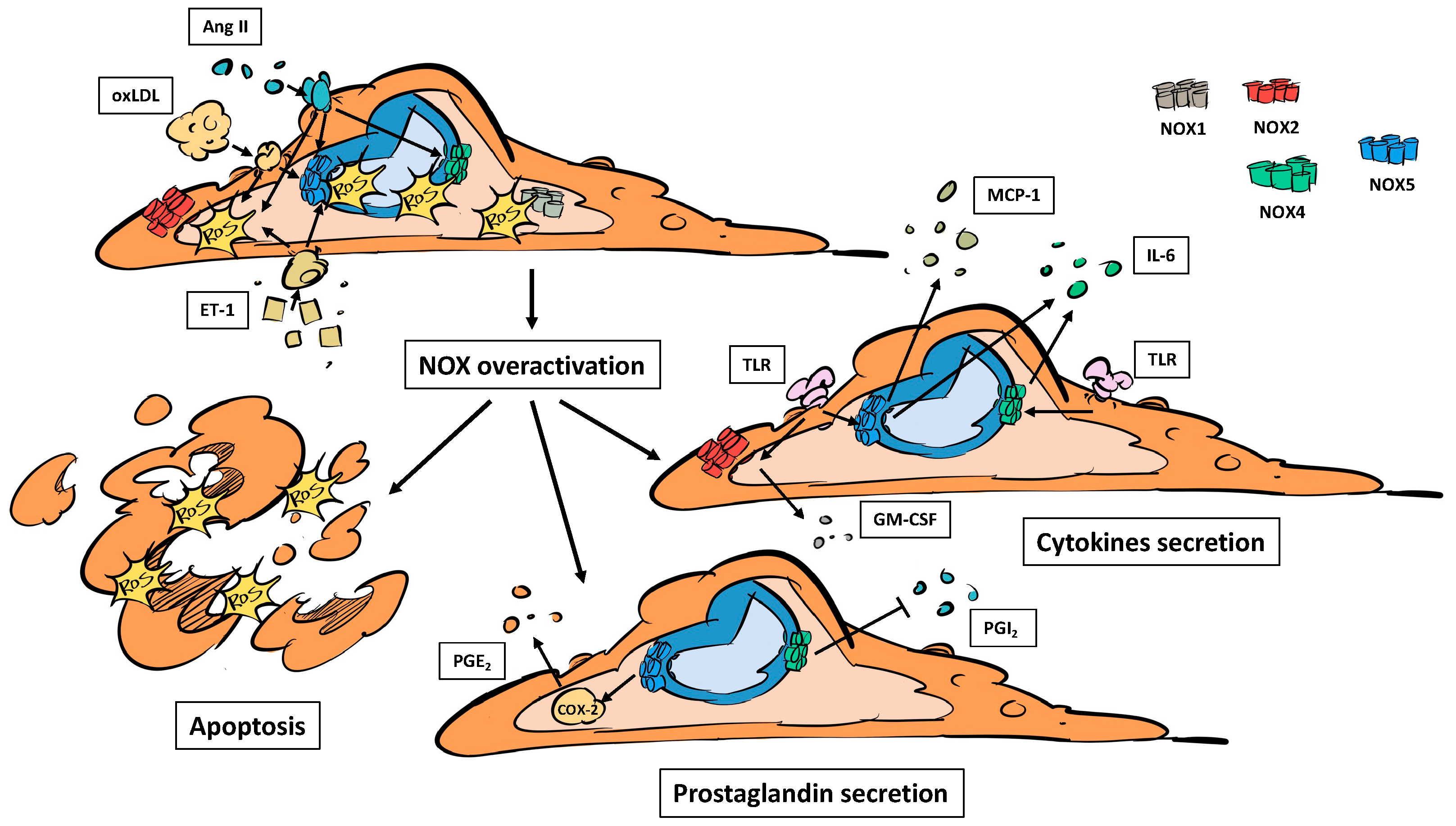
3.1. NADPH Oxidases, Endothelial Dysfunction and Inflammation
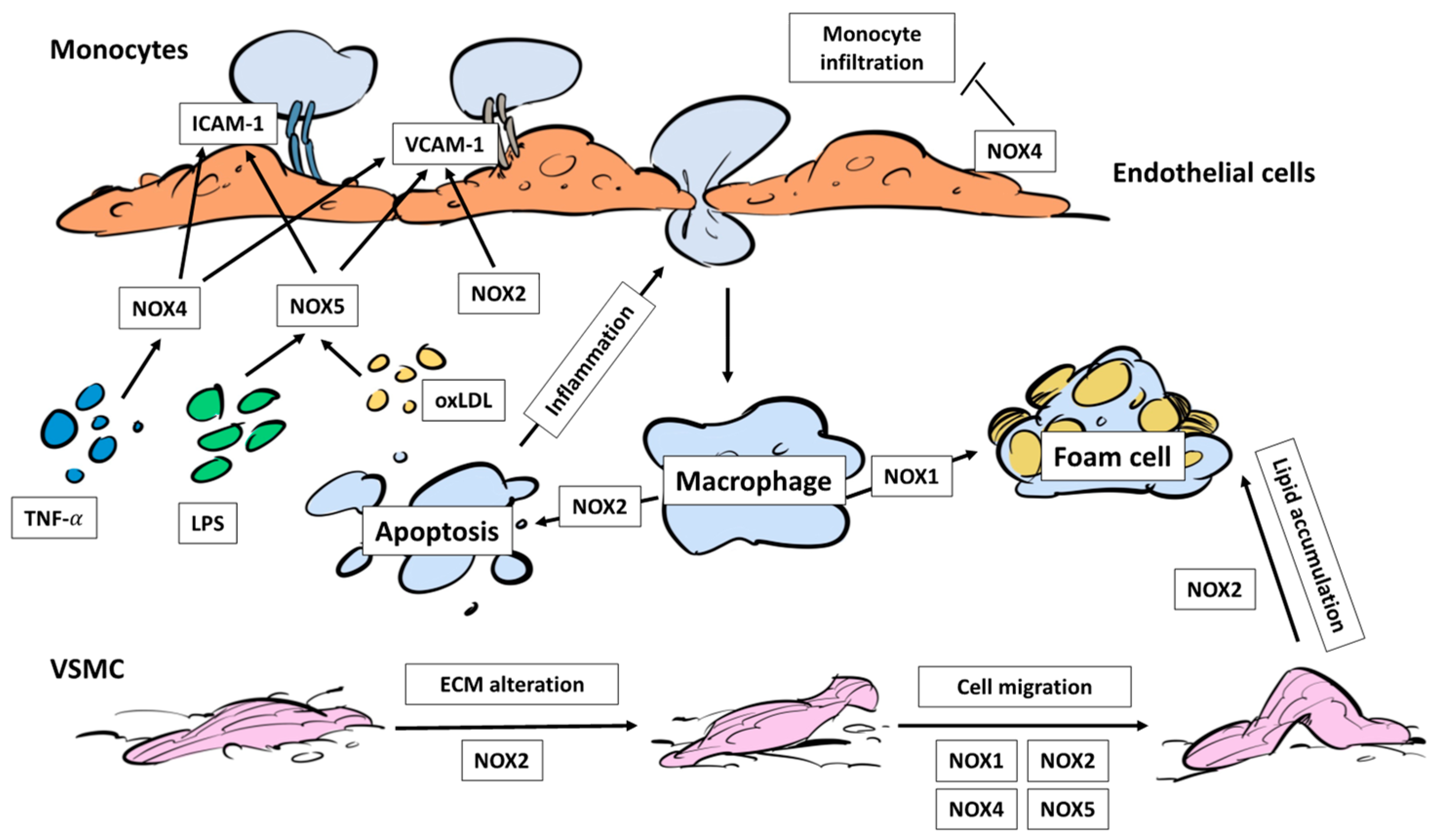
3.2. NADPH Oxidases, Immune Cell Infiltration and Foam Cells
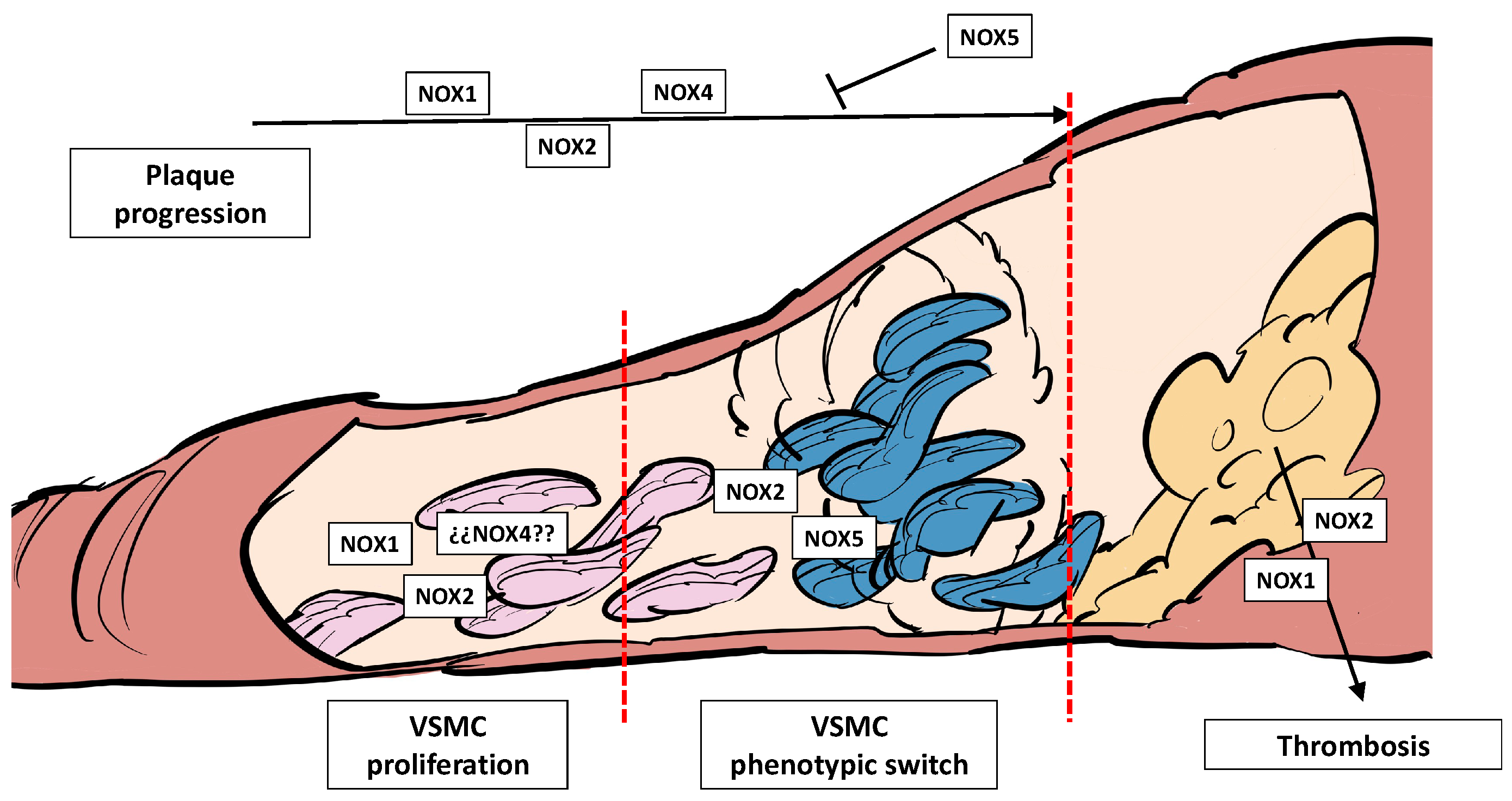
3.3. NADPH Oxidases, Plaque Development, Plaque Rupture, and Thrombosis
4. NADPH Oxidases in Thrombosis and Stroke
4.1. NADPH Oxidases, BBB Disruption and Stroke
4.2. NADPH Oxidases and Immune Infiltration in the Brain
4.3. NADPH Oxidases and Ischemia-Reperfusion Injury
5. Conclusions
6. Future Perspectives
- (i)
- More cell type-specific knock-out/knock-in in vivo models would improve the current knowledge. Most of the reviewed studies demonstrate that each individual NOX homolog plays a specific role in each cell type. Therefore, understanding which specific NOX is playing the pathological effect in each stage of the disease would help to address the situation;
- (ii)
- More integrative studies that delve deep into the interconnection between different NOXs or their paracrine effect in other cell types should be performed. As atherothrombosis involves different cell types in its pathogenesis, it should be interesting to use cocultures, organoids, or organ-on-chip devices mimicking the vessel wall to contextualize the effect of NOXs in a more complex biological system;
- (iii)
- There is an urgent need to develop isoform-specific NOX inhibitors and study these enzymes as potential therapeutical targets in CVDs [146]. These molecules have demonstrated efficacy in different diseases in vivo. For instance, NCATS-SM7270, a NOX2 inhibitor, protects from traumatic brain injury [147]. In fact, GKT137831, a NOX1/NOX4 inhibitor has reached phase 2 clinical trials, used to slow down diabetic kidney disease [148]. The combination of NOX-specific inhibitors with a cell-specific drug delivery system applied in target stages of the disease could be a promising therapeutic strategy not only in atherothrombosis but also in other CVDs.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xing, Y.; Lin, X. Challenges and Advances in the Management of Inflammation in Atherosclerosis. J. Adv. Res. 2024; online ahead of print. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxidative Med. Cell. Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [PubMed]
- Shioi, A.; Ikari, Y. Plaque Calcification During Atherosclerosis Progression and Regression. J. Atheroscler. Thromb. 2018, 25, 294–303. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, A.J.P.O.; De Oliveira, J.C.P.L.; Da Silva Pontes, L.V.; De Souza Júnior, J.F.; Gonçalves, T.A.F.; Dantas, S.H.; De Almeida Feitosa, M.S.; Silva, A.O.; De Medeiros, I.A. ROS: Basic Concepts, Sources, Cellular Signaling, and Its Implications in Aging Pathways. Oxid. Med. Cell. Longev. 2022, 2022, 1225578. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Ogboo, B.C.; Grabovyy, U.V.; Maini, A.; Scouten, S.; van der Vliet, A.; Mattevi, A.; Heppner, D.E. Architecture of the NADPH Oxidase Family of Enzymes. Redox Biol. 2022, 52, 102298. [Google Scholar] [CrossRef]
- Touyz, R.M.; Briones, A.M. Reactive Oxygen Species and Vascular Biology: Implications in Human Hypertension. Hypertens. Res. 2011, 34, 5–14. [Google Scholar] [CrossRef]
- Bánfi, B.; Malgrange, B.; Knisz, J.; Steger, K.; Dubois-Dauphin, M.; Krause, K.H. NOX3, a Superoxide-Generating NADPH Oxidase of the Inner Ear. J. Biol. Chem. 2004, 279, 46065–46072. [Google Scholar] [CrossRef]
- Bánfi, B.; Maturana, A.; Jaconi, S.; Arnaudeau, S.; Laforge, T.; Sinha, B.; Ligeti, E.; Demaurex, N.; Krause, K.H. A Mammalian H+ Channel Generated through Alternative Splicing of the NADPH Oxidase Homolog NOH-1. Science 2000, 287, 138–142. [Google Scholar] [CrossRef]
- Kobayashi, S.; Nojima, Y.; Shibuya, M.; Maru, Y. Nox1 Regulates Apoptosis and Potentially Stimulates Branching Morphogenesis in Sinusoidal Endothelial Cells. Exp. Cell Res. 2004, 300, 455–462. [Google Scholar] [CrossRef]
- Lassègue, B.; Sorescu, D.; Szöcs, K.; Yin, Q.Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel Gp91(Phox) Homologues in Vascular Smooth Muscle Cells: Nox1 Mediates Angiotensin II-Induced Superoxide Formation and Redox-Sensitive Signaling Pathways. Circ. Res. 2001, 88, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.K.; Choi, Y.G.; Baik, J.Y.; Han, S.Y.; Jeong, D.W.; Bae, Y.S.; Kim, N.; Lee, S.Y. A Crucial Role for Reactive Oxygen Species in RANKL-Induced Osteoclast Differentiation. Blood 2005, 106, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Griendling, K.K. Nox Proteins in Signal Transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct Subcellular Localizations of Nox1 and Nox4 in Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef]
- Niu, X.L.; Madamanchi, N.R.; Vendrov, A.E.; Tchivilev, I.; Rojas, M.; Madamanchi, C.; Brandes, R.P.; Krause, K.H.; Humphries, J.; Smith, A.; et al. Nox Activator 1: A Potential Target for Modulation of Vascular Reactive Oxygen Species in Atherosclerotic Arteries. Circulation 2010, 121, 549–559. [Google Scholar] [CrossRef]
- Tabet, F.; Schiffrin, E.L.; Callera, G.E.; He, Y.; Yao, G.; Östman, A.; Kappert, K.; Tonks, N.K.; Touyz, R.M. Redox-Sensitive Signaling by Angiotensin II Involves Oxidative Inactivation and Blunted Phosphorylation of Protein Tyrosine Phosphatase SHP-2 in Vascular Smooth Muscle Cells from SHR. Circ. Res. 2008, 103, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.C.; Daiber, A.; Kleschyov, A.L.; Mülsch, A.; Sydow, K.; Schulz, E.; Chen, K.; Keaney, J.F.; Lassègue, B.; Walter, U.; et al. Differential Effects of Diabetes on the Expression of the Gp91phox Homologues Nox1 and Nox4. Free Radic. Biol. Med. 2005, 39, 381–391. [Google Scholar] [CrossRef]
- Borregaard, N.; Heiple, J.M.; Simons, E.R.; Clark, R.A. Subcellular Localization of the B-Cytochrome Component of the Human Neutrophil Microbicidal Oxidase: Translocation during Activation. J. Cell Biol. 1983, 97, 52–61. [Google Scholar] [CrossRef]
- Van Buul, J.D.; Fernandez-Borja, M.; Anthony, E.C.; Hordijk, P.L. Expression and Localization of NOX2 and NOX4 in Primary Human Endothelial Cells. Antioxid. Redox Signal. 2005, 7, 308–317. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, L.; Yuan, W.; Sun, L.; Xia, Z.; Zhang, Z.; Yao, W. Diabetes Aggravates Myocardial Ischaemia Reperfusion Injury via Activating Nox2-Related Programmed Cell Death in an AMPK-Dependent Manner. J. Cell. Mol. Med. 2020, 24, 6670–6679. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Bartimoccia, S.; Nocella, C.; Di Santo, S.; Loffredo, L.; Illuminati, G.; Lombardi, E.; Boz, V.; Del Ben, M.; De Marco, L.; et al. LDL Oxidation by Platelets Propagates Platelet Activation via an Oxidative Stress-Mediated Mechanism. Atherosclerosis 2014, 237, 108–116. [Google Scholar] [CrossRef]
- Geiszt, M.; Kopp, J.B.; Várnai, P.; Leto, T.L. Identification of Renox, an NAD(P)H Oxidase in Kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar] [CrossRef]
- Ellmark, S.H.M.; Dusting, G.J.; Ng Tang Fui, M.; Guzzo-Pernell, N.; Drummond, G.R. The Contribution of Nox4 to NADPH Oxidase Activity in Mouse Vascular Smooth Muscle. Cardiovasc. Res. 2005, 65, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kitazono, T.; Ooboshi, H.; Iyama, T.; Han, Y.H.; Takada, J.; Wakisaka, M.; Ibayashi, S.; Utsumi, H.; Iida, M. Nox4 as the Major Catalytic Component of an Endothelial NAD(P)H Oxidase. Circulation 2004, 109, 227–233. [Google Scholar] [CrossRef]
- Lee, C.F.; Qiao, M.; Schröder, K.; Zhao, Q.; Asmis, R. Nox4 Is a Novel Inducible Source of Reactive Oxygen Species in Monocytes and Macrophages and Mediates Oxidized Low Density Lipoprotein-Induced Macrophage Death. Circ. Res. 2010, 106, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Stevenson, M.D.; Vendrov, A.E.; Hayami, T.; Robidoux, J.; Xiao, H.; Zhang, Y.Y.; Eitzman, D.T.; Runge, M.S.; Madamanchi, N.R. Increased Mitochondrial NADPH Oxidase 4 (NOX4) Expression in Aging Is a Causative Factor in Aortic Stiffening. Redox Biol. 2019, 26, 101288. [Google Scholar] [CrossRef]
- Perrotta, I.; Sciangula, A.; Perrotta, E.; Donato, G.; Cassese, M. Ultrastructural Analysis and Electron Microscopic Localization of Nox4 in Healthy and Atherosclerotic Human Aorta. Ultrastruct. Pathol. 2011, 35, 1–6. [Google Scholar] [CrossRef]
- Rajaram, R.D.; Dissard, R.; Jaquet, V.; De Seigneux, S. Potential Benefits and Harms of NADPH Oxidase Type 4 in the Kidneys and Cardiovascular System. Nephrol. Dial. Transplant. 2019, 34, 567–576. [Google Scholar] [CrossRef]
- Bánfi, B.; Molnár, G.; Maturana, A.; Steger, K.; Hegedûs, B.; Demaurex, N.; Krause, K.H. A Ca2+-Activated NADPH Oxidase in Testis, Spleen, and Lymph Nodes. J. Biol. Chem. 2001, 276, 37594–37601. [Google Scholar] [CrossRef]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; et al. Calcium Dependent NOX5 NADPH Oxidase Contributes to Vascular Oxidative Stress in Human Coronary Artery Disease. J. Am. Coll. Cardiol. 2008, 52, 1803. [Google Scholar] [CrossRef]
- Pai, W.Y.; Lo, W.Y.; Hsu, T.; Peng, C.T.; Wang, H.J. Angiotensin-(1-7) Inhibits Thrombin-Induced Endothelial Phenotypic Changes and Reactive Oxygen Species Production via NADPH Oxidase 5 Downregulation. Front. Physiol. 2017, 8, 994. [Google Scholar] [CrossRef] [PubMed]
- Petsophonsakul, P.; Burgmaier, M.; Willems, B.; Heeneman, S.; Stadler, N.; Gremse, F.; Reith, S.; Burgmaier, K.; Kahles, F.; Marx, N.; et al. Nicotine Promotes Vascular Calcification via Intracellular Ca2+-Mediated, NOX5-Induced Oxidative Stress, and Extracellular Vesicle Release in Vascular Smooth Muscle Cells. Cardiovasc. Res. 2022, 118, 2196–2210. [Google Scholar] [CrossRef]
- Manea, A.; Manea, S.A.; Gan, A.M.; Constantin, A.; Fenyo, I.M.; Raicu, M.; Muresian, H.; Simionescu, M. Human Monocytes and Macrophages Express NADPH Oxidase 5; a Potential Source of Reactive Oxygen Species in Atherosclerosis. Biochem. Biophys. Res. Commun. 2015, 461, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Anagnostopoulou, A.; Rios, F.; Montezano, A.C.; Camargo, L.L. NOX5: Molecular Biology and Pathophysiology. Exp. Physiol. 2019, 104, 605. [Google Scholar] [CrossRef]
- Anagnostopoulou, A.; Camargo, L.L.; Rodrigues, D.; Montezano, A.C.; Touyz, R.M. Importance of Cholesterol-Rich Microdomains in the Regulation of Nox Isoforms and Redox Signaling in Human Vascular Smooth Muscle Cells. Sci. Rep. 2020, 10, 17818. [Google Scholar] [CrossRef] [PubMed]
- Marzaioli, V.; Hurtado-Nedelec, M.; Pintard, C.; Tlili, A.; Marie, J.C.; Monteiro, R.C.; Gougerot-Pocidalo, M.A.; Dang, P.M.C.; El-Benna, J. NOX5 and P22phox Are 2 Novel Regulators of Human Monocytic Differentiation into Dendritic Cells. Blood 2017, 130, 1734–1745. [Google Scholar] [CrossRef]
- Richter, S.M.; Massman, L.C.; Stuehr, D.J.; Sweeny, E.A. Functional Interactions between NADPH Oxidase 5 and Actin. Front. Cell. Dev. Biol. 2023, 11, 1116833. [Google Scholar] [CrossRef]
- Marqués, J.; Cortés, A.; Pejenaute, Á.; Zalba, G. Implications of NADPH Oxidase 5 in Vascular Diseases. Int. J. Biochem. Cell Biol. 2020, 128, 105851. [Google Scholar] [CrossRef]
- Heitzer, T.; Baldus, S.; Von Kodolitsch, Y.; Rudolph, V.; Meinertz, T. Systemic Endothelial Dysfunction as an Early Predictor of Adverse Outcome in Heart Failure. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1174–1179. [Google Scholar] [CrossRef]
- Heitzer, T.; Schlinzig, T.; Krohn, K.; Meinertz, T.; Münzel, T. Endothelial Dysfunction, Oxidative Stress, and Risk of Cardiovascular Events in Patients with Coronary Artery Disease. Circulation 2001, 104, 2673–2678. [Google Scholar] [CrossRef] [PubMed]
- Yubero-Serrano, E.M.; Fernandez-Gandara, C.; Garcia-Rios, A.; Rangel-Zuñiga, O.A.; Gutierrez-Mariscal, F.M.; Torres-Peña, J.D.; Marin, C.; Lopez-Moreno, J.; Castaño, J.P.; Delgado-Lista, J.; et al. Mediterranean Diet and Endothelial Function in Patients with Coronary Heart Disease: An Analysis of the CORDIOPREV Randomized Controlled Trial. PLoS Med. 2020, 17, e1003282. [Google Scholar] [CrossRef] [PubMed]
- Pedralli, M.L.; Marschner, R.A.; Kollet, D.P.; Neto, S.G.; Eibel, B.; Tanaka, H.; Lehnen, A.M. Different Exercise Training Modalities Produce Similar Endothelial Function Improvements in Individuals with Prehypertension or Hypertension: A Randomized Clinical Trial Exercise, Endothelium and Blood Pressure. Sci. Rep. 2020, 10, 7628. [Google Scholar] [CrossRef]
- Li, M.; Liu, X.; He, Y.; Zheng, Q.; Wang, M.; Wu, Y.; Zhang, Y.; Wang, C. Celastrol Attenuates Angiotensin II Mediated Human Umbilical Vein Endothelial Cells Damage through Activation of Nrf2/ERK1/2/Nox2 Signal Pathway. Eur. J. Pharmacol. 2017, 797, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.Z.; Cheng, L.M.; Chen, X.F.; Li, Y.J.; Li, X.L.; Guan, Y.Y.; Du, Y.H. ClC-3 Promotes Angiotensin II-Induced Reactive Oxygen Species Production in Endothelial Cells by Facilitating Nox2 NADPH Oxidase Complex Formation. Acta Pharmacol. Sin. 2018, 39, 1725–1734. [Google Scholar] [CrossRef]
- Hong, O.K.; Lee, S.S.; Yoo, S.J.; Choi, S.H.; Lee, M.K.; Cha, B.Y.; Kim, M.K.; Baek, K.H.; Song, K.H.; Kwon, H.S. Effects of DA-9801 on the Inflammation and Apoptosis Induced by Angiotensin II in Human Dermal Microvascular Endothelial Cells. J. Pharmacol. Sci. 2021, 145, 52–59. [Google Scholar] [CrossRef]
- Montezano, A.C.; Burger, D.; Paravicini, T.M.; Chignalia, A.Z.; Yusuf, H.; Almasri, M.; He, Y.; Callera, G.E.; He, G.; Krause, K.H.; et al. Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 5 (NOX5) Regulation by Angiotensin II and Endothelin-1 Is Mediated via Calcium/Calmodulin-Dependent, Rac-1-Independent Pathways in Human Endothelial Cells. Circ. Res. 2010, 106, 1363. [Google Scholar] [CrossRef]
- Marqués, J.; Cortés, A.; Pejenaute, Á.; Ansorena, E.; Abizanda, G.; Prósper, F.; Martínez-Irujo, J.J.; de Miguel, C.; Zalba, G. Induction of Cyclooxygenase-2 by Overexpression of the Human NADPH Oxidase 5 (NOX5) Gene in Aortic Endothelial Cells. Cells 2020, 9, 637. [Google Scholar] [CrossRef]
- Su, E.; Zhao, L.; Yang, X.; Zhu, B.; Liu, Y.; Zhao, W.; Wang, X.; Qi, D.; Zhu, L.; Gao, C. Aggravated Endothelial Endocrine Dysfunction and Intimal Thickening of Renal Artery in High-Fat Diet-Induced Obese Pigs Following Renal Denervation. BMC Cardiovasc. Disord. 2020, 20, 176. [Google Scholar] [CrossRef]
- Valente, A.J.; Irimpen, A.M.; Siebenlist, U.; Chandrasekar, B. OxLDL Induces Endothelial Dysfunction and Death via TRAF3IP2: Inhibition by HDL3 and AMPK Activators. Free Radic. Biol. Med. 2014, 70, 117–128. [Google Scholar] [CrossRef]
- Chen, B.; Zhao, J.; Zhang, S.; Wu, W.; Qi, R. Aspirin Inhibits the Production of Reactive Oxygen Species by Downregulating Nox4 and Inducible Nitric Oxide Synthase in Human Endothelial Cells Exposed to Oxidized Low-Density Lipoprotein. J. Cardiovasc. Pharmacol. 2012, 59, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Li, C.; Gao, H.; Wu, Q.; Shi, J.; Chen, X. Dihydrotanshinone I Attenuates Atherosclerosis in ApoE-Deficient Mice: Role of NOX4/NF-ΚB Mediated Lectin-Like Oxidized LDL Receptor-1 (LOX-1) of the Endothelium. Front. Pharmacol. 2016, 7, 418. [Google Scholar] [CrossRef] [PubMed]
- Langbein, H.; Brunssen, C.; Hofmann, A.; Cimalla, P.; Brux, M.; Bornstein, S.R.; Deussen, A.; Koch, E.; Morawietz, H. NADPH Oxidase 4 Protects against Development of Endothelial Dysfunction and Atherosclerosis in LDL Receptor Deficient Mice. Eur. Heart J. 2016, 37, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.F.; Alves, J.V.; Silva-Neto, J.A.; Costa, R.M.; Neves, K.B.; Alves-Lopes, R.; Carmargo, L.L.; Rios, F.J.; Montezano, A.C.; Touyz, R.M.; et al. Lysophosphatidylcholine Induces Oxidative Stress in Human Endothelial Cells via NOX5 Activation-Implications in Atherosclerosis. Clin. Sci. 2021, 135, 1845–1858. [Google Scholar] [CrossRef]
- Sipkens, J.A.; Hahn, N.; van den Brand, C.S.; Meischl, C.; Cillessen, S.A.G.M.; Smith, D.E.C.; Juffermans, L.J.M.; Musters, R.J.P.; Roos, D.; Jakobs, C.; et al. Homocysteine-Induced Apoptosis in Endothelial Cells Coincides with Nuclear NOX2 and Peri-Nuclear NOX4 Activity. Cell Biochem. Biophys. 2013, 67, 341–352. [Google Scholar] [CrossRef]
- Cortés, A.; Pejenaute, Á.; Marqués, J.; Izal, Í.; Cenoz, S.; Ansorena, E.; Martínez-Irujo, J.J.; Miguel, C.; Zalba, G. Nadph Oxidase 5 Induces Changes in the Unfolded Protein Response in Human Aortic Endothelial Cells and in Endothelial-Specific Knock-in Mice. Antioxidants 2021, 10, 194. [Google Scholar] [CrossRef]
- Marqués, J.; Fernández-Irigoyen, J.; Ainzúa, E.; Martínez-Azcona, M.; Cortés, A.; Roncal, C.; Orbe, J.; Santamaría, E.; Zalba, G. NADPH Oxidase 5 (NOX5) Overexpression Promotes Endothelial Dysfunction via Cell Apoptosis, Migration, and Metabolic Alterations in Human Brain Microvascular Endothelial Cells (HCMEC/D3). Antioxidants 2022, 11, 2147. [Google Scholar] [CrossRef]
- Schuett, J.; Schuett, H.; Oberoi, R.; Koch, A.K.; Pretzer, S.; Luchtefeld, M.; Schieffer, B.; Grote, K. NADPH Oxidase NOX2 Mediates TLR2/6-Dependent Release of GM-CSF from Endothelial Cells. FASEB J. 2017, 31, 2612–2624. [Google Scholar] [CrossRef]
- Kim, J.; Seo, M.; Kim, S.K.; Bae, Y.S. Flagellin-Induced NADPH Oxidase 4 Activation Is Involved in Atherosclerosis. Sci. Rep. 2016, 6, 25437. [Google Scholar] [CrossRef]
- Yuan, S.; Hahn, S.A.; Miller, M.P.; Sanker, S.; Calderon, M.J.; Sullivan, M.; Dosunmu-Ogunbi, A.M.; Fazzari, M.; Li, Y.; Reynolds, M.; et al. Cooperation between CYB5R3 and NOX4 via Coenzyme Q Mitigates Endothelial Inflammation. Redox Biol. 2021, 47, 102166. [Google Scholar] [CrossRef]
- Hou, H.; Qin, X.; Li, G.; Cui, Z.; Zhang, J.; Dong, B.; Wang, Z.; Zhao, H. Nrf2-Mediated Redox Balance Alleviates LPS-Induced Vascular Endothelial Cell Inflammation by Inhibiting Endothelial Cell Ferroptosis. Sci. Rep. 2024, 14, 3335. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Dai, A.; Holterman, C.E.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R.; Jandeleit-Dahm, K.A.M. Endothelial or Vascular Smooth Muscle Cell-Specific Expression of Human NOX5 Exacerbates Renal Inflammation, Fibrosis and Albuminuria in the Akita Mouse. Diabetologia 2019, 62, 1712–1726. [Google Scholar] [CrossRef] [PubMed]
- Marques, P.; Collado, A.; Escudero, P.; Rius, C.; González, C.; Servera, E.; Piqueras, L.; Sanz, M.J. Cigarette Smoke Increases Endothelial CXCL16-Leukocyte CXCR6 Adhesion in Vitro and in Vivo. Potential Consequences in Chronic Obstructive Pulmonary Disease. Front. Immunol. 2017, 8, 1766. [Google Scholar] [CrossRef]
- Tai, Y.; Zhao, C.; Zhang, L.; Tang, S.; Jia, X.; Tong, H.; Liu, R.; Tang, C.; Gao, J. Celecoxib Reduces Hepatic Vascular Resistance in Portal Hypertension by Amelioration of Endothelial Oxidative Stress. J. Cell. Mol. Med. 2021, 25, 10389–10402. [Google Scholar] [CrossRef] [PubMed]
- González-Sierra, M.; Quevedo-Rodríguez, A.; Romo-Cordero, A.; González-Chretien, G.; Quevedo-Abeledo, J.C.; de Vera-González, A.; González-Delgado, A.; Martín-González, C.; González-Gay, M.Á.; Ferraz-Amaro, I. Relationship of Blood Inflammatory Composite Markers with Cardiovascular Risk Factors and Subclinical Atherosclerosis in Patients with Rheumatoid Arthritis. Life 2023, 13, 1469. [Google Scholar] [CrossRef]
- Chou, W.C.; Tsai, K.L.; Hsieh, P.L.; Wu, C.H.; Jou, I.M.; Tu, Y.K.; Ma, C.H. Galectin-3 Facilitates Inflammation and Apoptosis in Chondrocytes through Upregulation of the TLR-4-Mediated Oxidative Stress Pathway in TC28a2 Human Chondrocyte Cells. Environ. Toxicol. 2022, 37, 478–488. [Google Scholar] [CrossRef]
- Tsai, M.H.; Chi, M.C.; Hsu, J.F.; Lee, I.T.; Lin, K.M.; Fang, M.L.; Lee, M.H.; Lee, C.W.; Liu, J.F. Urban Particulate Matter Enhances ROS/IL-6/COX-II Production by Inhibiting MicroRNA-137 in Synovial Fibroblast of Rheumatoid Arthritis. Cells 2020, 9, 1378. [Google Scholar] [CrossRef]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New Insights into Oxidative Stress and Inflammation during Diabetes Mellitus-Accelerated Atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef]
- Goel, R.; Bhat, S.A.; Hanif, K.; Nath, C.; Shukla, R. Perindopril Attenuates Lipopolysaccharide-Induced Amyloidogenesis and Memory Impairment by Suppression of Oxidative Stress and RAGE Activation. ACS Chem. Neurosci. 2016, 7, 206–217. [Google Scholar] [CrossRef]
- Singh, S.; Bhowmick, D.C.; Pany, S.; Joe, M.; Zaghlula, N.; Jeremic, A.M. Apoptosis Signal Regulating Kinase-1 and NADPH Oxidase Mediate Human Amylin Evoked Redox Stress and Apoptosis in Pancreatic Beta-Cells. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1721–1733. [Google Scholar] [CrossRef]
- Murdoch, C.E.; Chaubey, S.; Zeng, L.; Yu, B.; Ivetic, A.; Walker, S.J.; Vanhoutte, D.; Heymans, S.; Grieve, D.J.; Cave, A.C.; et al. Endothelial NADPH Oxidase-2 Promotes Interstitial Cardiac Fibrosis and Diastolic Dysfunction through Proinflammatory Effects and Endothelial-Mesenchymal Transition. J. Am. Coll. Cardiol. 2014, 63, 2734–2741. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.J.; Zhao, M.X.; Liu, T.Y.; Ren, X.S.; Chen, Q.; Li, Y.H.; Kang, Y.M.; Zhu, G.Q. Salusin-β Induces Foam Cell Formation and Monocyte Adhesion in Human Vascular Smooth Muscle Cells via MiR155/NOX2/NFκB Pathway. Sci. Rep. 2016, 6, 23596. [Google Scholar] [CrossRef]
- Wang, M.; Murdoch, C.E.; Brewer, A.C.; Ivetic, A.; Evans, P.; Shah, A.M.; Zhang, M. Endothelial NADPH Oxidase 4 Protects against Angiotensin II-Induced Cardiac Fibrosis and Inflammation. ESC Heart Fail. 2021, 8, 1427–1437. [Google Scholar] [CrossRef]
- Xia, F.; Wang, C.; Jin, Y.; Liu, Q.; Meng, Q.; Liu, K.; Sun, H. Luteolin Protects HUVECs from TNF-α-Induced Oxidative Stress and Inflammation via Its Effects on the Nox4/ROS-NF-ΚB and MAPK Pathways. J. Atheroscler. Thromb. 2014, 21, 768–783. [Google Scholar] [CrossRef]
- Escudero, P.; De Marañón, A.M.; Collado, A.; Gonzalez-Navarro, H.; Hermenegildo, C.; Peiró, C.; Piqueras, L.; Sanz, M.J. Combined Sub-Optimal Doses of Rosuvastatin and Bexarotene Impair Angiotensin II-Induced Arterial Mononuclear Cell Adhesion through Inhibition of NOX5 Signaling Pathways and Increased RXR/PPARα and RXR/PPARγ Interactions. Antioxid Redox Signal. 2015, 22, 901–920. [Google Scholar] [CrossRef] [PubMed]
- Cortés, A.; Marqués, J.; Pejenaute, Á.; Ainzúa, E.; Ansorena, E.; Abizanda, G.; Prósper, F.; de Miguel, C.; Zalba, G. Endothelial NOX5 Overexpression Induces Changes in the Cardiac Gene Profile: Potential Impact in Myocardial Infarction? J. Physiol. Biochem. 2023, 79, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Albadawi, H.; Siddiquee, Z.; Stone, J.M.; Panchenko, M.P.; Watkins, M.T.; Stone, J.R. Altered Vascular Activation Due to Deficiency of the NADPH Oxidase Component P22phox. Cardiovasc. Pathol. 2014, 23, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.Q.; Li, X.Z.; Zhu, R.; Ge, R.; Wei, H.; Shi, H.W.; Wang, Z.; Yang, C.; Yang, Y.W.; Lu, X.J.; et al. Asprosin Contributes to Vascular Remodeling in Hypertensive Rats via Superoxide Signaling. J. Hypertens. 2024, 42, 1427–1439. [Google Scholar] [CrossRef]
- Valente, A.J.; Yoshida, T.; Murthy, S.N.; Sakamuri, S.S.V.P.; Katsuyama, M.; Clark, R.A.; Delafontaine, P.; Chandrasekar, B. Angiotensin II Enhances AT1-Nox1 Binding and Stimulates Arterial Smooth Muscle Cell Migration and Proliferation through AT1, Nox1, and Interleukin-18. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, 282–296. [Google Scholar] [CrossRef]
- Moraes, J.A.; Frony, A.C.; Dias, A.M.; Renovato-Martins, M.; Rodrigues, G.; Marcinkiewicz, C.; Assreuy, J.; Barja-Fidalgo, C. Alpha1beta1 and Integrin-Linked Kinase Interact and Modulate Angiotensin II Effects in Vascular Smooth Muscle Cells. Atherosclerosis 2015, 243, 477–485. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.F.; Li, X.X.; Xu, M. Contribution of Acid Sphingomyelinase to Angiotensin II-Induced Vascular Adventitial Remodeling via Membrane Rafts/Nox2 Signal Pathway. Life Sci. 2019, 219, 303–310. [Google Scholar] [CrossRef]
- Quesada, I.M.; Lucero, A.; Amaya, C.; Meijles, D.N.; Cifuentes, M.E.; Pagano, P.J.; Castro, C. Selective Inactivation of NADPH Oxidase 2 Causes Regression of Vascularization and the Size and Stability of Atherosclerotic Plaques. Atherosclerosis 2015, 242, 469–475. [Google Scholar] [CrossRef]
- Wang, H.F.; Yu, Q.Q.; Zheng, R.F.; Xu, M. Inhibition of Vascular Adventitial Remodeling by Netrin-1 in Diabetic Rats. J. Endocrinol. 2020, 244, 445–458. [Google Scholar] [CrossRef]
- Wu, J.H.; Zhang, L.; Nepliouev, I.; Brian, L.; Huang, T.; Snow, K.P.; Schickling, B.M.; Hauser, E.R.; Miller, F.J.; Freedman, N.J.; et al. Drebrin Attenuates Atherosclerosis by Limiting Smooth Muscle Cell Transdifferentiation. Cardiovasc. Res. 2022, 118, 772–784. [Google Scholar] [CrossRef]
- Csányi, G.; Feck, D.M.; Ghoshal, P.; Singla, B.; Lin, H.; Nagarajan, S.; Meijles, D.N.; Al Ghouleh, I.; Cantu-Medellin, N.; Kelley, E.E.; et al. CD47 and Nox1 Mediate Dynamic Fluid-Phase Macropinocytosis of Native LDL. Antioxid. Redox Signal. 2017, 26, 886–901. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Pagler, T.A.; Seimon, T.A.; Thorp, E.; Welch, C.L.; Witztum, J.L.; Tabas, I.; Tall, A.R. ABCA1 and ABCG1 Protect against Oxidative Stress-Induced Macrophage Apoptosis during Efferocytosis. Circ. Res. 2010, 106, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Han, W.; Villar, V.A.M.; Yang, Y.; Lu, Q.; Lee, H.; Li, F.; Quinn, M.T.; Gildea, J.J.; Felder, R.A.; et al. Unique Role of NADPH Oxidase 5 in Oxidative Stress in Human Renal Proximal Tubule Cells. Redox Biol. 2014, 2, 570–579. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Sumida, A.; Canugovi, C.; Lozhkin, A.; Hayami, T.; Madamanchi, N.R.; Runge, M.S. NOXA1-Dependent NADPH Oxidase Regulates Redox Signaling and Phenotype of Vascular Smooth Muscle Cell during Atherogenesis. Redox Biol. 2019, 21, 101063. [Google Scholar] [CrossRef] [PubMed]
- Al Ghouleh, I.; Rodríguez, A.; Pagano, P.J.; Csányi, G. Proteomic Analysis Identifies an NADPH Oxidase 1 (Nox1)-Mediated Role for Actin-Related Protein 2/3 Complex Subunit 2 (ARPC2) in Promoting Smooth Muscle Cell Migration. Int. J. Mol. Sci. 2013, 14, 20220–20235. [Google Scholar] [CrossRef]
- Abhijit, S.; Bhaskaran, R.; Narayanasamy, A.; Chakroborty, A.; Manickam, N.; Dixit, M.; Mohan, V.; Balasubramanyam, M. Hyperinsulinemia-Induced Vascular Smooth Muscle Cell (VSMC) Migration and Proliferation Is Mediated by Converging Mechanisms of Mitochondrial Dysfunction and Oxidative Stress. Mol. Cell. Biochem. 2013, 373, 95–105. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, J.; Wang, H. PGC-1α Limits Angiotensin II-Induced Rat Vascular Smooth Muscle Cells Proliferation via Attenuating NOX1-Mediated Generation of Reactive Oxygen Species. Biosci. Rep. 2015, 35, e00252. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, C.; Li, L.; Zhou, H.J.; Li, F.; Zhang, H.; Yu, L.; Chen, Y.; Min, W. Endothelial AIP1 Regulates Vascular Remodeling by Suppressing NADPH Oxidase-2. Front. Physiol. 2018, 9, 396. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, D.C.; Wosniak, J.; Gonçalves, R.C.; Tanaka, L.Y.; Fernandes, C.G.; Zanatta, D.B.; de Mattos, A.B.M.; Strauss, B.E.; Laurindo, F.R.M. PDIA1 Acts as Master Organizer of NOX1/NOX4 Balance and Phenotype Response in Vascular Smooth Muscle. Free Radic. Biol. Med. 2021, 162, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Aguado, A.; Fischer, T.; Rodríguez, C.; Manea, A.; Martínez-González, J.; Touyz, R.M.; Hernanz, R.; Alonso, M.J.; Dixon, D.A.; Briones, A.M.; et al. Hu Antigen R Is Required for NOX-1 but Not NOX-4 Regulation by Inflammatory Stimuli in Vascular Smooth Muscle Cells. J. Hypertens. 2016, 34, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Camargo, L.L.; Montezano, A.C.; Hussain, M.; Wang, Y.; Zou, Z.; Rios, F.J.; Neves, K.B.; Alves-Lopes, R.; Awan, F.R.; Guzik, T.J.; et al. Central Role of C-Src in NOX5- Mediated Redox Signalling in Vascular Smooth Muscle Cells in Human Hypertension. Cardiovasc. Res. 2022, 118, 1359–1373. [Google Scholar] [CrossRef]
- Furmanik, M.; Chatrou, M.; Van Gorp, R.; Akbulut, A.; Willems, B.; Schmidt, H.; Van Eys, G.; Bochaton-Piallat, M.L.; Proudfoot, D.; Biessen, E.; et al. Reactive Oxygen-Forming NOX5 Links Vascular Smooth Muscle Cell Phenotypic Switching and Extracellular Vesicle-Mediated Vascular Calcification. Circ. Res. 2020, 127, 911–927. [Google Scholar] [CrossRef]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; De Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH Oxidase 1 Plays a Key Role in Diabetes Mellitus-Accelerated Atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef]
- Ouerd, S.; Idris-Khodja, N.; Trindade, M.; Ferreira, N.S.; Berillo, O.; Coelho, S.C.; Neves, M.F.; Jandeleit-Dahm, K.A.; Paradis, P.; Schiffrin, E.L. Endothelium-Restricted Endothelin-1 Overexpression in Type 1 Diabetes Worsens Atherosclerosis and Immune Cell Infiltration via NOX1. Cardiovasc. Res. 2021, 117, 1144–1153. [Google Scholar] [CrossRef]
- Serino, A.; Zhao, Y.; Hwang, J.; Cullen, A.; Deeb, C.; Akhavan, N.; Arjmandi, B.; Salazar, G. Gender Differences in the Effect of Blackberry Supplementation in Vascular Senescence and Atherosclerosis in ApoE−/− Mice. J. Nutr. Biochem. 2020, 80, 108375. [Google Scholar] [CrossRef]
- Liu, J.; Sun, Q.; Sun, M.; Lin, L.; Ren, X.; Li, T.; Xu, Q.; Sun, Z.; Duan, J. Melatonin Alleviates PM2.5-Triggered Macrophage M1 Polarization and Atherosclerosis via Regulating NOX2-Mediated Oxidative Stress Homeostasis. Free Radic. Biol. Med. 2022, 181, 166–179. [Google Scholar] [CrossRef]
- Schürmann, C.; Rezende, F.; Kruse, C.; Yasar, Y.; Löwe, O.; Fork, C.; Van De Sluis, B.; Bremer, R.; Weissmann, N.; Shah, A.M.; et al. The NADPH Oxidase Nox4 Has Anti-Atherosclerotic Functions. Eur. Heart J. 2015, 36, 3447–3456. [Google Scholar] [CrossRef]
- Yu, W.; Xiao, L.; Que, Y.; Li, S.; Chen, L.; Hu, P.; Xiong, R.; Seta, F.; Chen, H.; Tong, X. Smooth Muscle NADPH Oxidase 4 Promotes Angiotensin II-Induced Aortic Aneurysm and Atherosclerosis by Regulating Osteopontin. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165912. [Google Scholar] [CrossRef] [PubMed]
- Vlad, M.L.; Manea, S.A.; Lazar, A.G.; Raicu, M.; Muresian, H.; Simionescu, M.; Manea, A. Histone Acetyltransferase-Dependent Pathways Mediate Upregulation of NADPH Oxidase 5 in Human Macrophages under Inflammatory Conditions: A Potential Mechanism of Reactive Oxygen Species Overproduction in Atherosclerosis. Oxid. Med. Cell. Longev. 2019, 2019, 3201062. [Google Scholar] [CrossRef]
- Ho, F.; Watson, A.M.D.; Elbatreek, M.H.; Kleikers, P.W.M.; Khan, W.; Sourris, K.C.; Dai, A.; Jha, J.; Schmidt, H.H.H.W.; Jandeleit-Dahm, K.A.M. Endothelial Reactive Oxygen-Forming NADPH Oxidase 5 Is a Possible Player in Diabetic Aortic Aneurysm but Not Atherosclerosis. Sci. Rep. 2022, 12, 11570. [Google Scholar] [CrossRef]
- Petheő, G.L.; Kerekes, A.; Mihálffy, M.; Donkó, Á.; Bodrogi, L.; Skoda, G.; Baráth, M.; Hoffmann, O.I.; Szeles, Z.; Balázs, B.; et al. Disruption of the NOX5 Gene Aggravates Atherosclerosis in Rabbits. Circ. Res. 2021, 128, 1320–1322. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.I.; Ahn, M.J.; Cho, H.H.; Cho, M.; Jun, J.H.; Kang, B.G.; Lim, S.Y.; Yoo, S.J.; Kim, M.R.; Kim, H.S.; et al. ANGPTL4 Stabilizes Atherosclerotic Plaques and Modulates the Phenotypic Transition of Vascular Smooth Muscle Cells through KLF4 Downregulation. Exp. Mol. Med. 2023, 55, 426–442. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.Y.; Zhao, W.X.; Li, F.D.; Guo, P.R.; Fan, Q.; Wu, X.F. NOX2 Inhibition Stabilizes Vulnerable Plaques by Enhancing Macrophage Efferocytosis via MertK/PI3K/AKT Pathway. Redox Biol. 2023, 64, 102763. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.; Frank, F.; Wolk, S.; Busch, A.; Klimova, A.; Sabarstinski, P.; Gerlach, M.; Egorov, D.; Kopaliani, I.; Weinert, S.; et al. NOX4 MRNA Correlates with Plaque Stability in Patients with Carotid Artery Stenosis. Redox Biol. 2022, 57, 102473. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Tarafdar, A.; Celikag, M.; Patinha, D.; Gulacsy, C.E.; Hounslea, E.; Warren, Z.; Ferreira, B.; Koeners, M.P.; Caggiano, L.; et al. NADPH Oxidase 1 Is a Novel Pharmacological Target for the Development of an Antiplatelet Drug without Bleeding Side Effects. FASEB J. 2020, 34, 13959–13977. [Google Scholar] [CrossRef]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential Roles of the NADPH-Oxidase 1 and 2 in Platelet Activation and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 846–854. [Google Scholar] [CrossRef]
- Sonkar, V.K.; Kumar, R.; Jensen, M.; Wagner, B.A.; Sharathkumar, A.A.; Miller, F.J.; Fasano, M.B.; Lentz, S.R.; Buettner, G.R.; Dayal, S. Nox2 NADPH Oxidase Is Dispensable for Platelet Activation or Arterial Thrombosis in Mice. Blood Adv. 2019, 3, 1272–1284. [Google Scholar] [CrossRef]
- Vara, D.; Mailer, R.K.; Tarafdar, A.; Wolska, N.; Heestermans, M.; Konrath, S.; Spaeth, M.; Renné, T.; Schröder, K.; Pula, G. NADPH Oxidases Are Required for Full Platelet Activation In Vitro and Thrombosis In Vivo but Dispensable for Plasma Coagulation and Hemostasis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 683. [Google Scholar] [CrossRef]
- Zhang, B.; Li, J. Phoenixin-14 Protects Human Brain Vascular Endothelial Cells against Oxygen-Glucose Deprivation/Reoxygenation (OGD/R)-Induced Inflammation and Permeability. Arch. Biochem. Biophys. 2020, 682, 108275. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.S.; Cha, E.H.; Ha, E.; Park, B.; Seo, J.H. GKT136901 Protects Primary Human Brain Microvascular Endothelial Cells against Methamphetamine-Induced Blood-Brain Barrier Dysfunction. Life Sci. 2020, 256, 117917. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, M.; Younger, D.; Ravula, A.R.; Alay, E.; Rama Rao, K.V.; Chandra, N. Synergistic Role of Oxidative Stress and Blood-Brain Barrier Permeability as Injury Mechanisms in the Acute Pathophysiology of Blast-Induced Neurotrauma. Sci. Rep. 2019, 9, 7717. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwarz, T.; Geis, C.; Kraft, P.; et al. Post-Stroke Inhibition of Induced NADPH Oxidase Type 4 Prevents Oxidative Stress and Neurodegeneration. PLoS Biol. 2010, 8, e1000479. [Google Scholar] [CrossRef]
- Jackman, K.A.; Miller, A.A.; Drummond, G.R.; Sobey, C.G. Importance of NOX1 for Angiotensin II-Induced Cerebrovascular Superoxide Production and Cortical Infarct Volume Following Ischemic Stroke. Brain Res. 2009, 1286, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Alwjwaj, M.; Kadir, R.R.A.; Bayraktutan, U. Outgrowth Endothelial Progenitor Cells Restore Cerebral Barrier Function Following Ischaemic Damage: The Impact of NOX2 Inhibition. Eur. J. Neurosci. 2022, 55, 1658–1670. [Google Scholar] [CrossRef]
- Namyen, J.; Permpoonputtana, K.; Nopparat, C.; Tocharus, J.; Tocharus, C.; Govitrapong, P. Protective Effects of Melatonin on Methamphetamine-Induced Blood-Brain Barrier Dysfunction in Rat Model. Neurotox. Res. 2020, 37, 640–660. [Google Scholar] [CrossRef]
- Yang, F.; Wang, Z.; Wei, X.; Han, H.; Meng, X.; Zhang, Y.; Shi, W.; Li, F.; Xin, T.; Pang, Q.; et al. NLRP3 Deficiency Ameliorates Neurovascular Damage in Experimental Ischemic Stroke. J. Cereb. Blood Flow Metab. 2014, 34, 660–667. [Google Scholar] [CrossRef]
- Casas, A.I.; Geuss, E.; Kleikers, P.W.M.; Mencl, S.; Herrmann, A.M.; Buendia, I.; Egea, J.; Meuth, S.G.; Lopez, M.G.; Kleinschnitz, C.; et al. NOX4-Dependent Neuronal Autotoxicity and BBB Breakdown Explain the Superior Sensitivity of the Brain to Ischemic Damage. Proc. Natl. Acad. Sci. USA 2017, 114, 12315–12320. [Google Scholar] [CrossRef]
- Cortés, A.; Solas, M.; Pejenaute, Á.; Abellanas, M.A.; Garcia-Lacarte, M.; Aymerich, M.S.; Marqués, J.; Ramírez, M.J.; Zalba, G. Expression of Endothelial NOX5 Alters the Integrity of the Blood-Brain Barrier and Causes Loss of Memory in Aging Mice. Antioxidants 2021, 10, 1311. [Google Scholar] [CrossRef]
- Qiu, Y.M.; Zhang, C.L.; Chen, A.Q.; Wang, H.L.; Zhou, Y.F.; Li, Y.N.; Hu, B. Immune Cells in the BBB Disruption After Acute Ischemic Stroke: Targets for Immune Therapy? Front. Immunol. 2021, 12, 678744. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Choi, J.H.; Oh, J.; Lee, Y.H.; In, J.G.; Chang, B.J.; Nah, S.Y.; Cho, I.H. Rg3-Enriched Korean Red Ginseng Extract Inhibits Blood-Brain Barrier Disruption in an Animal Model of Multiple Sclerosis by Modulating Expression of NADPH Oxidase 2 and 4. J. Ginseng. Res. 2021, 45, 433–441. [Google Scholar] [CrossRef]
- Lelli, A.; Gervais, A.; Colin, C.; Chéret, C.; de Almodovar, C.R.; Carmeliet, P.; Krause, K.H.; Boillée, S.; Mallat, M. The NADPH Oxidase Nox2 Regulates VEGFR1/CSF-1R-Mediated Microglial Chemotaxis and Promotes Early Postnatal Infiltration of Phagocytes in the Subventricular Zone of the Mouse Cerebral Cortex. Glia 2013, 61, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Haddad, Y.; Yun, H.J.; Geng, X.; Ding, Y. Induced Inflammatory and Oxidative Markers in Cerebral Microvasculature by Mentally Depressive Stress. Mediat. Inflamm. 2023, 2023, 4206316. [Google Scholar] [CrossRef]
- Li, R.; Yuan, Q.; Su, Y.; Chopp, M.; Yan, T.; Chen, J. Immune Response Mediates the Cardiac Damage after Subarachnoid Hemorrhage. Exp. Neurol. 2020, 323, 113093. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Davaanyam, D.; Seol, S.I.; Lee, H.K.; Lee, H.; Lee, J.K. Adenosine Triphosphate Accumulated Following Cerebral Ischemia Induces Neutrophil Extracellular Trap Formation. Int. J. Mol. Sci. 2020, 21, 7668. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, X.; Liu, L.; Liu, S.; Wang, Z.; Zhang, B.; Fan, B.; Yang, F.; Huang, S.; Jiang, F.; et al. TIPE2, a Novel Regulator of Immunity, Protects against Experimental Stroke. J. Biol. Chem. 2012, 287, 32546–32555. [Google Scholar] [CrossRef]
- Jin, R.; Song, Z.; Yu, S.; Piazza, A.; Nanda, A.; Penninger, J.M.; Granger, D.N.; Li, G. Phosphatidylinositol-3-Kinase Gamma Plays a Central Role in Blood-Brain Barrier Dysfunction in Acute Experimental Stroke. Stroke 2011, 42, 2033–2044. [Google Scholar] [CrossRef]
- Tuo, Y.H.; Liu, Z.; Chen, J.W.; Wang, Q.Y.; Li, S.L.; Li, M.C.; Dai, G.; Wang, J.S.; Zhang, Y.L.; Feng, L.; et al. NADPH Oxidase Inhibitor Improves Outcome of Mechanical Reperfusion by Suppressing Hemorrhagic Transformation. J. Neurointerv. Surg. 2017, 9, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Jian, Z.; Jin, T.; Li, Y.; Zeng, Z.; Zhang, X.; Xiong, X.; Gu, L. NOX2-Mediated Reactive Oxygen Species Are Double-Edged Swords in Focal Cerebral Ischemia in Mice. J. Neuroinflammation 2022, 19, 184. [Google Scholar] [CrossRef]
- Choi, D.H.; Kim, J.H.; Lee, K.H.; Kim, H.Y.; Kim, Y.S.; Choi, W.S.; Lee, J. Role of Neuronal NADPH Oxidase 1 in the Peri-Infarct Regions after Stroke. PLoS ONE 2015, 10, e0116814. [Google Scholar] [CrossRef]
- McCann, S.K.; Dusting, G.J.; Roulston, C.L. Nox2 Knockout Delays Infarct Progression and Increases Vascular Recovery through Angiogenesis in Mice Following Ischaemic Stroke with Reperfusion. PLoS ONE 2014, 9, e110602. [Google Scholar] [CrossRef] [PubMed]
- Zuo, M.L.; Wang, A.P.; Song, G.L.; Yang, Z.B. MiR-652 Protects Rats from Cerebral Ischemia/Reperfusion Oxidative Stress Injury by Directly Targeting NOX2. Biomed. Pharmacother. 2020, 124, 109860. [Google Scholar] [CrossRef]
- Liu, H.; Wei, X.; Kong, L.; Liu, X.; Liu, X.; Cheng, L.; Yan, S.; Zhang, X.; Chen, L. NOD2 Is Involved in the Inflammatory Response after Cerebral Ischemia-Reperfusion Injury and Triggers NADPH Oxidase 2-Derived Reactive Oxygen Species. Int. J. Biol. Sci. 2015, 11, 525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Han, H.; Zhou, Y.; Liu, Z.; Ma, T.; Cao, X. MicroRNA-454 Modulates the Oxidative Stress and Neuronal Apoptosis after Cerebral Ischemia/Reperfusion Injury via Targeting NADPH Oxidase 4 (NOX4). J. Biochem. Mol. Toxicol. 2022, 36, e23153. [Google Scholar] [CrossRef]
- Suzuki, Y.; Hattori, K.; Hamanaka, J.; Murase, T.; Egashira, Y.; Mishiro, K.; Ishiguro, M.; Tsuruma, K.; Hirose, Y.; Tanaka, H.; et al. Pharmacological Inhibition of TLR4-NOX4 Signal Protects against Neuronal Death in Transient Focal Ischemia. Sci. Rep. 2012, 2, 896. [Google Scholar] [CrossRef]
- Hu, Z.Y.; Yang, Z.B.; Zhang, R.; Luo, X.J.; Peng, J. The Protective Effect of Vitexin Compound B-1 on Rat Cerebral I/R Injury through a Mechanism Involving Modulation of MiR-92b/NOX4 Pathway. CNS Neurol. Disord. Drug Targets 2023, 22, 137–147. [Google Scholar] [CrossRef]
- Dai, Y.; Zhang, H.; Zhang, J.; Yan, M. Isoquercetin Attenuates Oxidative Stress and Neuronal Apoptosis after Ischemia/Reperfusion Injury via Nrf2-Mediated Inhibition of the NOX4/ROS/NF-ΚB Pathway. Chem. Biol. Interact. 2018, 284, 32–40. [Google Scholar] [CrossRef]
- Lu, P.; Zhang, C.C.; Zhang, X.M.; Li, H.G.; Luo, A.L.; Tian, Y.K.; Xu, H. Down-Regulation of NOX4 by Betulinic Acid Protects against Cerebral Ischemia-Reperfusion in Mice. J. Huazhong Univ. Sci. Technol. Med. Sci. 2017, 37, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.Y.; Li, M.; Feng, X.; Wang, J.; Cao, L.; Shen, X.K.; Chen, J.; Sun, M.; Sheng, R.; Han, F.; et al. Combined NADPH and the NOX Inhibitor Apocynin Provides Greater Anti-Inflammatory and Neuroprotective Effects in a Mouse Model of Stroke. Free Radic. Biol. Med. 2017, 104, 333–345. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Feng, D.; Liu, Y.; Xu, M.; Gao, A.; Tian, F.; Zhang, L.; Cui, Y.; Wang, Z.; et al. Alterations in the Time Course of Expression of the Nox Family in the Brain in a Rat Experimental Cerebral Ischemia and Reperfusion Model: Effects of Melatonin. J. Pineal Res. 2014, 57, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Casas, A.I.; Kleikers, P.W.M.; Geuss, E.; Langhauser, F.; Adler, T.; Busch, D.H.; Gailus-Durner, V.; De Angelis, M.H.; Egea, J.; Lopez, M.G.; et al. Calcium-Dependent Blood-Brain Barrier Breakdown by NOX5 Limits Postreperfusion Benefit in Stroke. J. Clin. Investig. 2019, 129, 1772. [Google Scholar] [CrossRef] [PubMed]
- Dao, V.T.V.; Elbatreek, M.H.; Altenhöfer, S.; Casas, A.I.; Pachado, M.P.; Neullens, C.T.; Knaus, U.G.; Schmidt, H.H.H.W. Isoform-Selective NADPH Oxidase Inhibitor Panel for Pharmacological Target Validation. Free Radic. Biol. Med. 2020, 148, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Mason, H.; Rai, G.; Kozyr, A.; De Jonge, N.; Gliniewicz, E.; Berg, L.J.; Wald, G.; Dorrier, C.; Henderson, M.J.; Zakharov, A.; et al. Development of an Improved and Specific Inhibitor of NADPH Oxidase 2 to Treat Traumatic Brain Injury. Redox Biol. 2023, 60, 102611. [Google Scholar] [CrossRef]
- Reutens, A.T.; Jandeleit-Dahm, K.; Thomas, M.; Salim, A.; De Livera, A.M.; Bach, L.A.; Colman, P.G.; Davis, T.M.E.; Ekinci, E.I.; Fulcher, G.; et al. A Physician-Initiated Double-Blind, Randomised, Placebo-Controlled, Phase 2 Study Evaluating the Efficacy and Safety of Inhibition of NADPH Oxidase with the First-in-Class Nox-1/4 Inhibitor, GKT137831, in Adults with Type 1 Diabetes and Persistently Elevated Urinary Albumin Excretion: Protocol and Statistical Considerations. Contemp. Clin. Trials 2020, 90, 105892. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marqués, J.; Zalba, G. Vascular NADPH Oxidases and Atherothrombotic Stroke. Stresses 2024, 4, 558-574. https://doi.org/10.3390/stresses4030036
Marqués J, Zalba G. Vascular NADPH Oxidases and Atherothrombotic Stroke. Stresses. 2024; 4(3):558-574. https://doi.org/10.3390/stresses4030036
Chicago/Turabian StyleMarqués, Javier, and Guillermo Zalba. 2024. "Vascular NADPH Oxidases and Atherothrombotic Stroke" Stresses 4, no. 3: 558-574. https://doi.org/10.3390/stresses4030036
APA StyleMarqués, J., & Zalba, G. (2024). Vascular NADPH Oxidases and Atherothrombotic Stroke. Stresses, 4(3), 558-574. https://doi.org/10.3390/stresses4030036