
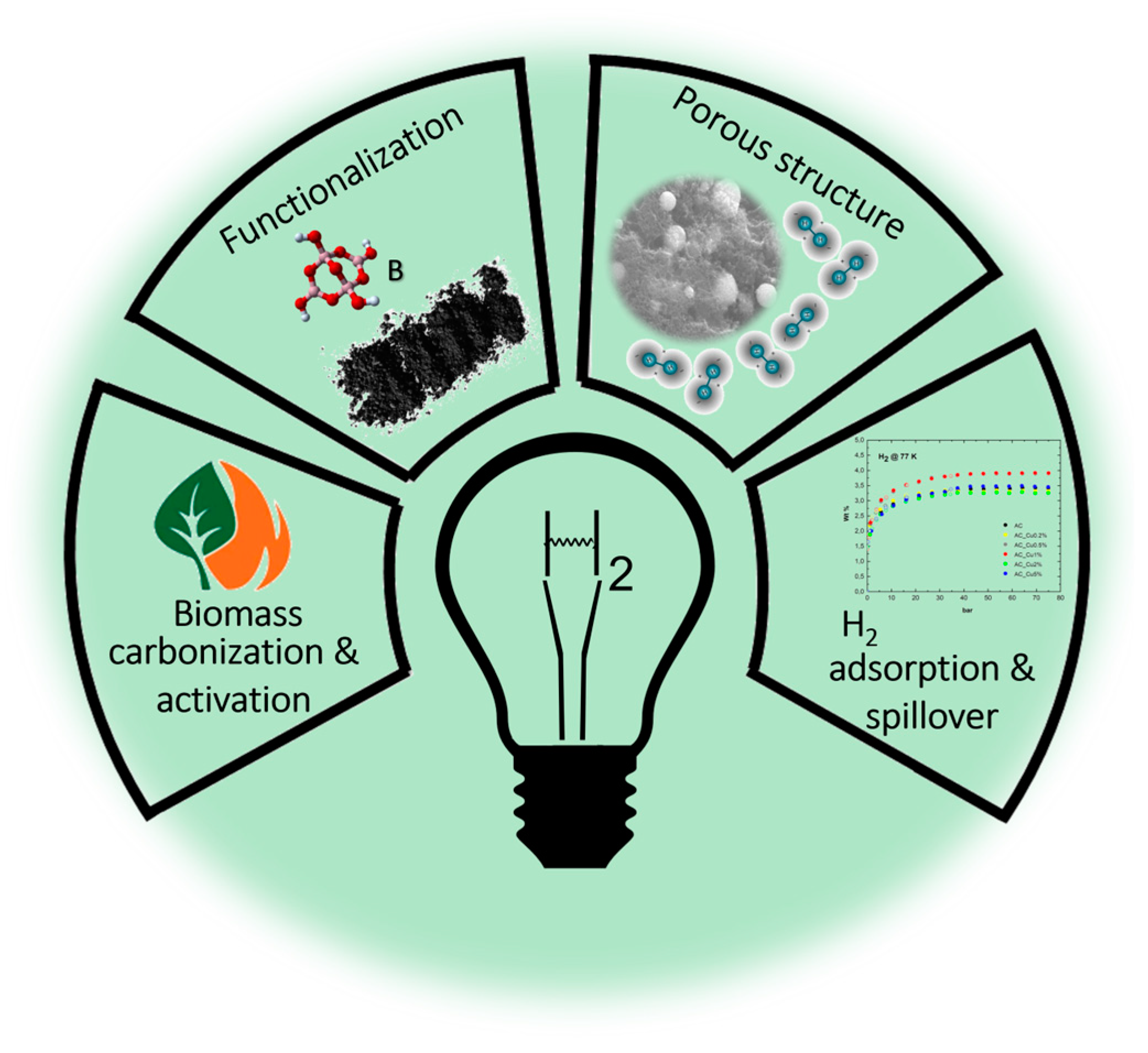
Boronation of Biomass-Derived Materials for Hydrogen Storage

 ,
, 
 , ,
, ,  ,
, 
Abstract
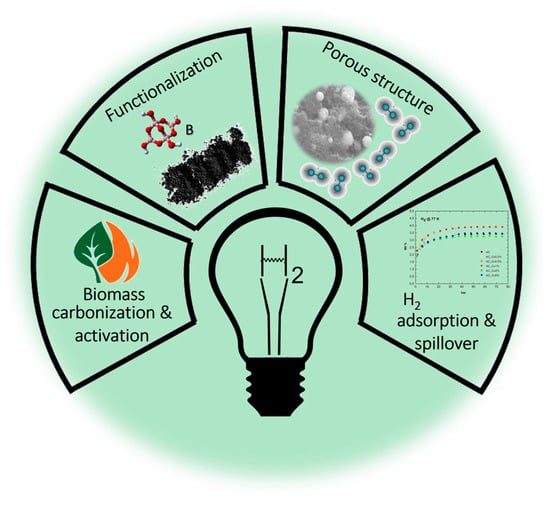
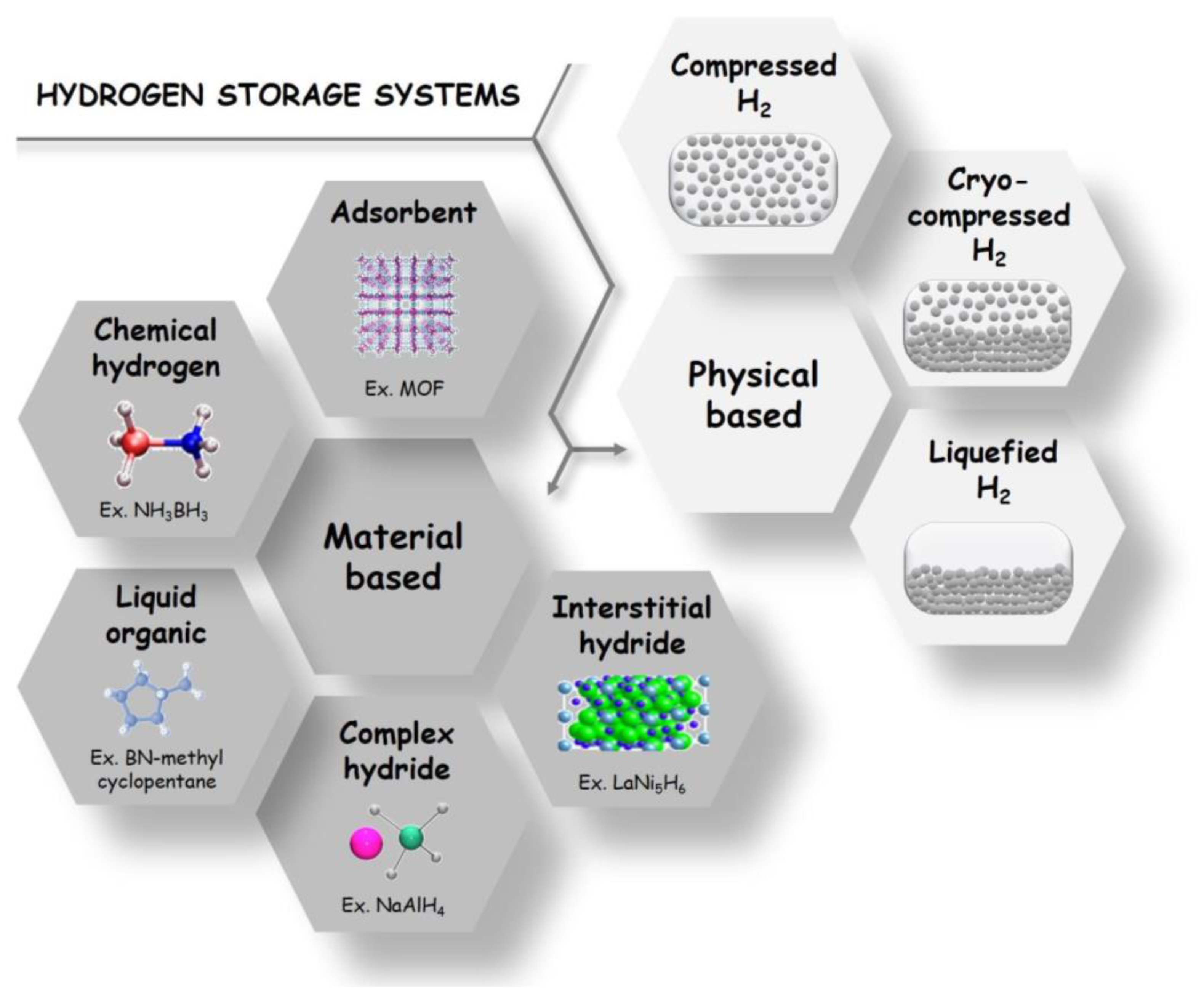
1. Hydrogen Storage and Delivery: State-of-the-Art Technology Overview
1.1. Compressed Gas
1.2. Cryogenic Liquid
1.3. Cryo-Compressed Gas
1.4. Material-Based H2 Storage
1.4.1. Intermetallic Compounds as H2 Storage Systems
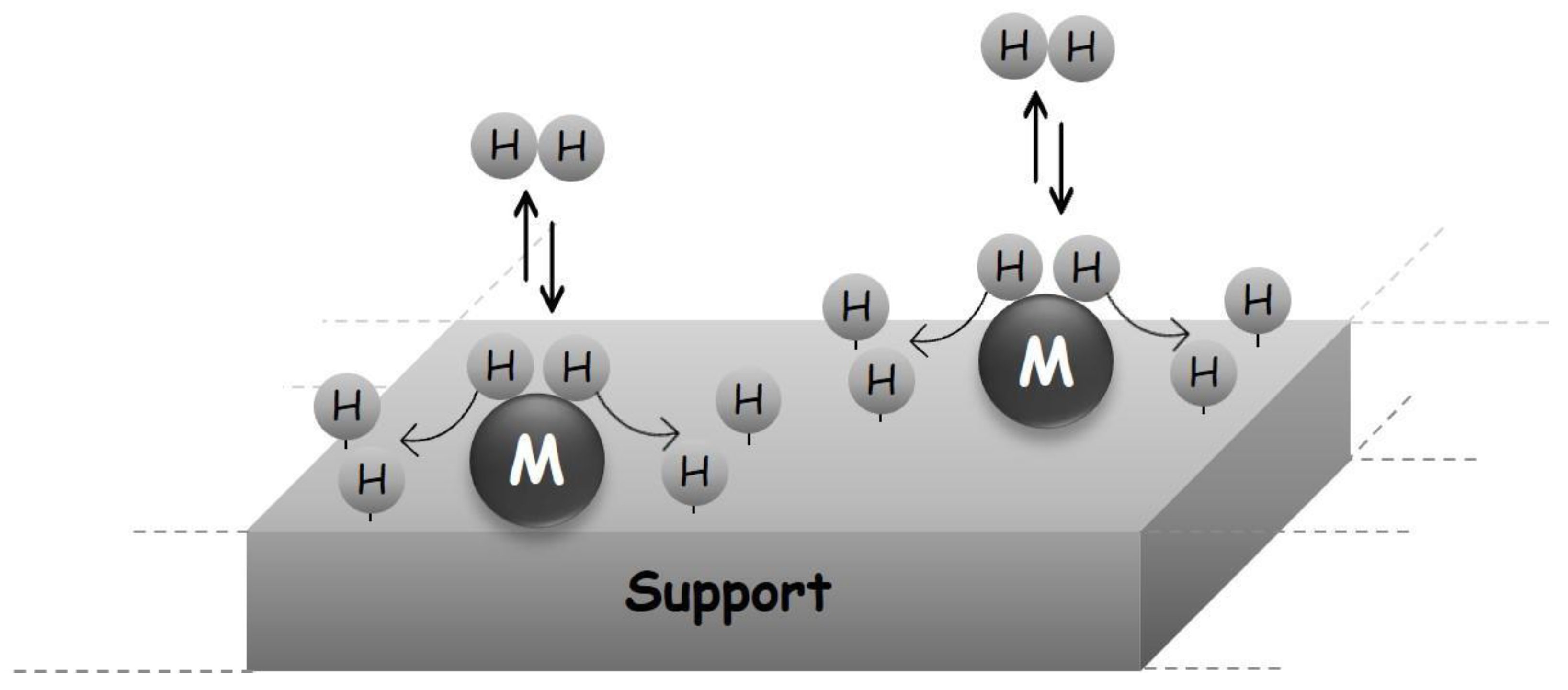
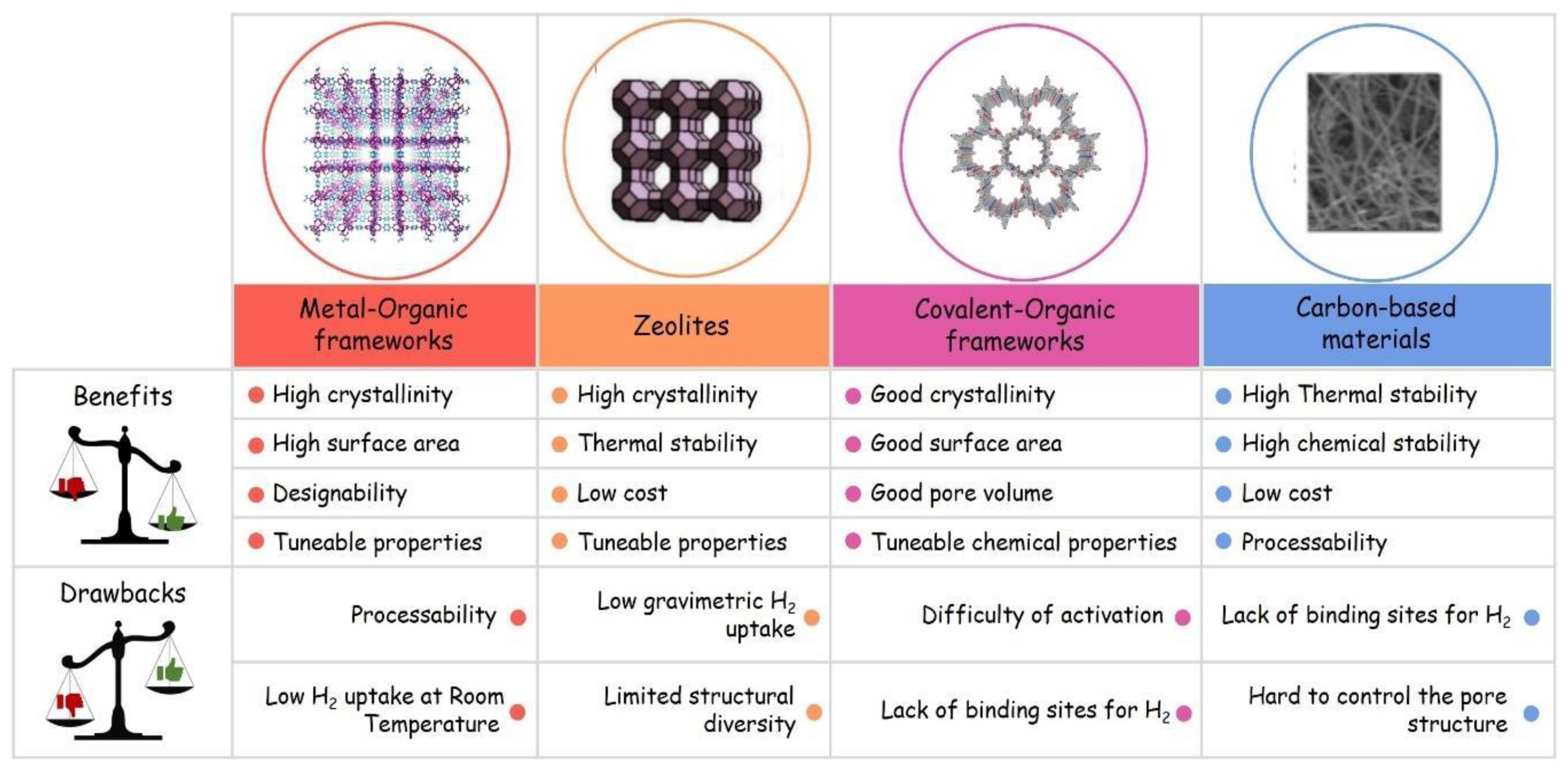
1.4.2. Porous Solids as H2 Storage Systems
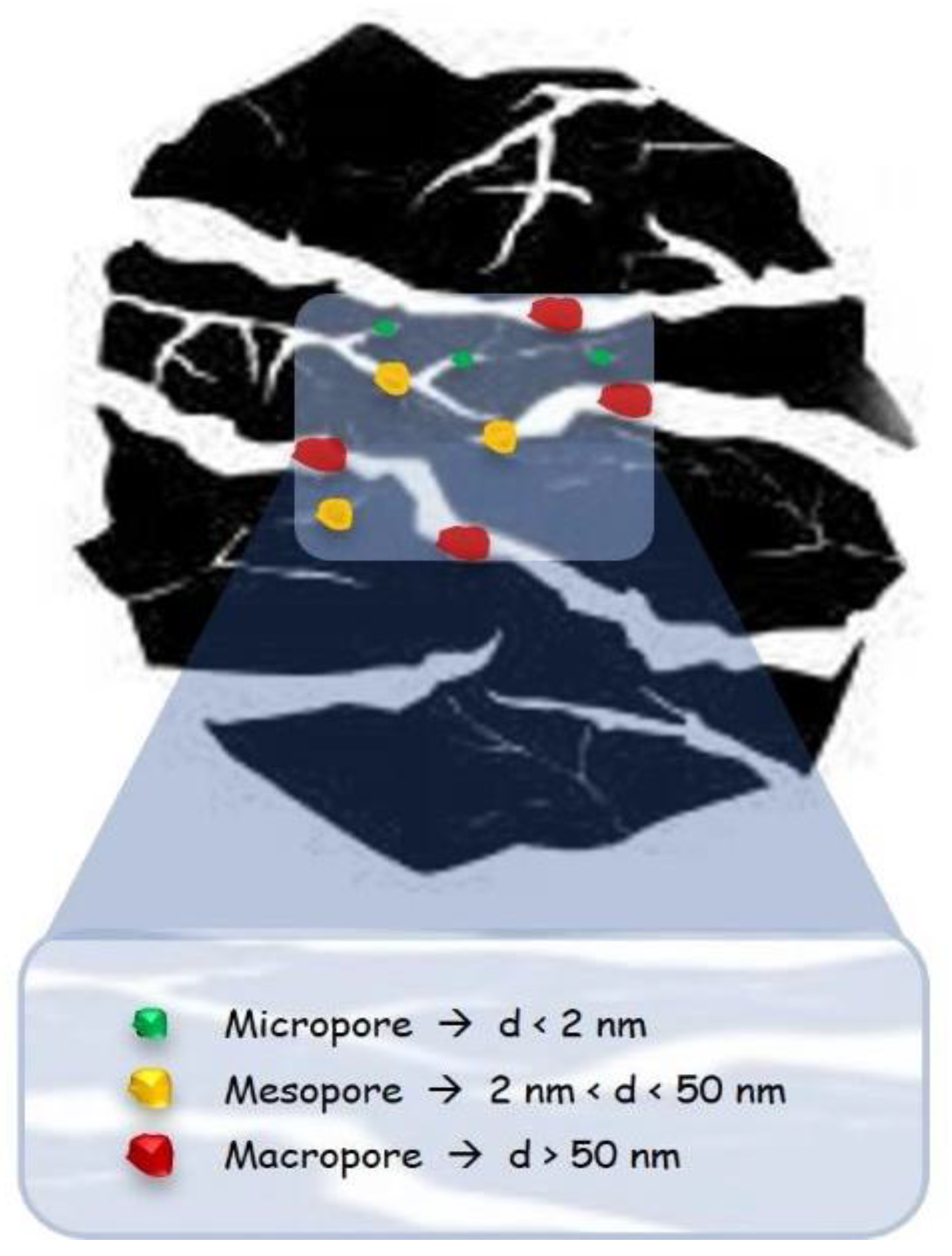
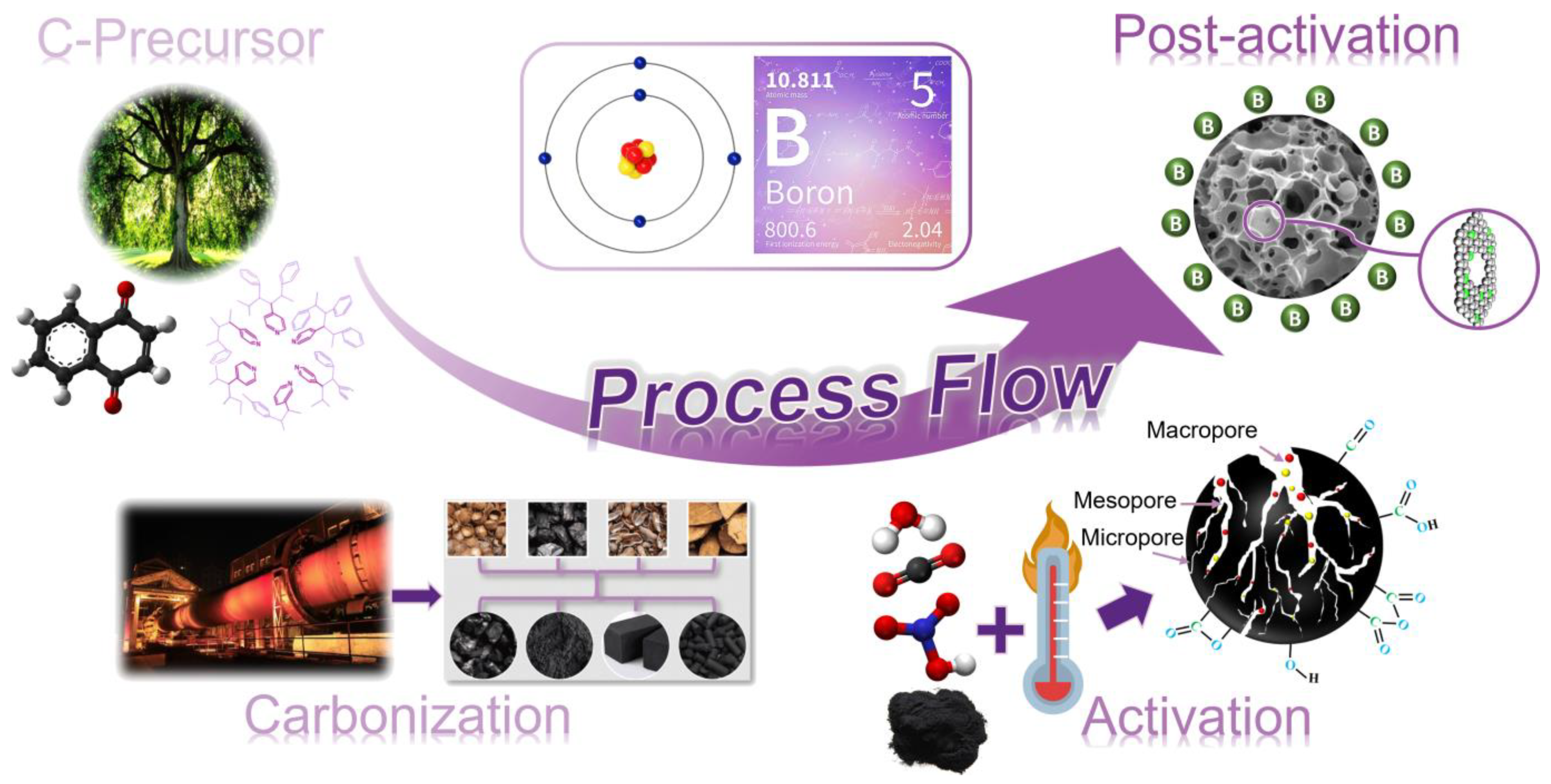
2. Biomass-Derived Carbonaceous Material
2.1. Activated Carbons (ACs)
2.1.1. ACs by Physical Activation
2.1.2. ACs by Chemical Activation (KOH)
2.1.3. ACs by Alternative Chemical Activation
2.1.4. Doped or Modified ACs
3. Boronation Methods
3.1. Classical Methods
3.2. Substitution Techniques
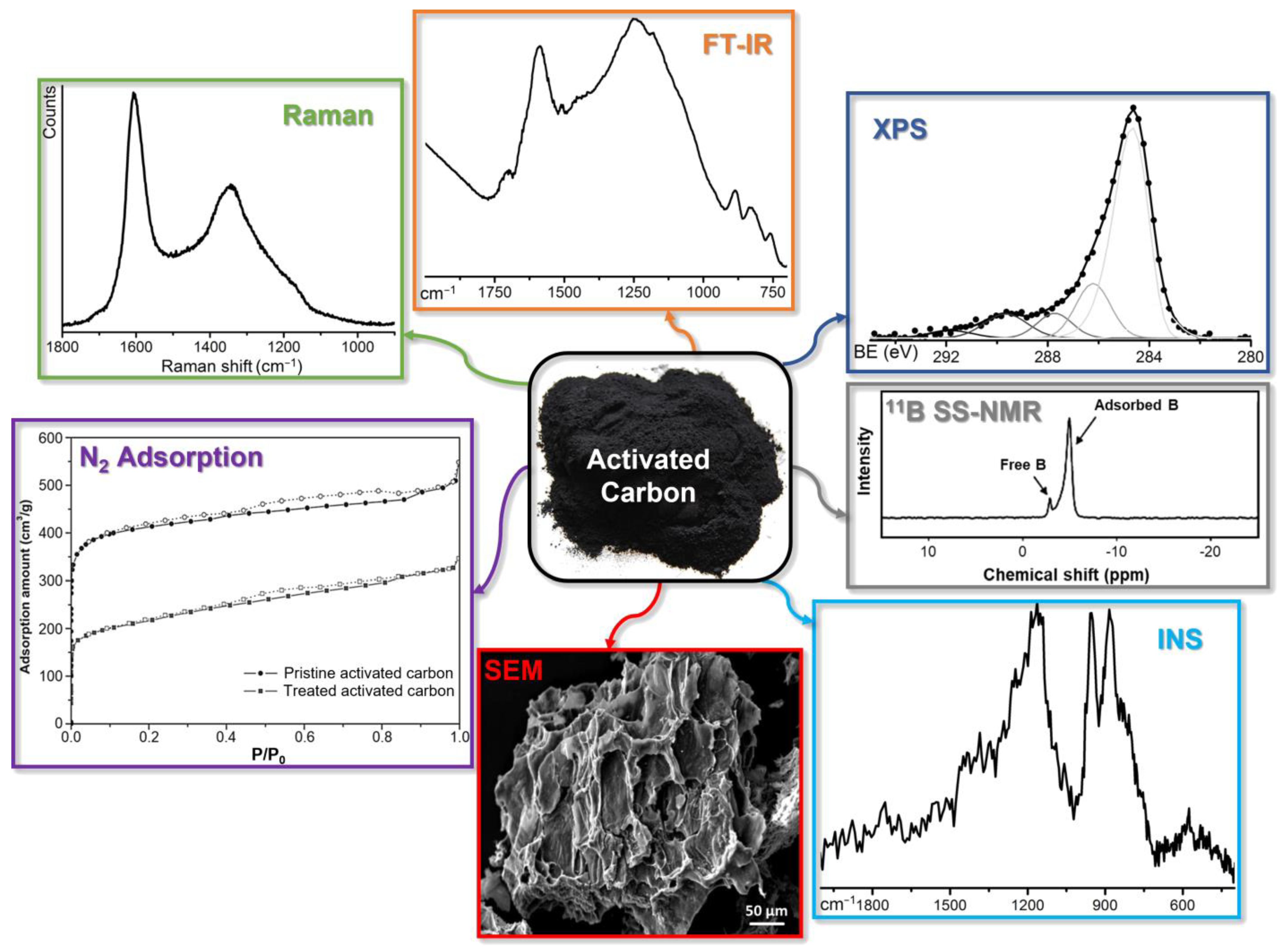
4. Characterization Techniques for Boron-Doped ACs: An Overview
5. Summary and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Moradi, R.; Groth, K.M. Hydrogen storage and delivery: Review of the state of the art technologies and risk and reliability analysis. Int. J. Hydrogen Energy 2019, 44, 12254–12269. [Google Scholar] [CrossRef]
- Ahluwalia, R.K.; Huaa, T.Q.; Peng, J.K.; Lasher, S.; McKenney, K.; Sinha, J.; Gardiner, M. Technical assessment of cryo-compressed hydrogen storage tank systems for automotive applications. Int. J. Hydrogen Energy 2010, 35, 4171–4184. [Google Scholar] [CrossRef]
- Demirdoven, N.; Deutch, J. Hybrid cars now, fuel cell cars later. Science 2004, 305, 974–976. [Google Scholar] [CrossRef]
- Bockris, J.O.M. The hydrogen economy: Its history. Int. J. Hydrogen Energy 2013, 38, 2579–2588. [Google Scholar] [CrossRef]
- Global Trends and Outlook for Hydrogen; IEA Hydrogen: Paris, France, 2017; Available online: https://www.ieahydrogen.org/other-relevant-reports/ (accessed on 10 December 2022).
- Schlapbach, L.; Zuttel, A. Hydrogen-storage materials for mobile applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- EG&G Technical Services, I. Fuel Cell Handbook, 7th ed.; U.S. Department of Energy—Office of Fossil Energy, National Energy Technology Laboratory: Morgantown, WV, USA, 2004. [Google Scholar]
- O’Hayre, R.; Cha, S.-W.; Colella, W.; Prinz, F.B. (Eds.) Fuel Cell Fundamentals; John Wiley & Sons: New York, NY, USA, 2005. [Google Scholar]
- Larminie, J.; Dicks, A. Medium and High Temperature Fuel Cells. In Fuel Cell Systems Explained, 2nd ed.; Larminie, J., Dicks, A., Eds.; John Wiley & Sons: Chichester, UK, 2003; pp. 163–228. [Google Scholar]
- Zuttel, A. Hydrogen storage methods. Naturwissenschaften 2004, 91, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Züttel, A. Materials for hydrogen storage. Mater. Today 2003, 6, 24–33. [Google Scholar] [CrossRef]
- Zivar, D.; Kumar, S.; Foroozesh, J. Underground hydrogen storage: A comprehensive review. Int. J. Hydrogen Energy 2021, 46, 23436–23462. [Google Scholar] [CrossRef]
- Karakilcik, H.; Karakilcik, M. Underground Large-Scale Hydrogen Storage. In Accelerating the Transition to a 100% Renewable Energy Era; Uyar, T.S., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 375–392. [Google Scholar]
- Fichtner, M. Nanotechnological aspects in materials for hydrogen storage. Adv. Eng. Mater. 2005, 7, 443–455. [Google Scholar] [CrossRef]
- Friedrich, K.A.; Büchi, F.N.; Li, Z.P.; Kiesgen, G.; Leinhos, D.C.; Rottengruber, H.S.; Bowman, R.C., Jr.; Ratnakumar, B.V. Applications. In Hydrogen as a Future Energy Carrier; Züttel, A., Borgschulte, A., Schlapbach, L., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, DE, USA, 2008; pp. 335–414. [Google Scholar]
- Steinfeld, A. Solar hydrogen production via a two-step water-splitting thermochemical cycle based on Zn/ZnO redox reactions. Int. J. Hydrogen Energy 2002, 27, 611–619. [Google Scholar] [CrossRef]
- Xiao, B.; Yuan, Q.C. Nanoporous metal organic framework materials for hydrogen storage. Particuology 2009, 7, 129–140. [Google Scholar] [CrossRef]
- Zhou, L. Progress and problems in hydrogen storage methods. Renew. Sustain. Energy Rev. 2005, 9, 395–408. [Google Scholar] [CrossRef]
- Dubno, W. Low Cost, High Efficiency, High Pressure Hydrogen Storage. In 2007 Annual Progress Report for the DOE Hydrogen Program; U.S. Department of Energy: Washington, DC, USA, 2007; pp. 608–610. [Google Scholar]
- Eberle, U.; Arnold, G.; von Helmolt, R. Hydrogen storage in metal-hydrogen systems and their derivatives. J. Power Sources 2006, 154, 456–460. [Google Scholar] [CrossRef]
- Barthelemy, H.; Weber, M.; Barbier, F. Hydrogen storage: Recent improvements and industrial perspectives. Int. J. Hydrogen Energy 2017, 42, 7254–7262. [Google Scholar] [CrossRef]
- Aceves, S.M.; Berry, G.D.; Martinez-Frias, J.; Espinosa-Loza, F. Vehicular storage of hydrogen in insulated pressure vessels. Int. J. Hydrogen Energy 2006, 31, 2274–2283. [Google Scholar] [CrossRef]
- Aceves, S.M.; Berry, G.D. Thermodynamics of Insulated Pressure Vessels for Vehicular Hydrogen Storage. J. Energy Resour. Technol. 1998, 120, 137–142. [Google Scholar] [CrossRef]
- Liley, P.E.; Thomson, G.H.; Friend, D.G.; Daubert, T.E.; Buck, E. Physical and Chemical Data. In Perry’s Chemical Engineers’ Handbook, 7th ed.; Perry, R.H., Green, D.W., Eds.; McGraw-Hill: New York, NY, USA, 1997; pp. 244–245. [Google Scholar]
- McCarty, R.D. Chapter 1: Introduction. In Hydrogen: Its Technology and Implications—Hydrogen Properties Vol. III; Cox, K.E., Williamson, K.D., Jr., Eds.; CRC Press: Cleveland, OH, USA, 1975; pp. 25–26. [Google Scholar]
- Schlapbach, L. Hydrogen as a fuel and its storage for mobility and transport. MRS Bull. 2002, 27, 675–676. [Google Scholar] [CrossRef]
- Aceves, S.M.; Espinosa-Loza, F.; Ledesma-Orozco, E.; Ross, T.O.; Weisberg, A.H.; Brunner, T.C.; Kircher, O. High-density automotive hydrogen storage with cryogenic capable pressure vessels. Int. J. Hydrogen Energy 2010, 35, 1219–1226. [Google Scholar] [CrossRef]
- El-Eskandarany, M.S. (Ed.) Solid-state hydrogen storage nanomaterials for fuel cell applications. In Mechanical Alloying, 3rd ed.; William Andrew Publishing: Norwich, CT, USA, 2020; pp. 229–261. [Google Scholar]
- Zhang, J.; Yan, Y.; Zhang, C.; Xu, Z.; Li, X.; Zhao, G.; Ni, Z. Properties improvement of composite layer of cryo-compressed hydrogen storage vessel by polyethylene glycol modified epoxy resin. Int. J. Hydrogen Energy 2023, 48, 5576–5594. [Google Scholar] [CrossRef]
- Moreno-Blanco, J.; Petitpas, G.; Espinosa-Loza, F.; Elizalde-Blancas, F.; Martinez-Frias, J.; Aceves, S.M. The storage performance of automotive cryo-compressed hydrogen vessels. Int. J. Hydrogen Energy 2019, 44, 16841–16851. [Google Scholar] [CrossRef]
- Zhao, X.H.; Yan, Y.; Zhang, J.Q.; Zhang, F.Y.H.; Wang, Z.; Ni, Z.H. Analysis of multilayered carbon fiber winding of cryo-compressed hydrogen storage vessel. Int. J. Hydrogen Energy 2022, 47, 10934–10946. [Google Scholar] [CrossRef]
- Sdanghi, G.; Maranzana, G.; Celzard, A.; Fierro, V. Review of the current technologies and performances of hydrogen compression for stationary and automotive applications. Renew. Sustain. Energy Rev. 2019, 102, 150–170. [Google Scholar] [CrossRef]
- Chen, L.; Xiao, R.F.; Cheng, C.; Tian, G.; Chen, S.T.; Hou, Y. Thermodynamic analysis of the para-to-ortho hydrogen conversion in cryo-compressed hydrogen vessels for automotive applications. Int. J. Hydrogen Energy 2020, 45, 24928–24937. [Google Scholar] [CrossRef]
- Andersson, J.; Gronkvist, S. Large-scale storage of hydrogen. Int. J. Hydrogen Energy 2019, 44, 11901–11919. [Google Scholar] [CrossRef]
- Bonadio, L. FUELS—HYDROGEN STORAGE | Liquid. In Encyclopedia of Electrochemical Power Sources; Garche, J., Ed.; Elsevier: Amsterdam, The Netherland, 2009; pp. 421–439. [Google Scholar]
- Stetson, N.T.; McWhorter, S.; Ahn, C.C. Introduction to hydrogen storage. In Compendium of Hydrogen Energy; Gupta, R.B., Basile, A., Veziroğlu, T.N., Eds.; Woodhead Publishing: Sawston, UK, 2016; pp. 3–25. [Google Scholar]
- Baetcke, L.; Kaltschmitt, M. Hydrogen Storage for Mobile Application: Technologies and Their Assessment. In Hydrogen Supply Chains; Azzaro-Pantel, C., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 167–206. [Google Scholar]
- Rouquerol, F.; Rouquerol, J.; Sing, K. (Eds.) CHAPTER 1—Introduction. In Adsorption by Powders and Porous Solids; Academic Press: London, UK, 1999; pp. 1–26. [Google Scholar]
- Ren, J.W.; Musyoka, N.M.; Langmi, H.W.; Mathe, M.; Liao, S.J. Current research trends and perspectives on materials-based hydrogen storage solutions: A critical review. Int. J. Hydrogen Energy 2017, 42, 289–311. [Google Scholar] [CrossRef]
- Chiarello, G.; Maccallini, E.; Agostino, R.G.; Caruso, T.; Formoso, V.; Papagno, L.; Colavita, E.; Goldoni, A.; Larciprete, R.; Lizzit, S.; et al. Vibrational and electronic properties of hydrogen adsorbed on single-wall carbon nanotubes. Phys. Rev. B 2004, 69, 153409. [Google Scholar] [CrossRef]
- Preuster, P.; Papp, C.; Wasserscheid, P. Liquid Organic Hydrogen Carriers (LOHCs): Toward a Hydrogen-free Hydrogen Economy. Accounts Chem. Res. 2017, 50, 74–85. [Google Scholar] [CrossRef]
- Teichmann, D.; Arlt, W.; Wasserscheid, P. Liquid Organic Hydrogen Carriers as an efficient vector for the transport and storage of renewable energy. Int. J. Hydrogen Energy 2012, 37, 18118–18132. [Google Scholar] [CrossRef]
- Lototskyy, M.V.; Davids, M.W.; Tolj, I.; Klochko, Y.V.; Sekhar, B.S.; Chidziva, S.; Smith, F.; Swanepoel, D.; Pollet, B.G. Metal hydride systems for hydrogen storage and supply for stationary and automotive low temperature PEM fuel cell power modules. Int. J. Hydrogen Energy 2015, 40, 11491–11497. [Google Scholar] [CrossRef]
- Rusman, N.A.A.; Dahari, M. A review on the current progress of metal hydrides material for solid-state hydrogen storage applications. Int. J. Hydrogen Energy 2016, 41, 12108–12126. [Google Scholar] [CrossRef]
- Sakintuna, B.; Lamari-Darkrim, F.; Hirscher, M. Metal hydride materials for solid hydrogen storage: A review. Int. J. Hydrogen Energy 2007, 32, 1121–1140. [Google Scholar] [CrossRef]
- von Colbe, J.M.B.; Lozano, G.; Metz, O.; Bucherl, T.; Bormann, R.; Klassen, T.; Dornheim, M. Design, sorption behaviour and energy management in a sodium alanate-based lightweight hydrogen storage tank. Int. J. Hydrogen Energy 2015, 40, 2984–2988. [Google Scholar] [CrossRef]
- Zaluska, A.; Zaluski, L.; Ström-Olsen, J.O. Sodium alanates for reversible hydrogen storage. J. Alloys Compd. 2000, 298, 125–134. [Google Scholar] [CrossRef]
- Song, F.; Zhang, T.; Zhou, D.; Sun, P.; Lu, Z.; Bian, H.; Dang, J.; Gao, H.; Qian, Y.; Li, W.; et al. Charge Transfer of Interfacial Catalysts for Hydrogen Energy. ACS Mater. Lett. 2022, 4, 967–977. [Google Scholar] [CrossRef]
- Yang, X.; Chen, L.; Liu, H.; Kurihara, T.; Horike, S.; Xu, Q. Encapsulating Ultrastable Metal Nanoparticles within Reticular Schiff Base Nanospaces for Enhanced Catalytic Performance. Cell Rep. Phys. Sci. 2021, 2, 100289. [Google Scholar] [CrossRef]
- Yang, X.; Li, Z.; Kitta, M.; Tsumori, N.; Guo, W.; Zhang, Z.; Zhang, J.; Zou, R.; Xu, Q. Solid-solution alloy nanoclusters of the immiscible gold-rhodium system achieved by a solid ligand-assisted approach for highly efficient catalysis. Nano Res. 2020, 13, 105–111. [Google Scholar] [CrossRef]
- Sastri, M.V.C. Metal Hydrides: Fundamentals and Applications; Springer GmbH & Co. KG: New York, NY, USA, 1988; pp. 1–21. [Google Scholar]
- Yang, R.T. Sorbents for Applications. In Adsorbents: Fundamentals and Applications; Yang, R.T., Ed.; John Wiley & Sons: New York, NY, USA, 2003; pp. 280–381. [Google Scholar]
- Blackburn, J.L.; Parilla, P.A.; Gennett, T.; Hurst, K.E.; Dillon, A.C.; Heben, M.J. Measurement of the reversible hydrogen storage capacity of milligram Ti-6Al-4V alloy samples with temperature programmed desorption and volumetric techniques. J. Alloys Compd. 2008, 454, 483–490. [Google Scholar] [CrossRef]
- Jensen, J.O.; Vestbo, A.P.; Li, Q.; Bjerrum, N.J. The energy efficiency of onboard hydrogen storage. J. Alloys Compd. 2007, 446, 723–728. [Google Scholar] [CrossRef]
- Smith, M.B.; Bass, G.E., Jr. Heats and Free Energies of Formation of the Alkali Aluminum Hydrides and of Cesium Hydride. J. Chem. Eng. Data 1963, 8, 342–346. [Google Scholar] [CrossRef]
- Schweppe, F.; Martin, M.; Fromm, E. Hydrogen absorption of LaNi5 powders precovered with O2, CO, H2S, CO2 or N2. J. Alloys Compd. 1997, 253–254, 511–514. [Google Scholar] [CrossRef]
- Gross, K.J.; Thomas, G.J.; Jensen, C.M. Catalyzed alanates for hydrogen storage. J. Alloys Compd. 2002, 330, 683–690. [Google Scholar] [CrossRef]
- Zaluski, L.; Zaluska, A.; Tessier, P.; Ström-Olsen, J.O.; Schulz, R. Catalytic effect of Pd on hydrogen absorption in mechanically alloyed Mg2Ni, LaNi5 and FeTi. J. Alloys Compd. 1995, 217, 295–300. [Google Scholar] [CrossRef]
- Wu, T.I.; Wu, J.K. The effects of chemical additives on the hydrogen uptake behavior of Ti-6AI-4V alloy. Mater. Chem. Phys. 2003, 80, 150–156. [Google Scholar] [CrossRef]
- Prins, R. Hydrogen Spillover. Facts and Fiction. Chem. Rev. 2012, 112, 2714–2738. [Google Scholar] [CrossRef] [PubMed]
- Becher, M.; Haluska, M.; Hirscher, M.; Quintel, A.; Skakalova, V.; Dettlaff-Weglikovska, U.; Chen, X.; Hulman, M.; Choi, Y.; Roth, S.; et al. Hydrogen storage in carbon nanotubes. C. R. Phys. 2003, 4, 1055–1062. [Google Scholar] [CrossRef]
- Lachawiec, A.J.; Qi, G.S.; Yang, R.T. Hydrogen storage in nanostructured carbons by spillover: Bridge-building enhancement. Langmuir 2005, 21, 11418–11424. [Google Scholar] [CrossRef]
- Haluska, M.; Hirscher, M.; Becher, M.; Dettlaff-Weglikowska, U.; Chen, X.; Roth, S. Interaction of hydrogen isotopes with carbon nanostructures. Mater. Sci. Eng. B 2004, 108, 130–133. [Google Scholar] [CrossRef]
- Nijkamp, M.G.; Raaymakers, J.E.M.J.; van Dillen, A.J.; de Jong, K.P. Hydrogen storage using physisorption—Materials demands. Appl. Phys. A 2001, 72, 619–623. [Google Scholar] [CrossRef]
- Felderhoff, M.; Weidenthaler, C.; von Helmolt, R.; Eberle, U. Hydrogen storage: The remaining scientific and technological challenges. Phys. Chem. Chem. Phys. 2007, 9, 2643–2653. [Google Scholar] [CrossRef]
- Dong, J.X.; Wang, X.Y.; Xu, H.; Zhao, Q.; Li, J.P. Hydrogen storage in several microporous zeolites. Int. J. Hydrogen Energy 2007, 32, 4998–5004. [Google Scholar] [CrossRef]
- Policicchio, A.; Conte, G.; Stelitano, S.; Bonaventura, C.P.; Putz, A.M.; Ianasi, C.; Almasy, L.; Horvath, Z.E.; Agostino, R.G. Hydrogen storage performances for mesoporous silica synthesized with mixed tetraethoxysilane and methyltriethoxysilane precursors in acidic condition. Colloid Surf. A 2020, 601, 125040. [Google Scholar] [CrossRef]
- Policicchio, A.; Putz, A.M.; Conte, G.; Stelitano, S.; Bonaventura, C.P.; Ianasi, C.; Almasy, L.; Wacha, A.; Horvath, Z.E.; Agostino, R.G. Hydrogen storage performance of methyl-substituted mesoporous silica with tailored textural characteristics. J. Porous Mater. 2021, 28, 1049–1058. [Google Scholar] [CrossRef]
- Heinemann, N.; Alcalde, J.; Miocic, J.M.; Hangx, S.J.T.; Kallmeyer, J.; Ostertag-Henning, C.; Hassanpouryouzband, A.; Thaysen, E.M.; Strobel, G.J.; Schmidt-Hattenberger, C.; et al. Enabling large-scale hydrogen storage in porous media—The scientific challenges. Energy Environ. Sci. 2021, 14, 853–864. [Google Scholar] [CrossRef]
- Ziemianski, P.P.; Derkowski, A. Structural and textural control of high-pressure hydrogen adsorption on expandable and non-expandable clay minerals in geologic conditions. Int. J. Hydrogen Energy 2022, 47, 28794–28805. [Google Scholar] [CrossRef]
- Ma, S.Q.; Zhou, H.C. Gas storage in porous metal-organic frameworks for clean energy applications. Chem. Commun. 2010, 46, 44–53. [Google Scholar] [CrossRef]
- Lee, S.Y.; Park, S.J. Effect of platinum doping of activated carbon on hydrogen storage behaviors of metal-organic frameworks-5. Int. J. Hydrogen Energy 2011, 36, 8381–8387. [Google Scholar] [CrossRef]
- Svec, F.; Germain, J.; Frechet, J.M.J. Nanoporous Polymers for Hydrogen Storage. Small 2009, 5, 1098–1111. [Google Scholar] [CrossRef]
- Kopac, T.; Karaaslan, T. H2, He and Ar sorption on arc-produced cathode deposit consisting of multiwalled carbon nanotubes—Graphitic and diamond-like carbon. Int. J. Hydrogen Energy 2007, 32, 3990–3997. [Google Scholar] [CrossRef]
- Yurum, Y.; Taralp, A.; Veziroglu, T.N. Storage of hydrogen in nanostructured carbon materials. Int. J. Hydrogen Energy 2009, 34, 3784–3798. [Google Scholar] [CrossRef]
- Zhang, Q.F.; Uchaker, E.; Candelaria, S.L.; Cao, G.Z. Nanomaterials for energy conversion and storage. Chem. Soc. Rev. 2013, 42, 3127–3171. [Google Scholar] [CrossRef]
- Cychosz, K.A.; Thommes, M. Progress in the Physisorption Characterization of Nanoporous Gas Storage Materials. Engineering 2018, 4, 559–566. [Google Scholar] [CrossRef]
- Ho, T.M.; Howes, T.; Bhandari, B.R. Encapsulation of gases in powder solid matrices and their applications: A review. Powder Technol. 2014, 259, 87–108. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Dash, R.K.; Yushin, G.; Yildirim, T.; Laudisio, G.; Fischer, J.E. Tailoring of nanoscale porosity in carbide-derived carbons for hydrogen storage. J. Am. Chem. Soc. 2005, 127, 16006–16007. [Google Scholar] [CrossRef] [PubMed]
- Gregg, S.J.; Sing, K.S.W. (Eds.) Adsorption, Surface Area and Porosity, 2nd ed.; Academic Press: London, UK, 1982. [Google Scholar]
- Stelitano, S.; Conte, G.; Policicchio, A.; Aloise, A.; Desiderio, G.; Agostino, R.G. Pinecone-Derived Activated Carbons as an Effective Medium for Hydrogen Storage. Energies 2020, 13, 2237. [Google Scholar] [CrossRef]
- Kopac, T.; Kirca, Y. Effect of ammonia and boron modifications on the surface and hydrogen sorption characteristics of activated carbons from coal. Int. J. Hydrogen Energy 2020, 45, 10494–10506. [Google Scholar] [CrossRef]
- Gonzalez-Garcia, P. Activated carbon from lignocellulosics precursors: A review of the synthesis methods, characterization techniques and applications. Renew. Sustain. Energy Rev. 2018, 82, 1393–1414. [Google Scholar] [CrossRef]
- Kopac, T. Hydrogen storage characteristics of bio-based porous carbons of different origin: A comparative review. Int. J. Energy Res. 2021, 45, 20497–20523. [Google Scholar] [CrossRef]
- Babel, K.; Janasiak, D.; Jurewicz, K. Electrochemical hydrogen storage in activated carbons with different pore structures derived from certain lignocellulose materials. Carbon 2012, 50, 5017–5026. [Google Scholar] [CrossRef]
- Tascón, J.M.D. Chapter Two—Overview of Carbon Materials in Relation to Adsorption. In Adsorption by Carbons; Bottani, E.J., Tascón, J.M.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 15–49. [Google Scholar]
- Maulana Kusdhany, M.I.; Lyth, S.M. New insights into hydrogen uptake on porous carbon materials via explainable machine learning. Carbon 2021, 179, 190–201. [Google Scholar] [CrossRef]
- Tzimas, E.; Filiou, C.; Peteves, S.D.; Veyret, J.B. Hydrogen Storage: State-of-the-Art and Future Perspective; IE, I.f.E.-J., Ed.; JRC Publications Repository: Petten, The Netherlands, 2003. [Google Scholar]
- Savova, D.; Apak, E.; Ekinci, E.; Yardim, F.; Petrov, N.; Budinova, T.; Razvigorova, M.; Minkova, V. Biomass conversion to carbon adsorbents and gas. Biomass Bioener. 2001, 21, 133–142. [Google Scholar] [CrossRef]
- Bhatnagar, A.; Sillanpaa, M. Utilization of agro-industrial and municipal waste materials as potential adsorbents for water treatment—A review. Chem. Eng. J. 2010, 157, 277–296. [Google Scholar] [CrossRef]
- Akasaka, H.; Takahata, T.; Toda, I.; Ono, H.; Ohshio, S.; Himeno, S.; Kokubu, T.; Saitoh, H. Hydrogen storage ability of porous carbon material fabricated from coffee bean wastes. Int. J. Hydrogen Energy 2011, 36, 580–585. [Google Scholar] [CrossRef]
- Ariharan, A.; Viswanathan, B. Porous activated carbon material derived from sustainable bio-resource of peanut shell for H2 and CO2 storage applications. Indian J. Chem. Technol. 2018, 25, 140–149. [Google Scholar]
- Jin, H.; Lee, Y.S.; Hong, I. Hydrogen adsorption characteristics of activated carbon. Catal. Today 2007, 120, 399–406. [Google Scholar] [CrossRef]
- Sun, Y.; Webley, P.A. Preparation of activated carbons from corncob with large specific surface area by a variety of chemical activators and their application in gas storage. Chem. Eng. J. 2010, 162, 883–892. [Google Scholar] [CrossRef]
- Chen, H.B.; Wang, H.B.; Xue, Z.P.; Yang, L.F.; Xiao, Y.; Zheng, M.T.; Lei, B.F.; Liu, Y.L.; Sun, L.X. High hydrogen storage capacity of rice hull based porous carbon. Int. J. Hydrogen Energy 2012, 37, 18888–18894. [Google Scholar] [CrossRef]
- Bhat, V.V.; Contescu, C.I.; Gallego, N.C.; Baker, F.S. Atypical hydrogen uptake on chemically-activated, ultramicroporous carbon. Carbon 2010, 48, 1331–1340. [Google Scholar] [CrossRef]
- Zhao, W.; Luo, L.; Wang, H.; Fan, M. Synthesis of bamboo-based activated carbons with super-high specific surface area for hydrogen storage. BioResources 2017, 12, 1246–1262. [Google Scholar] [CrossRef]
- Chen, T.; Zhou, Y.; Luo, L.; Wu, X.; Li, Z.; Fan, M.; Zhao, W. Preparation and characterization of heteroatom self-doped activated biocarbons as hydrogen storage and supercapacitor electrode materials. Electrochim. Acta 2019, 325, 134941. [Google Scholar] [CrossRef]
- Balathanigaimani, M.S.; Haider, M.B.; Jha, D.; Kumar, R.; Lee, S.J.; Shim, W.G.; Shon, H.K.; Kim, S.C.; Moon, H. Nanostructured Biomass Based Carbon Materials from Beer Lees for Hydrogen Storage. J. Nanosci. Nanotechnol. 2018, 18, 2196–2199. [Google Scholar] [CrossRef]
- Xiao, Y.; Dong, H.W.; Long, C.; Zheng, M.T.; Lei, B.F.; Zhang, H.R.; Liu, Y.L. Melaleuca bark based porous carbons for hydrogen storage. Int. J. Hydrogen Energy 2014, 39, 11661–11667. [Google Scholar] [CrossRef]
- Nasruddin; Martin, A.; Alhamid, M.I.; Tampubolon, D. Adsorption Isotherms of Hydrogen on Granular Activated Carbon Derived From Coal and Derived From Coconut Shell. Heat Transf. Eng. 2017, 38, 403–408. [Google Scholar] [CrossRef]
- Hermosilla-Lara, G.; Momen, G.; Marty, P.H.; Le Neindre, B.; Hassouni, K. Hydrogen storage by adsorption on activated carbon: Investigation of the thermal effects during the charging process. Int. J. Hydrogen Energy 2007, 32, 1542–1553. [Google Scholar] [CrossRef]
- Rowlandson, J.L.; Edler, K.J.; Tian, M.; Ting, V.P. Toward Process-Resilient Lignin-Derived Activated Carbons for Hydrogen Storage Applications. ACS Sustain. Chem. Eng. 2020, 8, 2186–2195. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, H.; Chen, J.; Li, G.D.; Zhang, Y.; Chen, J.S. Preparation and gas storage of high surface area microporous carbon derived from biomass source cornstalks. Bioresour. Technol. 2008, 99, 4803–4808. [Google Scholar] [CrossRef]
- Bader, N.; Ouederni, A. Optimization of biomass-based carbon materials for hydrogen storage. J. Energy Storage 2016, 5, 77–84. [Google Scholar] [CrossRef]
- Samantaray, S.S.; Mangisetti, S.R.; Ramaprabhu, S. Investigation of room temperature hydrogen storage in biomass derived activated carbon. J. Alloys Compd. 2019, 789, 800–804. [Google Scholar] [CrossRef]
- Yang, R.; Liu, G.Q.; Li, M.; Zhang, J.C.; Hao, X.M. Preparation and N2, CO2 and H2 adsorption of super activated carbon derived from biomass source hemp (Cannabis sativa L.) stem. Microporous Mesoporous Mater. 2012, 158, 108–116. [Google Scholar] [CrossRef]
- Wang, J.C.; Senkovska, I.; Kaskel, S.; Liu, Q. Chemically activated fungi-based porous carbons for hydrogen storage. Carbon 2014, 75, 372–380. [Google Scholar] [CrossRef]
- Wrobel-Iwaniec, I.; Diez, N.; Gryglewicz, G. Chitosan-based highly activated carbons for hydrogen storage. Int. J. Hydrogen Energy 2015, 40, 5788–5796. [Google Scholar] [CrossRef]
- Hu, W.Q.; Huang, J.Y.; Yu, P.F.; Zheng, M.T.; Xiao, Y.; Dong, H.W.; Liang, Y.R.; Hu, H.; Liu, Y.L. Hierarchically Porous Carbon Derived from Neolamarckia cadamba for Electrochemical Capacitance and Hydrogen Storage. ACS Sustain. Chem. Eng. 2019, 7, 15385–15393. [Google Scholar] [CrossRef]
- Pedicini, R.; Maisano, S.; Chiodo, V.; Conte, G.; Policicchio, A.; Agostino, R.G. Posidonia Oceanica and Wood chips activated carbon as interesting materials for hydrogen storage. Int. J. Hydrogen Energy 2020, 45, 14038–14047. [Google Scholar] [CrossRef]
- Arshad, S.H.M.; Ngadi, N.; Aziz, A.A.; Amin, N.S.; Jusoh, M.; Wong, S. Preparation of activated carbon from empty fruit bunch for hydrogen storage. J. Energy Storage 2016, 8, 257–261. [Google Scholar] [CrossRef]
- Ganesan, A.; Mukherjee, R.; Raj, J.; Shaijumon, M.M. Nanoporous rice husk derived carbon for gas storage and high performance electrochemical energy storage. J. Porous Mater. 2014, 21, 839–847. [Google Scholar] [CrossRef]
- Huang, C.C.; Chen, H.M.; Chen, C.H. Hydrogen adsorption on modified activated carbon. Int. J. Hydrogen Energy 2010, 35, 2777–2780. [Google Scholar] [CrossRef]
- Blankenship, L.S.; Balahmar, N.; Mokaya, R. Oxygen-rich microporous carbons with exceptional hydrogen storage capacity. Nat. Commun. 2017, 8, 1545. [Google Scholar] [CrossRef] [PubMed]
- Abo-Shosha, M.H.; Ibrahim, N.A.; Allam, E.; El-Zairy, E. Preparation and characterization of polyacrylic acid/karaya gum and polyacrylic acid/tamarind seed gum adducts and utilization in textile printing. Carbohydr. Polym. 2008, 74, 241–249. [Google Scholar] [CrossRef]
- Ibrahim, N.A.; Abo-Shosha, M.H.; Allam, E.A.; El-Zairy, E.M. New thickening agents based on tamarind seed gum and karaya gum polysaccharides. Carbohydr. Polym. 2010, 81, 402–408. [Google Scholar] [CrossRef]
- Oomah, B.D.; Busson, M.; Godfrey, D.V.; Drover, J.C.G. Characteristics of hemp (Cannabis sativa L.) seed oil. Food Chem. 2002, 76, 33–43. [Google Scholar] [CrossRef]
- Yang, R.; Su, M.X.; Li, M.; Zhang, J.C.; Hao, X.M.; Zhang, H. One-pot process combining transesterification and selective hydrogenation for biodiesel production from starting material of high degree of unsaturation. Bioresour. Technol. 2010, 101, 5903–5909. [Google Scholar] [CrossRef]
- Yang, R.; Liu, G.Q.; Xu, X.H.; Li, M.; Zhang, J.C.; Hao, X.M. Surface texture, chemistry and adsorption properties of acid blue 9 of hemp (Cannabis sativa L.) bast-based activated carbon fibers prepared by phosphoric acid activation. Biomass Bioener. 2011, 35, 437–445. [Google Scholar] [CrossRef]
- Rosas, J.M.; Bedia, J.; Rodriguez-Mirasol, J.; Cordero, T. Preparation of hemp-derived activated carbon monoliths. adsorption of water vapor. Ind. Eng. Chem. Res. 2008, 47, 1288–1296. [Google Scholar] [CrossRef]
- Cheng, F.; Liang, J.; Zhao, J.; Tao, Z.; Chen, J. Biomass waste-derived microporous carbons with controlled texture and enhanced hydrogen uptake. Chem. Mater. 2008, 20, 1889–1895. [Google Scholar] [CrossRef]
- Plaza, M.G.; Gonzalez, A.S.; Pis, J.J.; Rubiera, F.; Pevida, C. Production of microporous biochars by single-step oxidation: Effect of activation conditions on CO2 capture. Appl. Energy 2014, 114, 551–562. [Google Scholar] [CrossRef]
- Chan, K.Y.; Van Zwieten, L.; Meszaros, I.; Downie, A.; Joseph, S. Agronomic values of greenwaste biochar as a soil amendment. Aust. J. Soil Res. 2007, 45, 629–634. [Google Scholar] [CrossRef]
- Pallares, J.; Gonzalez-Cencerrado, A.; Arauzo, I. Production and characterization of activated carbon from barley straw by physical activation with carbon dioxide and steam. Biomass Bioener. 2018, 115, 64–73. [Google Scholar] [CrossRef]
- Farma, R.; Deraman, M.; Awitdrus, A.; Talib, I.A.; Taer, E.; Basri, N.H.; Manjunatha, J.G.; Ishak, M.M.; Dollah, B.N.M.; Hashmi, S.A. Preparation of highly porous binderless activated carbon electrodes from fibres of oil palm empty fruit bunches for application in supercapacitors. Bioresour. Technol. 2013, 132, 254–261. [Google Scholar] [CrossRef]
- Tan, X.F.; Liu, S.B.; Liu, Y.G.; Gu, Y.L.; Zeng, G.M.; Hua, X.J.; Wang, X.; Liu, S.H.; Jiang, L.H. Biochar as potential sustainable precursors for activated carbon production: Multiple applications in environmental protection and energy storage. Bioresour. Technol. 2017, 227, 359–372. [Google Scholar] [CrossRef]
- Ahmed, M.B.; Zhou, J.L.; Ngo, H.H.; Guo, W.S.; Chen, M.F. Progress in the preparation and application of modified biochar for improved contaminant removal from water and wastewater. Bioresour. Technol. 2016, 214, 836–851. [Google Scholar] [CrossRef]
- Tan, X.F.; Liu, Y.G.; Gu, Y.L.; Xu, Y.; Zeng, G.M.; Hu, X.J.; Liu, S.B.; Wang, X.; Liu, S.M.; Li, J. Biochar-based nano-composites for the decontamination of wastewater: A review. Bioresour. Technol. 2016, 212, 318–333. [Google Scholar] [CrossRef]
- Rajapaksha, A.U.; Chen, S.S.; Tsang, D.C.W.; Zhang, M.; Vithanage, M.; Mandal, S.; Gao, B.; Bolan, N.S.; Ok, Y.S. Engineered/designer biochar for contaminant removal/immobilization from soil and water: Potential and implication of biochar modification. Chemosphere 2016, 148, 276–291. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, I.; Qureshi, K.; Kazi, R.A.; Ansari, A.K. Preparation and Characterisation of Chemically Activated Almond Shells by Optimization of Adsorption Parameters for Removal of Chromium VI from Aqueous Solutions. Int. J. Chem. Molec. Eng. 2007, 1, 105–110. [Google Scholar]
- Inomata, K.; Kanazawa, K.; Urabe, Y.; Hosono, H.; Araki, T. Natural gas storage in activated carbon pellets without a binder. Carbon 2002, 40, 87–93. [Google Scholar] [CrossRef]
- Marcilla, A.; Garcia-Garcia, S.; Asensio, M.; Conesa, J.A. Influence of thermal treatment regime on the density and reactivity of activated carbons from almond shells. Carbon 2000, 38, 429–440. [Google Scholar] [CrossRef]
- Rambabu, N.; Rao, B.; Surisetty, V.R.; Das, U.; Dalai, A.K. Production, characterization, and evaluation of activated carbons from de-oiled canola meal for environmental applications. Ind. Crop. Prod. 2015, 65, 572–581. [Google Scholar] [CrossRef]
- Aworn, A.; Thiravetyan, P.; Nakbanpote, W. Preparation and characteristics of agricultural waste activated carbon by physical activation having micro- and mesopores. J. Anal. Appl. Pyrolysis 2008, 82, 279–285. [Google Scholar] [CrossRef]
- Chang, C.F.; Chang, C.Y.; Tsai, W.T. Effects of burn-off and activation temperature on preparation of activated carbon from corn cob agrowaste by CO2 and steam. J. Colloid Interface Sci. 2000, 232, 45–49. [Google Scholar] [CrossRef]
- Bouchelta, C.; Medjram, M.S.; Zoubida, M.; Chekkat, F.A.; Ramdane, N.; Bellat, J.P. Effects of pyrolysis conditions on the porous structure development of date pits activated carbon. J. Anal. Appl. Pyrolysis 2012, 94, 215–222. [Google Scholar] [CrossRef]
- Lua, A.C.; Lau, F.Y.; Guo, J. Influence of pyrolysis conditions on pore development of oil-palm-shell activated carbons. J. Anal. Appl. Pyrolysis 2006, 76, 96–102. [Google Scholar] [CrossRef]
- Lua, A.C.; Yang, T.; Guo, J. Effects of pyrolysis conditions on the properties of activated carbons prepared from pistachio-nut shells. J. Anal. Appl. Pyrolysis 2004, 72, 279–287. [Google Scholar] [CrossRef]
- Loredo-Cancino, M.; Soto-Regalado, E.; Cerino-Cordova, F.J.; Garcia-Reyes, R.B.; Garcia-Leon, A.M.; Garza-Gonzalez, M.T. Determining optimal conditions to produce activated carbon from barley husks using single or dual optimization. J. Environ. Manag. 2013, 125, 117–125. [Google Scholar] [CrossRef]
- Cagnon, B.; Py, X.; Guillot, A.; Stoeckli, F. The effect of the carbonization/activation procedure on the microporous texture of the subsequent chars and active carbons. Microporous Mesoporous Mater. 2003, 57, 273–282. [Google Scholar] [CrossRef]
- Zhang, T.Y.; Walawender, W.P.; Fan, L.T.; Fan, M.; Daugaard, D.; Brown, R.C. Preparation of activated carbon from forest and agricultural residues through CO2 activation. Chem. Eng. J. 2004, 105, 53–59. [Google Scholar] [CrossRef]
- Xu, W.C.; Takahashi, K.; Matsuo, Y.; Hattori, Y.; Kumagai, M.; Ishiyama, S.; Kaneko, K.; Iijima, S. Investigation of hydrogen storage capacity of various carbon materials. Int. J. Hydrogen Energy 2007, 32, 2504–2512. [Google Scholar] [CrossRef]
- Pacula, A.; Mokaya, R. Synthesis and high hydrogen storage capacity of zeolite-like carbons nanocast using as-synthesized zeolite templates. J. Phys. Chem. C 2008, 112, 2764–2769. [Google Scholar] [CrossRef]
- Yang, Z.X.; Xia, Y.D.; Mokaya, R. Enhanced hydrogen storage capacity of high surface area zeolite-like carbon materials. J. Am. Chem. Soc. 2007, 129, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
- Sultana, A.I.; Saha, N.; Reza, M.T. Synopsis of Factors Affecting Hydrogen Storage in Biomass-Derived Activated Carbons. Sustainability 2021, 13, 1947. [Google Scholar] [CrossRef]
- Sevilla, M.; Mokaya, R. Energy storage applications of activated carbons: Supercapacitors and hydrogen storage. Energy Environ. Sci. 2014, 7, 1250–1280. [Google Scholar] [CrossRef]
- de la Casa-Lillo, M.A.; Lamari-Darkrim, F.; Cazorla-Amoros, D.; Linares-Solano, A. Hydrogen storage in activated carbons and activated carbon fibers. J. Phys. Chem. B 2002, 106, 10930–10934. [Google Scholar] [CrossRef]
- Carpetis, C.; Peschka, W. A study on hydrogen storage by use of cryoadsorbents. Int. J. Hydrogen Energy 1980, 5, 539–554. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, J.M.; Chen, Y.; Hao, X.M.; Jin, X.J. Characterization, preparation, and reaction mechanism of hemp stem based activated carbon. Results Phys. 2017, 7, 1628–1633. [Google Scholar] [CrossRef]
- Ding, F.; Yakobson, B.I. Challenges in hydrogen adsorptions: From physisorption to chemisorption. Front. Phys. 2011, 6, 142–150. [Google Scholar] [CrossRef]
- Sur, U.K. Graphene: A Rising Star on the Horizon of Materials Science. Int. J. Electrochem. 2012, 2012, 237689. [Google Scholar] [CrossRef]
- Zhao, X.B.; Xiao, B.; Fletcher, A.J.; Thomas, K.M. Hydrogen adsorption on functionalized nanoporous activated carbons. J. Phys. Chem. B 2005, 109, 8880–8888. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Srinivasan, M.P.; Ni, Y. Preparation of Mesoporous High-Surface-Area Activated Carbon. Adv. Mater. 2000, 12, 62–65. [Google Scholar] [CrossRef]
- Sevilla, M.; Fuertes, A.B.; Mokaya, R. High density hydrogen storage in superactivated carbons from hydrothermally carbonized renewable organic materials. Energy Environ. Sci. 2011, 4, 1400–1410. [Google Scholar] [CrossRef]
- Coromina, H.M.; Walsh, D.A.; Mokaya, R. Biomass-derived activated carbon with simultaneously enhanced CO2 uptake for both pre and post combustion capture applications. J. Mater. Chem. A 2016, 4, 280–289. [Google Scholar] [CrossRef]
- Wang, H.L.; Gao, Q.M.; Hu, J. High Hydrogen Storage Capacity of Porous Carbons Prepared by Using Activated Carbon. J. Am. Chem. Soc. 2009, 131, 7016–7022. [Google Scholar] [CrossRef]
- Schaefer, S.; Jeder, A.; Sdanghi, G.; Gadonneix, P.; Abdedayem, A.; Izquierdo, M.T.; Maranzana, G.; Ouederni, A.; Celzard, A.; Fierro, V. Oxygen-promoted hydrogen adsorption on activated and hybrid carbon materials. Int. J. Hydrogen Energy 2020, 45, 30767–30782. [Google Scholar] [CrossRef]
- Pellenq, R.J.M.; Marinelli, F.; Fuhr, J.D.; Fernandez-Alonso, F.; Refson, K. Strong physisorption site for H2 in K- and Li-doped porous carbons. J. Chem. Phys. 2008, 129, 224701. [Google Scholar] [CrossRef]
- Cabria, I.; Lopez, M.J.; Alonso, J.A. The optimum average nanopore size for hydrogen storage in carbon nanoporous materials. Carbon 2007, 45, 2649–2658. [Google Scholar] [CrossRef]
- Benard, P.; Chahine, R. Modeling of adsorption storage of hydrogen on activated carbons. Int. J. Hydrogen Energy 2001, 26, 849–855. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Portet, C.; Osswald, S.; Simmons, J.M.; Yidirim, T.; Laudisio, G.; Fischer, J.E. Importance of pore size in high-pressure hydrogen storage by porous carbons. Int. J. Hydrogen Energy 2009, 34, 6314–6319. [Google Scholar] [CrossRef]
- Masika, E.; Mokaya, R. Hydrogen Storage in High Surface Area Carbons with Identical Surface Areas but Different Pore Sizes: Direct Demonstration of the Effects of Pore Size. J. Phys. Chem. C 2012, 116, 25734–25740. [Google Scholar] [CrossRef]
- Dogan, M.; Sabaz, P.; Bicil, Z.; Kizilduman, B.K.; Turhan, Y. Activated carbon synthesis from tangerine peel and its use in hydrogen storage. J. Energy Inst. 2020, 93, 2176–2185. [Google Scholar] [CrossRef]
- Rossetti, I.; Ramis, G.; Gallo, A.; Di Michele, A. Hydrogen storage over metal-doped activated carbon. Int. J. Hydrogen Energy 2015, 40, 7609–7616. [Google Scholar] [CrossRef]
- Ya’aini, N.; Pillay, A.; Krishnan, G.; Ripin, A. Synthesis of activated carbon doped with transition metals for hydrogen storage. E3S Web Conf. 2019, 90, 01016. [Google Scholar] [CrossRef]
- Conte, G.; Policicchio, A.; De Luca, O.; Rudolf, P.; Desiderio, G.; Agostino, R.G. Copper-doped activated carbon from amorphous cellulose for hydrogen, methane and carbon dioxide storage. Int. J. Hydrogen Energy 2022, 47, 18384–18395. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Zhu, Y.W.; Liu, Q.Y.; Li, X.W. Preparation of porous carbon materials from biomass pyrolysis vapors for hydrogen storage. Appl. Energy 2022, 306, 118131. [Google Scholar] [CrossRef]
- Sawant, S.V.; Patwardhan, A.W.; Joshi, J.B.; Dasgupta, K. Boron doped carbon nanotubes: Synthesis, characterization and emerging applications—A review. Chem. Eng. J. 2022, 427, 131616. [Google Scholar] [CrossRef]
- Sawant, S.V.; Banerjee, S.; Patwardhan, A.W.; Joshi, J.B.; Dasgupta, K. Effect of in-situ boron doping on hydrogen adsorption properties of carbon nanotubes. Int. J. Hydrogen Energy 2019, 44, 18193–18204. [Google Scholar] [CrossRef]
- Sawant, S.V.; Yadav, M.D.; Banerjee, S.; Patwardhan, A.W.; Joshi, J.B.; Dasgupta, K. Hydrogen storage in boron-doped carbon nanotubes: Effect of dopant concentration. Int. J. Hydrogen Energy 2021, 46, 39297–39314. [Google Scholar] [CrossRef]
- Jeong, Y.; Chung, T.C.M. The synthesis and characterization of a super-activated carbon containing substitutional boron (BCX) and its applications in hydrogen storage. Carbon 2010, 48, 2526–2537. [Google Scholar] [CrossRef]
- Ariharan, A.; Viswanathan, B.; Nandhakumar, V. Hydrogen storage on boron substituted carbon materials. Int. J. Hydrogen Energy 2016, 41, 3527–3536. [Google Scholar] [CrossRef]
- Shcherban, N.; Filonenko, S.; Yaremov, P.; Dyadyun, V.; Bezverkhyy, I.; Ilyin, V. Boron-doped nanoporous carbons as promising materials for supercapacitors and hydrogen storage. J. Mater. Sci. 2017, 52, 1523–1533. [Google Scholar] [CrossRef]
- Bult, J.B.; Lee, J.; O’Neill, K.; Engtrakul, C.; Hurst, K.E.; Zhao, Y.F.; Simpson, L.J.; Parilla, P.A.; Gennett, T.; Blackburn, J.L. Manipulation of Hydrogen Binding Energy and Desorption Kinetics by Boron Doping of High Surface Area Carbon. J. Phys. Chem. C 2012, 116, 26138–26143. [Google Scholar] [CrossRef]
- Bhatia, S.K.; Myers, A.L. Optimum conditions for adsorptive storage. Langmuir 2006, 22, 1688–1700. [Google Scholar] [CrossRef]
- Minuto, F.D.; Policicchio, A.; Aloise, A.; Agostino, R.G. Liquid-like hydrogen in the micropores of commercial activated carbons. Int. J. Hydrogen Energy 2015, 40, 14562–14572. [Google Scholar] [CrossRef]
- Panchokarla, L.S.; Subrahmanyam, K.S.; Saha, S.K.; Govindaraj, A.; Krishnamurthy, H.R.; Waghmare, U.V.; Rao, C.N.R. Synthesis, Structure, and Properties of Boron- and Nitrogen-Doped Graphene. Adv. Mater. 2009, 21, 4726–4730. [Google Scholar] [CrossRef]
- Kim, Y.H.; Zhao, Y.F.; Williamson, A.; Heben, M.J.; Zhang, S.B. Nondissociative adsorption of H2 molecules in light-element-doped fullerenes. Phys. Rev. Lett. 2006, 96, 016102. [Google Scholar] [CrossRef]
- Dasgupta, K.; Joshi, J.B.; Banerjee, S. Fluidized bed synthesis of carbon nanotubes—A review. Chem. Eng. J. 2011, 171, 841–869. [Google Scholar] [CrossRef]
- Yadav, M.D.; Patwardhan, A.W.; Joshi, J.B.; Dasgupta, K. Kinetic study of multi-walled carbon nanotube synthesis by thermocatalytic decomposition of methane using floating catalyst chemical vapour deposition. Chem. Eng. J. 2019, 377, 119895. [Google Scholar] [CrossRef]
- Yadav, M.D.; Dasgupta, K.; Patwardhan, A.W.; Kaushal, A.; Joshi, J.B. Kinetic study of single-walled carbon nanotube synthesis by thermocatalytic decomposition of methane using floating catalyst chemical vapour deposition. Chem. Eng. Sci. 2019, 196, 91–103. [Google Scholar] [CrossRef]
- Ceragioli, H.J.; Peterlevitz, A.C.; Quispe, J.C.R.; Larena, A.; Pasquetto, M.P.; Sampaio, M.A.; Baranauskas, V. Synthesis and characterization of boron-doped carbon nanotubes. J. Phys. Conf. Ser. 2008, 100, 052029. [Google Scholar] [CrossRef]
- Ayala, P.; Rummeli, M.H.; Gemming, T.; Kauppinen, E.; Kuzmany, H.; Pichler, T. CVD growth of single-walled B-doped carbon nanotubes. Phys. Stat. Sol. B 2008, 245, 1935–1938. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, C.H.; Ba, D.C.; Liang, J. Influence of the gas composition on the synthesis of boron-doped carbon nanotubes by ECR-CVD. Vacuum 2007, 81, 579–582. [Google Scholar] [CrossRef]
- Chen, C.F.; Tsai, C.L.; Lin, C.L. The characterization of boron-doped carbon nanotube arrays. Diamond Relat. Mat. 2003, 12, 1500–1504. [Google Scholar] [CrossRef]
- Mondal, K.C.; Coville, N.J.; Witcomb, M.J.; Tejral, G.; Havel, J. Boron mediated synthesis of multiwalled carbon nanotubes by chemical vapor deposition. Chem. Phys. Lett. 2007, 437, 87–91. [Google Scholar] [CrossRef]
- Keqiang, C.; Erli, Z.; Jinfa, W.; Hansheng, Z.; Zuoyao, G.; Bangwei, Z. Microwave electron cyclotron resonance plasma for chemical vapor deposition and etching. J. Vac. Sci. Technol. A 1986, 4, 828–831. [Google Scholar] [CrossRef]
- Wang, Z.; Ba, D.C.; Liu, F.; Cao, P.J.; Yang, T.Z.; Gu, Y.S.; Gao, H.J. Synthesis and characterization of large area well-aligned carbon nanotubes by ECR-CVD without substrate bias. Vacuum 2005, 77, 139–144. [Google Scholar] [CrossRef]
- Ruiz-Soria, G.; Ayala, P.; Puchegger, S.; Kataura, H.; Yanagi, K.; Pichler, T. On the purification of CVD grown boron doped single-walled carbon nanotubes. Phys. Stat. Sol. B 2011, 248, 2504–2507. [Google Scholar] [CrossRef]
- Ishii, S.; Nagao, M.; Watanabe, T.; Tsuda, S.; Yamaguchi, T.; Takano, Y. Electrical properties of boron-doped MWNTs synthesized by hot-filament chemical vapor deposition. Phys. C 2009, 469, 1002–1004. [Google Scholar] [CrossRef]
- Preston, C.; Song, D.; Taillon, J.; Cumings, J.; Hu, L. Boron-doped few-walled carbon nanotubes: Novel synthesis and properties. Nanotechnology 2016, 27, 445601. [Google Scholar] [CrossRef] [PubMed]
- Iijima, S. Helical Microtubules of Graphitic Carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Stephan, O.; Ajayan, P.M.; Colliex, C.; Redlich, P.; Lambert, J.M.; Bernier, P.; Lefin, P. Doping Graphitic and Carbon Nanotube Structures with Boron and Nitrogen. Science 1994, 266, 1683–1685. [Google Scholar] [CrossRef] [PubMed]
- Maultzsch, J.; Reich, S.; Thomsen, C.; Webster, S.; Czerw, R.; Carroll, D.L.; Vieira, S.M.C.; Birkett, P.R.; Rego, C.A. Raman characterization of boron-doped multiwalled carbon nanotubes. Appl. Phys. Lett. 2002, 81, 2647–2649. [Google Scholar] [CrossRef]
- Babanejad, S.A.; Malekfar, R.; Ashrafi, F.; Hosseini, S.M.R.S. Production and study of boron and nitrogen-doped carbon nanotubes by arc discharge method using dispersive Raman back-scattering spectroscopy. Asian J. Chem. 2010, 22, 245–252. [Google Scholar]
- Carroll, D.L.; Redlich, P.; Ajayan, P.M.; Curran, S.; Roth, S.; Ruhle, M. Spatial variations in the electronic structure of pure and B-doped nanotubes. Carbon 1998, 36, 753–756. [Google Scholar] [CrossRef]
- Carroll, D.L.; Redlich, P.; Blase, X.; Charlier, J.C.; Curran, S.; Ajayan, P.M.; Roth, S.; Ruhle, M. Effects of nanodomain formation on the electronic structure of doped carbon nanotubes. Phys. Rev. Lett. 1998, 81, 2332–2335. [Google Scholar] [CrossRef]
- Wang, B.; Ma, Y.F.; Wu, Y.P.; Li, N.; Huang, Y.; Chen, Y.S. Direct and large scale electric arc discharge synthesis of boron and nitrogen doped single-walled carbon nanotubes and their electronic properties. Carbon 2009, 47, 2112–2115. [Google Scholar] [CrossRef]
- McGuire, K.; Gothard, N.; Gai, P.L.; Dresselhaus, M.S.; Sumanasekera, G.; Rao, A.M. Synthesis and Raman characterization of boron-doped single-walled carbon nanotubes. Carbon 2005, 43, 219–227. [Google Scholar] [CrossRef]
- Gai, P.L.; Stephan, O.; McGuire, K.; Rao, A.M.; Dresselhaus, M.S.; Dresselhaus, G.; Colliex, C. Structural systematics in boron-doped single wall carbon nanotubes. J. Mater. Chem. 2004, 14, 669–675. [Google Scholar] [CrossRef]
- Ayala, P.; Reppert, J.; Grobosch, M.; Knupfer, M.; Pichler, T.; Rao, A.M. Evidence for substitutional boron in doped single-walled carbon nanotubes. Appl. Phys. Lett. 2010, 96, 183110. [Google Scholar] [CrossRef]
- Liu, B.H.; Guo, W.Q.; Wang, H.Z.; Si, Q.S.; Zhao, Q.; Luo, H.C.; Ren, N.Q. B-doped graphitic porous biochar with enhanced surface affinity and electron transfer for efficient peroxydisulfate activation. Chem. Eng. J. 2020, 396, 125119. [Google Scholar] [CrossRef]
- Zhu, F.; Zhu, M.; Kang, L. B-doped activated carbon as a support for a high-performance Zn-based catalyst in acetylene acetoxylation. Green Energy Environ. 2022, 7, 221–228. [Google Scholar] [CrossRef]
- Yao, Y.H.; Huang, G.X.; Liu, Y.B.; Liu, Y.; Li, Y.Y.; Han, G.X.; Xing, B.L.; Liu, Q.R.; Jia, J.B.; Zhang, C.X. Facile synthesis of B/N co-doped porous carbon nanosheets with high heteroatom content and electrical conductivity for excellent-performance supercapacitors. Appl. Surf. Sci. 2022, 580, 152236. [Google Scholar] [CrossRef]
- Liu, Y.B.; Huang, G.X.; Li, Y.Y.; Yao, Y.H.; Liu, Q.R.; Xing, B.L.; Jia, J.B.; Zhang, C.X. Structural evolution of porous graphitic carbon nanosheets based on quinonyl decomposition for supercapacitor electrodes. Appl. Surf. Sci. 2021, 537, 147824. [Google Scholar] [CrossRef]
- Wu, Z.S.; Winter, A.; Chen, L.; Sun, Y.; Turchanin, A.; Feng, X.L.; Mullen, K. Three-Dimensional Nitrogen and Boron Co-doped Graphene for High-Performance All-Solid-State Supercapacitors. Adv. Mater. 2012, 24, 5130–5135. [Google Scholar] [CrossRef]
- Wee, J.H.; Kim, C.H.; Tojo, T.; Choi, G.B.; Yang, C.M.; Kim, Y.A. Boron-Doped Edges as Active Sites for Water Adsorption in Activated Carbons. Langmuir 2021, 37, 13179–13186. [Google Scholar] [CrossRef]
- Li, J.H.; Shi, C.; Bao, A.; Jia, J.C. Development of Boron-Doped Mesoporous Carbon Materials for Use in CO2 Capture and Electrochemical Generation of H2O2. ACS Omega 2021, 6, 8438–8446. [Google Scholar] [CrossRef]
- Shin, Y.; Park, S. Production of B-doped reduced graphene oxide using wet-process in tetrahydrofuran. Carbon Lett. 2021, 31, 887–893. [Google Scholar] [CrossRef]
- Ayala, P.; Plank, W.; Gruneis, A.; Kauppinen, E.I.; Rummeli, M.H.; Kuzmany, H.; Pichler, T. A one step approach to B-doped single-walled carbon nanotubes. J. Mater. Chem. 2008, 18, 5676–5681. [Google Scholar] [CrossRef]
- Lazzarini, A.; Colaiezzi, R.; Gabriele, F.; Crucianelli, M. Support-Activity Relationship in Heterogeneous Catalysis for Biomass Valorization and Fine-Chemicals Production. Materials 2021, 14, 6796. [Google Scholar] [CrossRef]
- Lazzarini, A.; Pellegrini, R.; Piovano, A.; Rudic, S.; Castan-Guerrero, C.; Torelli, P.; Chierotti, M.R.; Gobetto, R.; Lamberti, C.; Groppo, E. The effect of surface chemistry on the performances of Pd-based catalysts supported on activated carbons. Catal. Sci. Technol. 2017, 7, 4162–4172. [Google Scholar] [CrossRef]
- Lazzarini, A.; Piovano, A.; Pellegrini, R.; Leofanti, G.; Agostini, G.; Rudic, S.; Chierotti, M.R.; Gobetto, R.; Battiato, A.; Spoto, G.; et al. A comprehensive approach to investigate the structural and surface properties of activated carbons and related Pd-based catalysts. Catal. Sci. Technol. 2016, 6, 4910–4922. [Google Scholar] [CrossRef]
- Camilli, L.; Capista, D.; Eramo, P.; D’Archivio, A.A.; Maggi, M.A.; Lazzarini, A.; Crucianelli, M.; Passacantando, M. Synthesis of hydrophilic carbon nanotube sponge via post-growth thermal treatment. Nanotechnology 2022, 33, 245707. [Google Scholar] [CrossRef]
- Lazzarini, A.; Piovano, A.; Pellegrini, R.; Agostini, G.; Rudic, S.; Lamberti, C.; Groppo, E. Graphitization of activated carbons: A molecular-level investigation by INS, DRIFT, XRD and Raman techniques. Phys. Procedia 2016, 85, 20–26. [Google Scholar] [CrossRef]
- Hu, M.C.; Yao, Z.H.; Wang, X.Q. Characterization techniques for graphene-based materials in catalysis. AIMS Mater. Sci. 2017, 4, 755–788. [Google Scholar] [CrossRef]
- Bottari, G.; Herranz, M.A.; Wibmer, L.; Volland, M.; Rodriguez-Perez, L.; Guldi, D.M.; Hirsch, A.; Martin, N.; D’Souza, F.; Torres, T. Chemical functionalization and characterization of graphene-based materials. Chem. Soc. Rev. 2017, 46, 4464–4500. [Google Scholar] [CrossRef]
- Fraga, T.J.M.; Sobrinho, M.A.D.; Carvalho, M.N.; Ghislandi, M.G. State of the art: Synthesis and characterization of functionalized graphene nanomaterials. Nano Express 2020, 1, 022002. [Google Scholar] [CrossRef]
- Wani, T.U.; Mohi-ud-din, R.; Wani, T.A.; Mir, R.H.; Itoo, A.M.; Sheikh, F.A.; Khan, N.A.; Pottoo, F.H. Green Synthesis, Spectroscopic Characterization and Biomedical Applications of Carbon Nanotubes. Curr. Pharm. Biotechnol. 2021, 22, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Rathinavel, S.; Priyadharshini, K.; Panda, D. A review on carbon nanotube: An overview of synthesis, properties, functionalization, characterization, and the application. Mater. Sci. Eng. B 2021, 268, 115095. [Google Scholar] [CrossRef]
- Lazzarini, A. Activated carbons for applications in catalysis: The point of view of a physical-chemist. Rend. Lincei. Sci. Fis. Nat. 2017, 28, 29–42. [Google Scholar] [CrossRef]
- Panchakarla, L.S.; Govindaraj, A.; Rao, C.N.R. Boron- and nitrogen-doped carbon nanotubes and graphene. Inorg. Chim. Acta 2010, 363, 4163–4174. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Langmuir, I. The Adsorption of Gases on Plane Surfaces of Glass, Mica and Platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef]
- Thommes, M.; Cychosz, K.A.; Neimark, A.V. Advanced physical adsorption characterization of nanoporous carbons. In Novel Carbon Adsorbents; Tascòn, J.M.D., Ed.; Elsevier: Oxford, UK, 2012; pp. 107–145. [Google Scholar]
- Nguyen, V.T.; Do, D.D.; Nicholson, D. A new molecular model for water adsorption on graphitized carbon black. Carbon 2014, 66, 629–636. [Google Scholar] [CrossRef]
- Li, Z.Q.; Lu, C.J.; Xia, Z.P.; Zhou, Y.; Luo, Z. X-ray diffraction patterns of graphite and turbostratic carbon. Carbon 2007, 45, 1686–1695. [Google Scholar] [CrossRef]
- Li, K.C.; Bo, Z.; Yan, J.H.; Cen, K.F. Solid-state NMR Study of Ion Adsorption and Charge Storage in Graphene Film Supercapacitor Electrodes. Sci. Rep. 2016, 6, 39689. [Google Scholar] [CrossRef]
- Harris, R.K.; Wasylishen, R.E.; Duer, M.J. NMR Crystallography; Harris, R.K., Wasylishen, R.E., Duer, M.J., Eds.; John Wiley & Sons: Chichester, UK, 2009. [Google Scholar]
- Chierotti, M.R.; Gobetto, R. NMR crystallography: The use of dipolar interactions in polymorph and co-crystal investigation. Crystengcomm 2013, 15, 8599–8612. [Google Scholar] [CrossRef]
- Lee, S.I.; Saito, K.; Kanehashi, K.; Hatakeyama, M.; Mitani, S.; Yoon, S.H.; Korai, Y.; Mochida, I. 11B NMR study of the BF4- anion in activated carbons at various stages of charge of EDLCs in organic electrolyte. Carbon 2006, 44, 2578–2586. [Google Scholar] [CrossRef]
- Fulik, N.; Hippauf, F.; Leistenschneider, D.; Paasch, S.; Kaskel, S.; Brunner, E.; Borchardt, L. Electrolyte mobility in supercapacitor electrodes—Solid state NMR studies on hierarchical and narrow pore sized carbons. Energy Storage Mater. 2018, 12, 183–190. [Google Scholar] [CrossRef]
- Mark, L.O.; Dorn, R.W.; McDermott, W.P.; Agbi, T.O.; Altvater, N.R.; Jansen, J.; Lebron-Rodriguez, E.A.; Cendejas, M.C.; Rossini, A.J.; Hermans, I. Highly Selective Carbon-Supported Boron for Oxidative Dehydrogenation of Propane. ChemCatChem 2021, 13, 3611–3618. [Google Scholar] [CrossRef]
- Puziy, A.M.; Poddubnaya, O.I.; Socha, R.P.; Gurgul, J.; Wisniewski, M. XPS and NMR studies of phosphoric acid activated carbons. Carbon 2008, 46, 2113–2123. [Google Scholar] [CrossRef]
- Cao, E.P.; Chen, Z.M.; Wu, H.; Yu, P.; Wang, Y.; Xiao, F.; Chen, S.; Du, S.C.; Xie, Y.; Wu, Y.Q.; et al. Boron-Induced Electronic-Structure Reformation of CoP Nanoparticles Drives Enhanced pH-Universal Hydrogen Evolution. Angew. Chem. Int. Edit. 2020, 59, 4154–4160. [Google Scholar] [CrossRef] [PubMed]
- Matsoso, B.J.; Ranganathan, K.; Mutuma, B.K.; Lerotholi, T.; Jones, G.; Coville, N.J. Synthesis and characterization of boron carbon oxynitride films with tunable composition using methane, boric acid and ammonia. New J. Chem. 2017, 41, 9497–9504. [Google Scholar] [CrossRef]
- Sheng, Z.H.; Gao, H.L.; Bao, W.J.; Wang, F.B.; Xia, X.H. Synthesis of boron doped graphene for oxygen reduction reaction in fuel cells. J. Mater. Chem. 2012, 22, 390–395. [Google Scholar] [CrossRef]
- Fujisawa, K.; Cruz-Silva, R.; Yang, K.S.; Kim, Y.A.; Hayashi, T.; Endo, M.; Terrones, M.; Dresselhaus, M.S. Importance of open, heteroatom-decorated edges in chemically doped-graphene for supercapacitor applications. J. Mater. Chem. A 2014, 2, 9532–9540. [Google Scholar] [CrossRef]
- Lin, Y.M.; Zhu, Y.S.; Zhang, B.S.; Kim, Y.A.; Endo, M.; Su, D.S. Boron-doped onion-like carbon with enriched substitutional boron: The relationship between electronic properties and catalytic performance. J. Mater. Chem. A 2015, 3, 21805–21814. [Google Scholar] [CrossRef]
- Schlögl, R. Carbons. In Handbook of Heterogeneous Catalysis; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, DE, USA, 2008; pp. 357–427. [Google Scholar]
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electron-phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 143, 47–57. [Google Scholar] [CrossRef]
- Endo, M.; Hayashi, T.; Hong, S.H.; Enoki, T.; Dresselhaus, M.S. Scanning tunneling microscope study of boron-doped highly oriented pyrolytic graphite. J. Appl. Phys. 2001, 90, 5670–5674. [Google Scholar] [CrossRef]
- Atkins, P.; de Paula, J. (Eds.) Elements of Physical Chemistry, 5th ed.; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Deshpande, S.V.; Gulari, E.; Harris, S.J.; Weiner, A.M. Filament activated chemical vapor deposition of boron carbide coatings. Appl. Phys. Lett. 1994, 65, 1757–1759. [Google Scholar] [CrossRef]
- Romanos, J.; Beckner, M.; Stalla, D.; Tekeei, A.; Suppes, G.; Jalisatgi, S.; Lee, M.; Hawthorne, F.; Robertson, J.D.; Firlej, L.; et al. Infrared study of boron-carbon chemical bonds in boron-doped activated carbon. Carbon 2013, 54, 208–214. [Google Scholar] [CrossRef]
- Cote, A.P.; Benin, A.I.; Ockwig, N.W.; O’Keeffe, M.; Matzger, A.J.; Yaghi, O.M. Porous, crystalline, covalent organic frameworks. Science 2005, 310, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Aoqui, S.; Miyata, H.; Ohshima, T.; Ikegami, T.; Ebihara, K. Preparation of boron carbide thin film by pulsed KrF excimer laser deposition process. Thin Solid Films 2002, 407, 126–131. [Google Scholar] [CrossRef]
- Sankaran, M.; Viswanathan, B. Hydrogen storage in boron substituted carbon nanotubes. Carbon 2007, 45, 1628–1635. [Google Scholar] [CrossRef]
- Podder, J.; Rusop, M.; Soga, T.; Jimbo, T. Boron doped amorphous carbon thin films grown by r.f. PECVD under different partial pressure. Diam. Relat. Mat. 2005, 14, 1799–1804. [Google Scholar] [CrossRef]
- Zheng, M.; Xu, Z.N.; Fu, M.L. Boron-doped activated carbon catalyzed reduction of dilute nitrite acid in oxidative esterification reaction in the coal to ethylene glycol process. J. Environ. Chem. Eng. 2022, 10, 107932. [Google Scholar] [CrossRef]
- Dong, G.H.; Sun, B.B.; Su, T.; Hao, L.J.; Chai, D.F.; Zhang, W.Z.; Zhang, Z.F.; Zhao, M.; Li, J.L. In Situ Construction of Hierarchical Nanoflower-Like MnO2/Biomass-Based Boron-Doped Carbon Spheres for Oxygen Evolution Reaction. J. Electrochem. Soc. 2022, 169, 054508. [Google Scholar] [CrossRef]
- Piovano, A.; Lazzarini, A.; Pellegrini, R.; Leofanti, G.; Agostini, G.; Rudic, S.; Bugaev, A.L.; Lamberti, C.; Groppo, E. Progress in the Characterization of the Surface Species in Activated Carbons by means of INS Spectroscopy Coupled with Detailed DFT Calculations. Adv. Condens. Matter Phys. 2015, 2015, 803267. [Google Scholar] [CrossRef]
- Albers, P.W.; Pietsch, J.; Krauter, J.; Parker, S.F. Investigations of activated carbon catalyst supports from different natural sources. Phys. Chem. Chem. Phys. 2003, 5, 1941–1949. [Google Scholar] [CrossRef]
- Sears, V.F. Neutron scattering lengths and cross sections. Neutron News 1992, 3, 26–37. [Google Scholar] [CrossRef]
- Glendinning, S.G.; El-Kadi, S.; Nelson, C.E.; Pedroni, R.S.; Purser, F.O.; Walter, R.L.; Beyerle, A.G.; Gould, C.R.; Seagondollar, L.W.; Thambidurai, P. Elastic and Inelastic Neutron Cross Sections for Boron-10 and Boron-11. Nucl. Sci. Eng. 1982, 80, 256–262. [Google Scholar] [CrossRef]
- Gompf, F.; Reichardt, W.; Schober, H.; Renker, B.; Buchgeister, M. Lattice vibrations and electron-phonon coupling in superconducting quarternary borocarbides: An inelastic neutron-scattering investigation. Phys. Rev. B 1997, 55, 9058–9066. [Google Scholar] [CrossRef]
- Liu, Y.; Brown, C.M.; Blackburn, J.L.; Neumann, D.A.; Gennett, T.; Simpson, L.; Parilla, P.; Dillon, A.C.; Heben, M. Inelastic neutron scattering of H2 adsorbed on boron substituted single walled carbon nanotubes. J. Alloys Compd. 2007, 446, 368–372. [Google Scholar] [CrossRef]
- Carosso, M.; Lazzarini, A.; Piovano, A.; Pellegrini, R.; Morandi, S.; Manzoli, M.; Vitillo, J.G.; Ruiz, M.J.; Lamberti, C.; Groppo, E. Looking for the active hydrogen species in a 5wt% Pt/C catalyst: A challenge for inelastic neutron scattering. Faraday Discuss. 2018, 208, 227–242. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal(s) | Hydride | Capacity (wt%) | Peq (MPa) | T (K) | ΔH (kJ/mol) | Refs. |
|---|---|---|---|---|---|---|
| Pd | PdH2 | 0.56 | 0.002 | 298 | 41 | [51,52] |
| Mg | MgH2 | 7.6 | 0.19 | 573 | 75.4 | [51,52,54] |
| Ti | TiH2 | 4.0 | 0.09 | 909 | 125.6 | [51,52] |
| V | VH2 | 2.1 | 0.37 | 313 | 58.6 | [51,52] |
| FeTi | FeTiH1.94 | 1.89 | 0.5 | 303 | 28 | [51,52,54] |
| LaNi5 | LaNi5H6 | 1.37 | 0.2 | 298 | 31 | [51,52,54] |
| Mg2Ni | Mg2NiH4 | 3.59 | 0.1 | 555 | 65 | [51,52,54] |
| NaAl | NaAlH4 | 8.0 | 9 | 403 | 113 | [51,52,54,55] |
| Ti0.9Al0.06V0.04 | Ti0.9Al0.06V0.04H2 | 3.8 | 0.1 | 300 | NR | [51,52,53] |
| Precursor | BET Surface Area (m2/g) | Activation Method | P (bar) | T (K) | H2 Storage (wt%) | Refs. |
|---|---|---|---|---|---|---|
| Coffee-bean wastes | 2070 | KOH | 120 40 | 77 298 | 4.0 0.6 | [91] |
| Peanut shell | 1726 | KOH | 100 | 298 | 1.2 | [92] |
| Coconut shell | 2800 | KOH | 100 | 297 | 0.8 | [93] |
| Corncob | 3012 | KOH | 1 50 | 77 298 | 2.0 0.4 | [94] |
| Rice hull | 3969 | NaOH/ΔT | 12 | 77 | 7.7 | [95] |
| Biomass wood | 2450 | H3PO4/KOH | 20 | 298 | 0.8 | [96] |
| Bamboo | 3148 | KOH | 1 40 | 77 | 2.7 6.5 | [97] |
| Sword-bean shells | 2838 | KOH/ΔT | 1 40 | 77 | 2.6 5.7 | [98] |
| Beer lees | 1927 | KOH | 1 | 77 | 2.9 | [99] |
| Melaleuca bark | 3170 | KOH | 10 | 77 | 4.1 | [100] |
| Coconut shells | 415 | O2/573 K/6 h | 40 100 | 298 | 0.3 0.5 | [101,102] |
| Lignin | 1000 | CO2/1273 K/1 h | 1 | 77 | 1.8 | [103] |
| Cornstalks | 3200 | KOH | 40 | 77 | 4.4 | [104] |
| Olive stones | 1269 | KOH | 200 | 77 | 6.0 | [105] |
| Palmyra sprouts | 2090 | KOH | 15 | 298 | 1.1 | [106] |
| Cannabissativa L. | 3241 | KOH | 1 | 77 | 3.3 | [107] |
| Fungi-based chars | 2500 | KOH | 1 35 | 77 | 2.4 4.7 | [108] |
| Chitosan | 3066 | KOH | 1 40 | 77 | 2.9 5.6 | [109] |
| Neolamarckia cadamba | 3462 | KOH | 1 | 77 | 2.8 | [110] |
| Posidonia oceanica | 2800 | KOH | 80 | 77 | 6.3 | [111] |
| Pinecones | 1173 | KOH | 1 80 | 77 | 1.6 5.5 | [81] |
| Empty fruit bunch | 687 | KOH/CO2 | 19 | 77 | 2.1 | [112] |
| Rice husk | 1490 | H3PO4/ΔT | 10 | 77 | 1.8 | [113] |
| Lychee trunk | 3400 | KOH KOH–Pd (10 wt%) | 1 60 | 77 303 | 2.9 0.5 | [114] |
| Celluloseacetate | 3800 | HTC | 20 30 | 77 | 8.1 8.9 | [115] |
| Entry | Carbon Type | Synthesis | Boron Doping | Boron Amount (wt%) | Refs. |
|---|---|---|---|---|---|
| 1 | MWCNTs | MP-CVD | B(OCH3)3; in situ | n.q. † | [186] |
| 2 | CVD | BF3-MeOH; in situ | <0.5 | [187] | |
| 3 | ECR-CVD | B2H6; in situ | 4.2–38.9 | [185] | |
| 4 | CVD | 2.8 | [192] | ||
| 5 | SWCNTs | HW-CVD | C9H21BO3; in situ | n.q. † | [184] |
| 6 | n.q. † | [211] | |||
| 7 | HV-CVD | <0.5 | [190] | ||
| 8 | MWCNTs | HF-CVD | B2O3/EtOH; in situ | n.q. † | [183] |
| 9 | H3BO3/MeOH; in situ | 1.0 | [191] | ||
| 10 | MWCNTs | Arc Discharge | Elemental B | 2.0 | [194] |
| 11 | 0.5–4.0 | [195] | |||
| 12 | n.q. † | [196] | |||
| 13 | SWCNTs | Arc Discharge | BC4N | 1.0–5.0 | [197,198] |
| 14 | CoNiB alloy | 0.97–1.2 | [199] | ||
| 15 | SWCNTs | Laser Ablation | Elemental B | 0.5–10.0 | [200,201] |
| 16 | 4.5 | [202] | |||
| 17 | Activated Carbon | Solid-Phase Substitution | H3BO3; before Carbonization | 4.3 | [203] |
| 18 | 1.3 | [209] | |||
| 19 | B2O3; post-activation | 1.0–6.0 | [205] | ||
| 20 | 1.4–1.8 | [208] | |||
| 21 | H3BO3; post-activation | 2.0 | [204] | ||
| 22 | B(OEt)3; before carbonization | 12.7 | [173] | ||
| 23 | Graphite oxide | Liquid-Phase Substitution | BH3-THF; post-activation | 0.9–2.4 | [210] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazzarini, A.; Marino, A.; Colaiezzi, R.; De Luca, O.; Conte, G.; Policicchio, A.; Aloise, A.; Crucianelli, M. Boronation of Biomass-Derived Materials for Hydrogen Storage. Compounds 2023, 3, 244-279. https://doi.org/10.3390/compounds3010020
Lazzarini A, Marino A, Colaiezzi R, De Luca O, Conte G, Policicchio A, Aloise A, Crucianelli M. Boronation of Biomass-Derived Materials for Hydrogen Storage. Compounds. 2023; 3(1):244-279. https://doi.org/10.3390/compounds3010020
Chicago/Turabian StyleLazzarini, Andrea, Alessia Marino, Roberta Colaiezzi, Oreste De Luca, Giuseppe Conte, Alfonso Policicchio, Alfredo Aloise, and Marcello Crucianelli. 2023. "Boronation of Biomass-Derived Materials for Hydrogen Storage" Compounds 3, no. 1: 244-279. https://doi.org/10.3390/compounds3010020
APA StyleLazzarini, A., Marino, A., Colaiezzi, R., De Luca, O., Conte, G., Policicchio, A., Aloise, A., & Crucianelli, M. (2023). Boronation of Biomass-Derived Materials for Hydrogen Storage. Compounds, 3(1), 244-279. https://doi.org/10.3390/compounds3010020