
New Polymers In Silico Generation and Properties Prediction

 , and
, and
Abstract
:
1. Introduction
- (1)
- Most of the published works using neural networks cannot be reproduced because the detailed configuration of the NN, e.g., the activation function weights, is not provided;
- (2)
- If a structural fragment is missing from the regression model, then its contribution to the property is assumed to be zero. In this respect, Bicerano’s models differ favorably from Askadskii’s [12] or Van Crevelen’s [13] models, where the absence of an increment for an atom with nearest neighbors [12] or a structural fragment [13] makes property prediction impossible;
- (3)
- Bicerano’s approach uses the similar models to predict a large number of properties. This simplifies the program code;
- (4)
- High computational speed, which is especially important when processing large amounts of data. The number of fragments calculated from the 2D structure is simply substituted into the equation with coefficients taken from Ref. [11];
- (5)
- A very high-quality presentation of sample calculations for the created models. For example, the tables in Ref. [11] contain not only the final results but also the intermediate calculated data: the number of fragments used in the model in the SRU structures and some intermediate parameters. This makes debugging the code much easier.
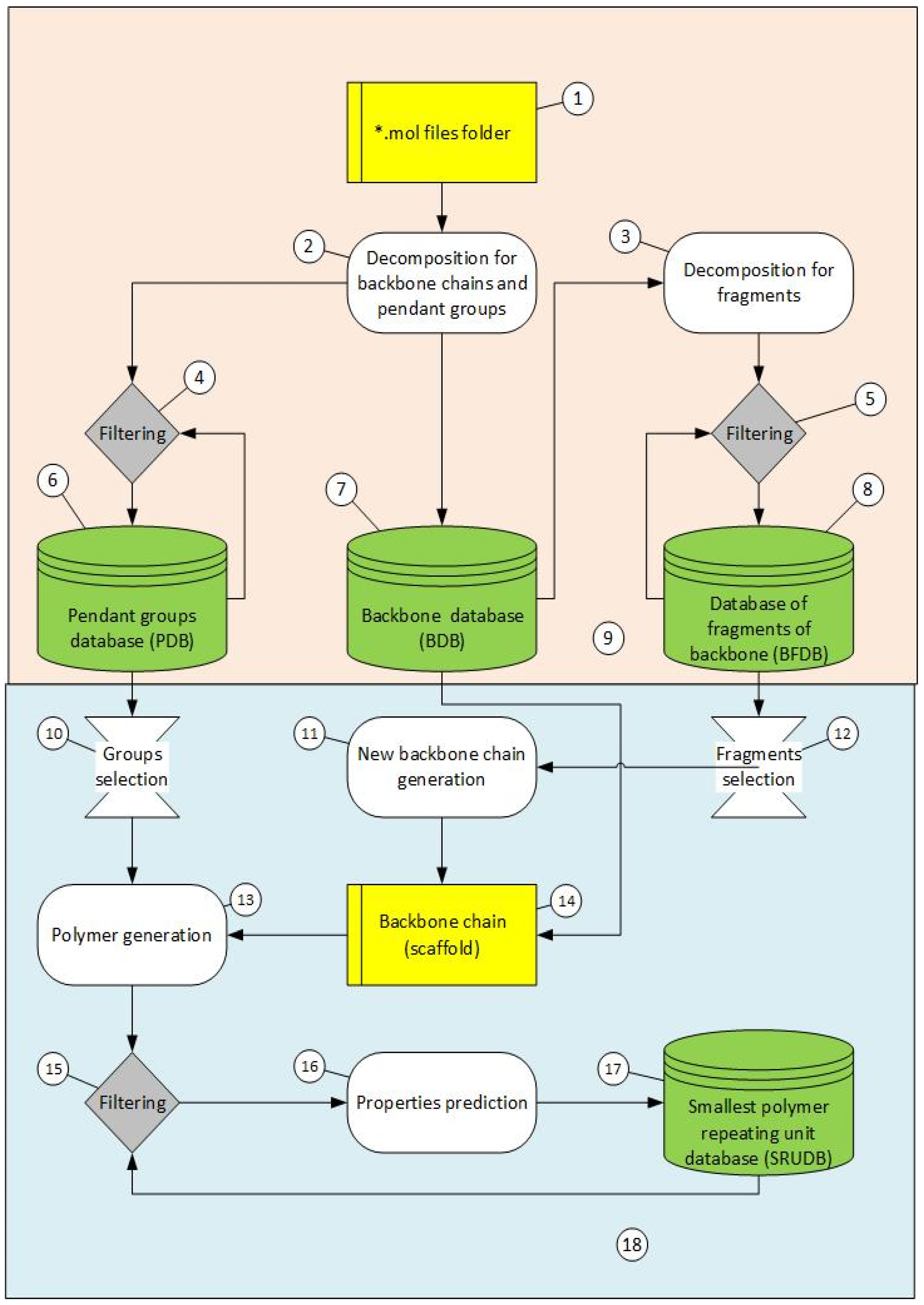
2. Materials and Methods
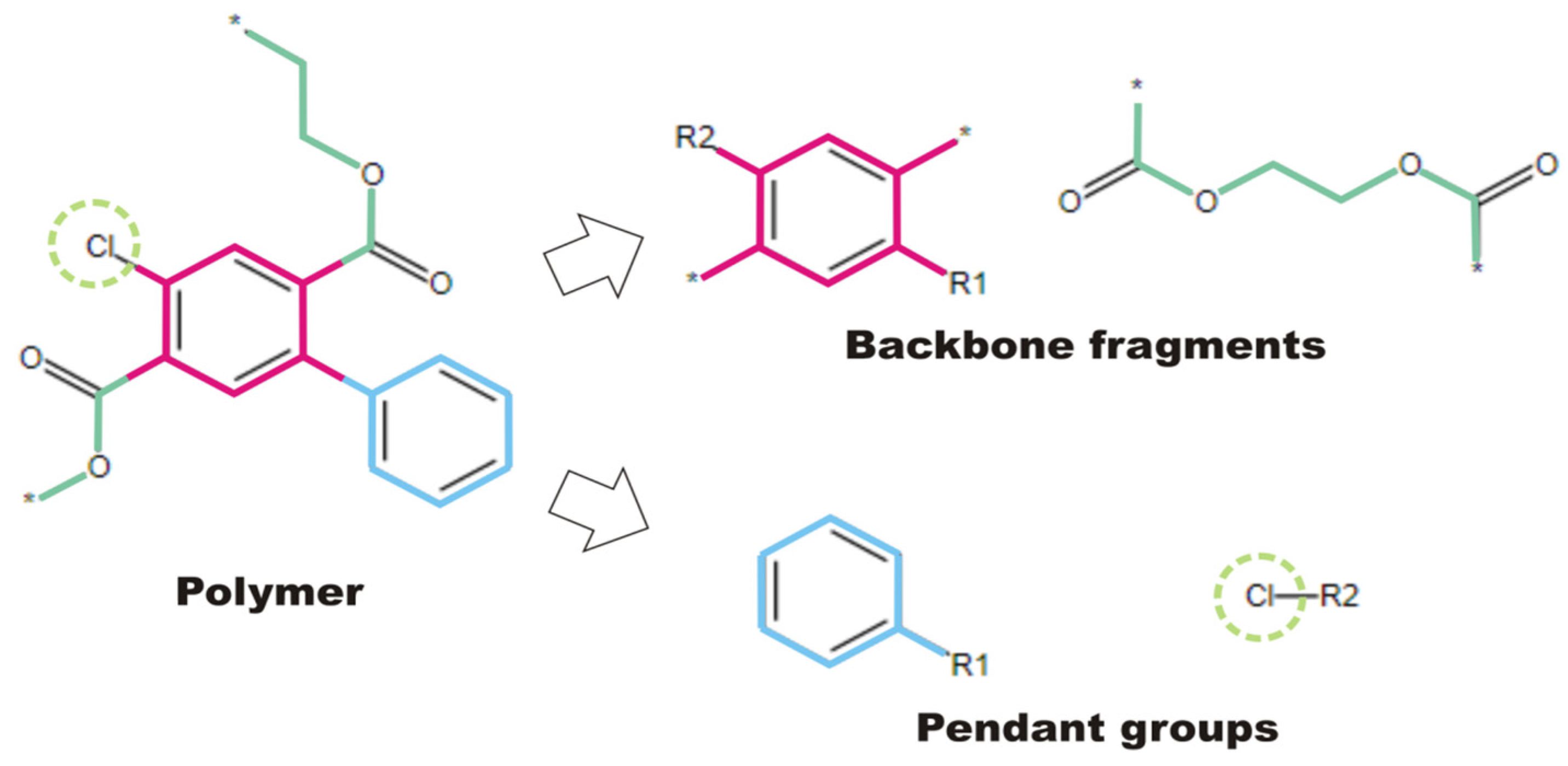
2.1. Creation of FDBs
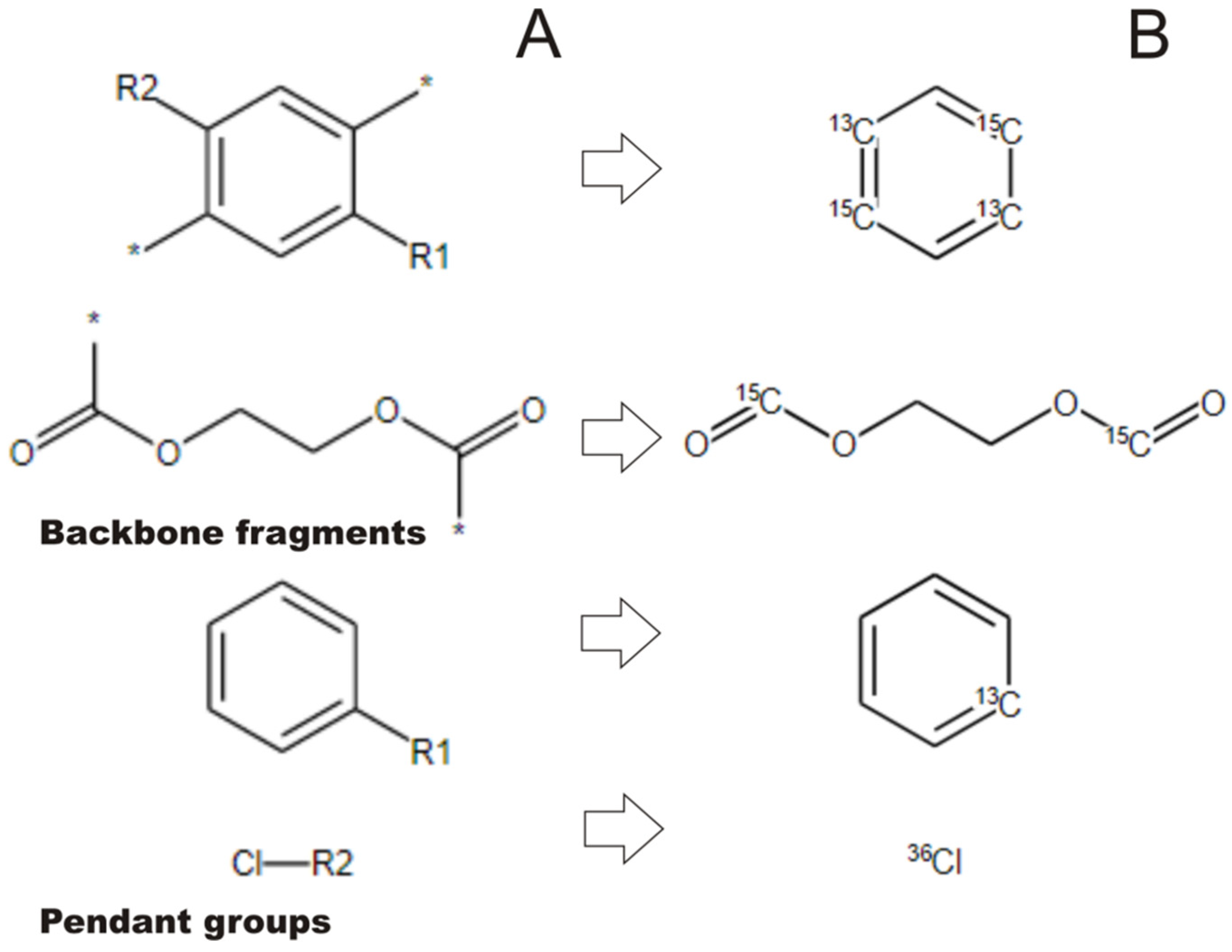
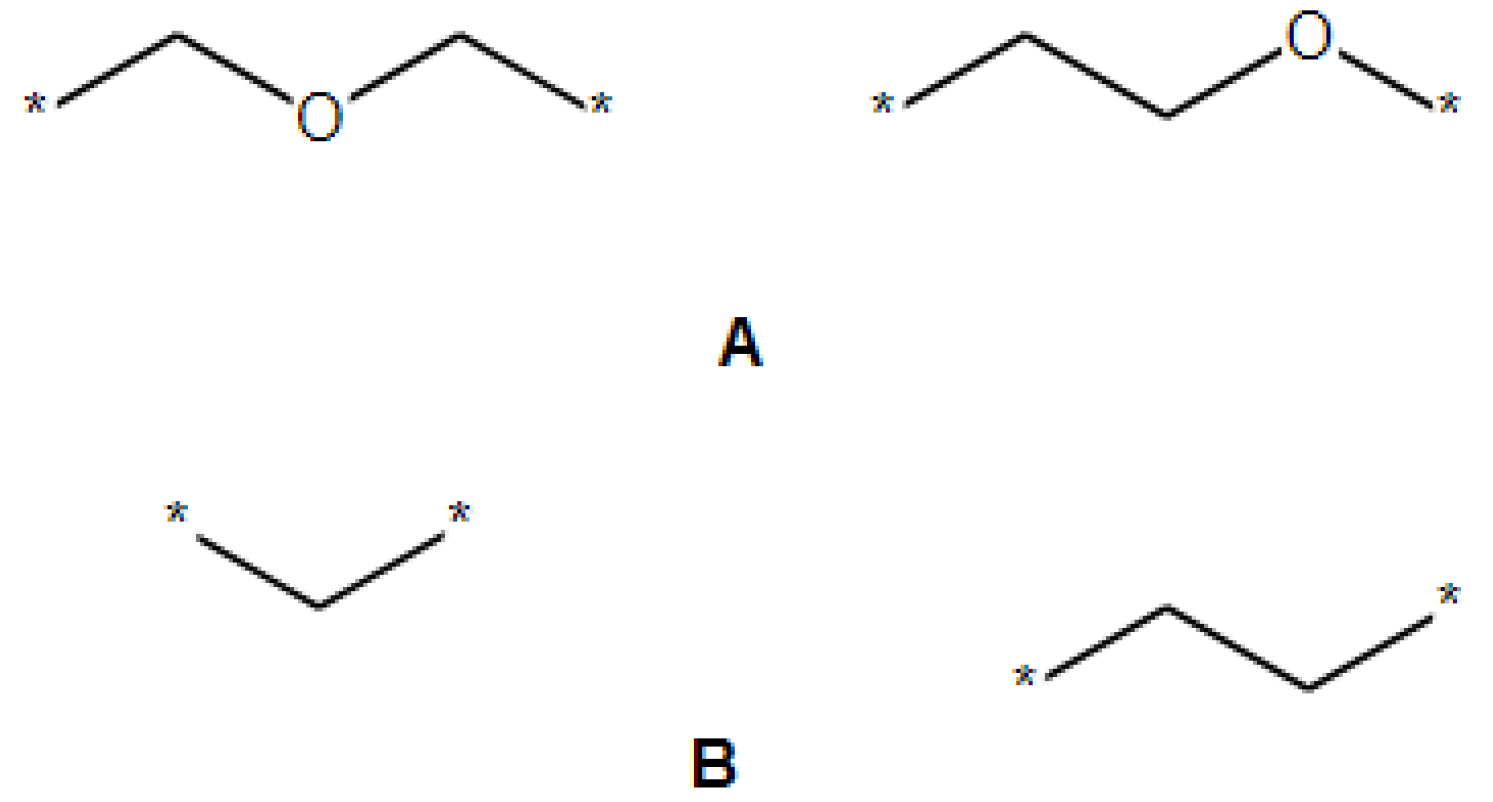
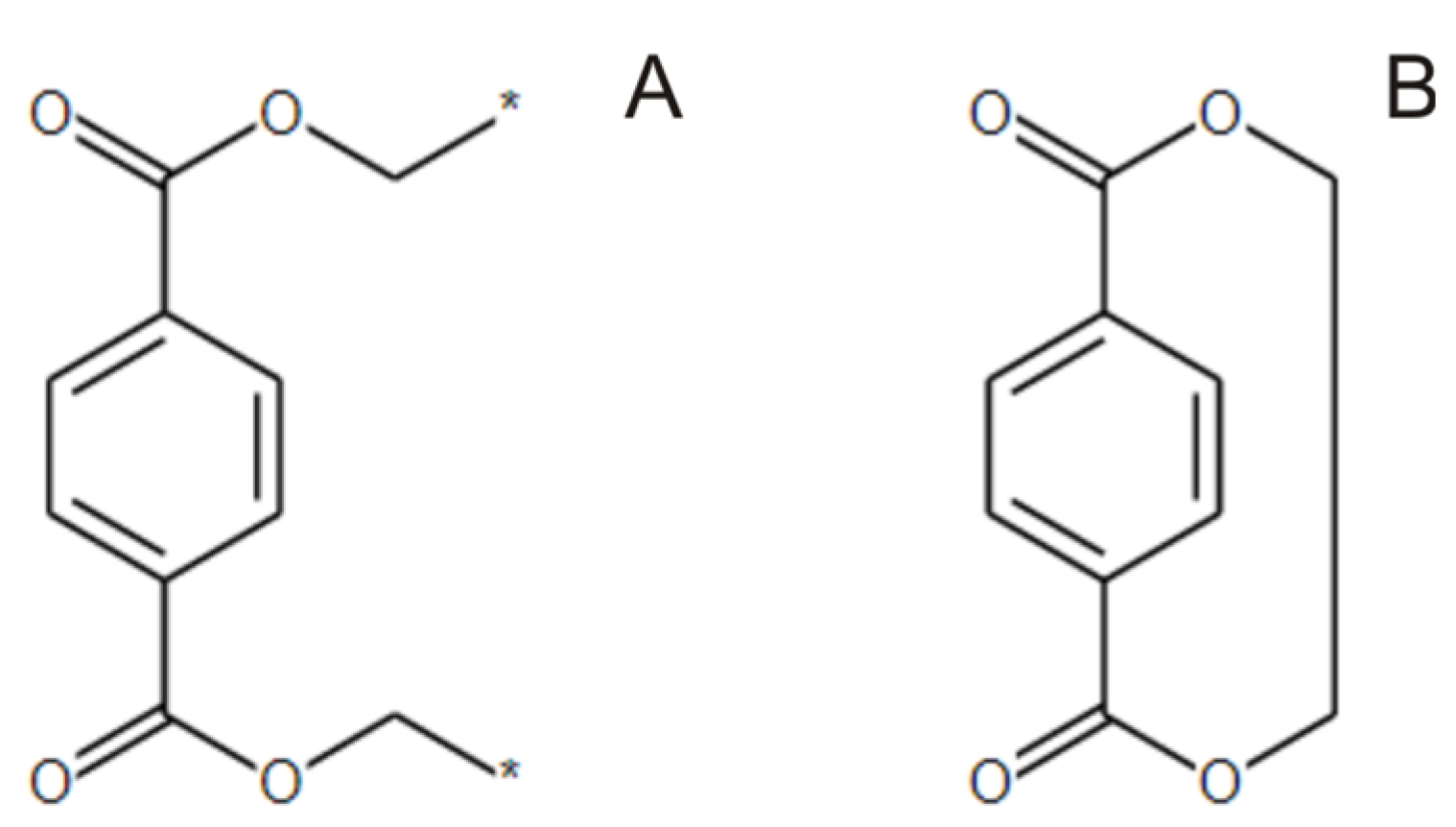
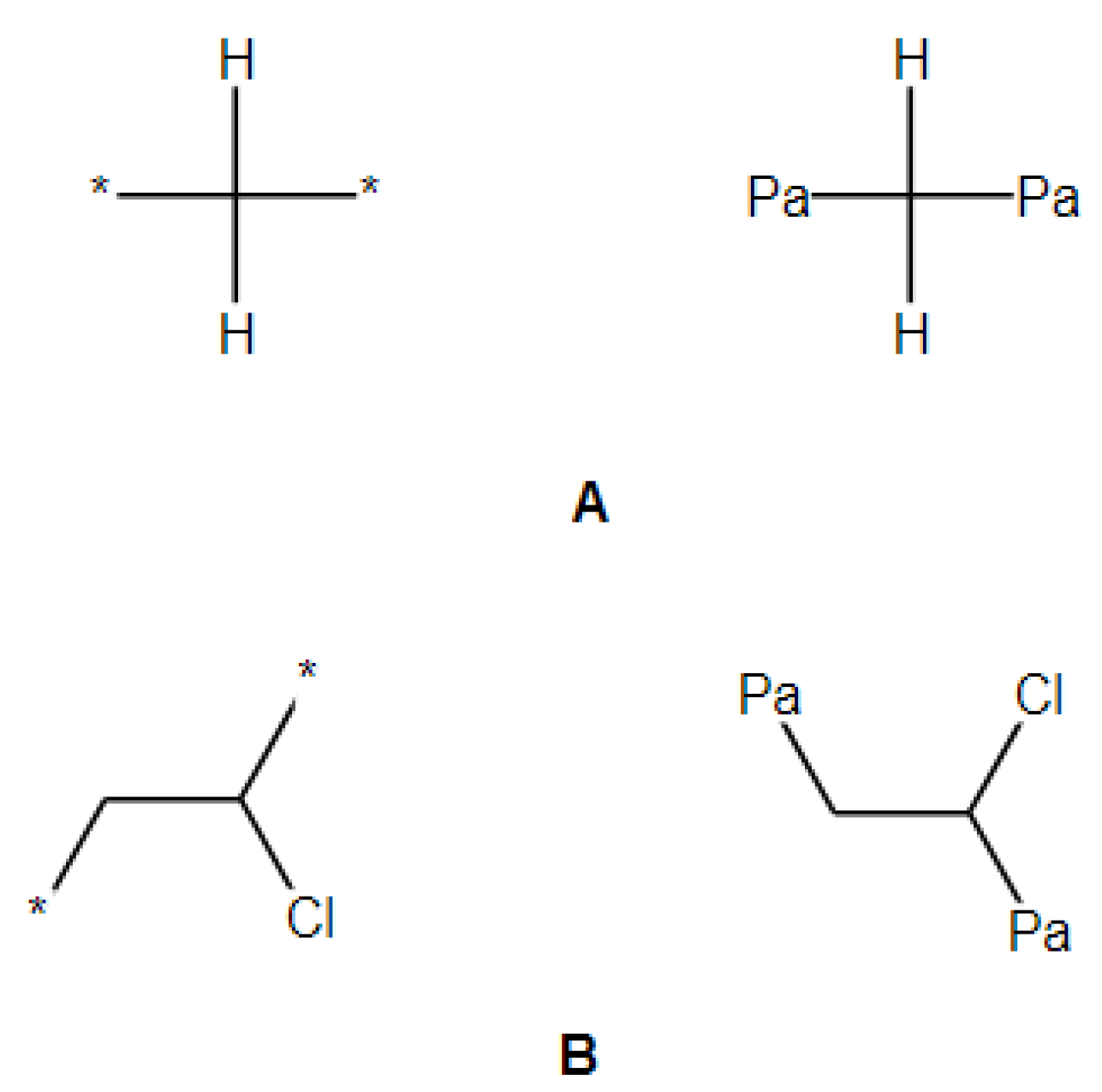
2.2. Transforming Chemical Structures before Searching for Duplicates
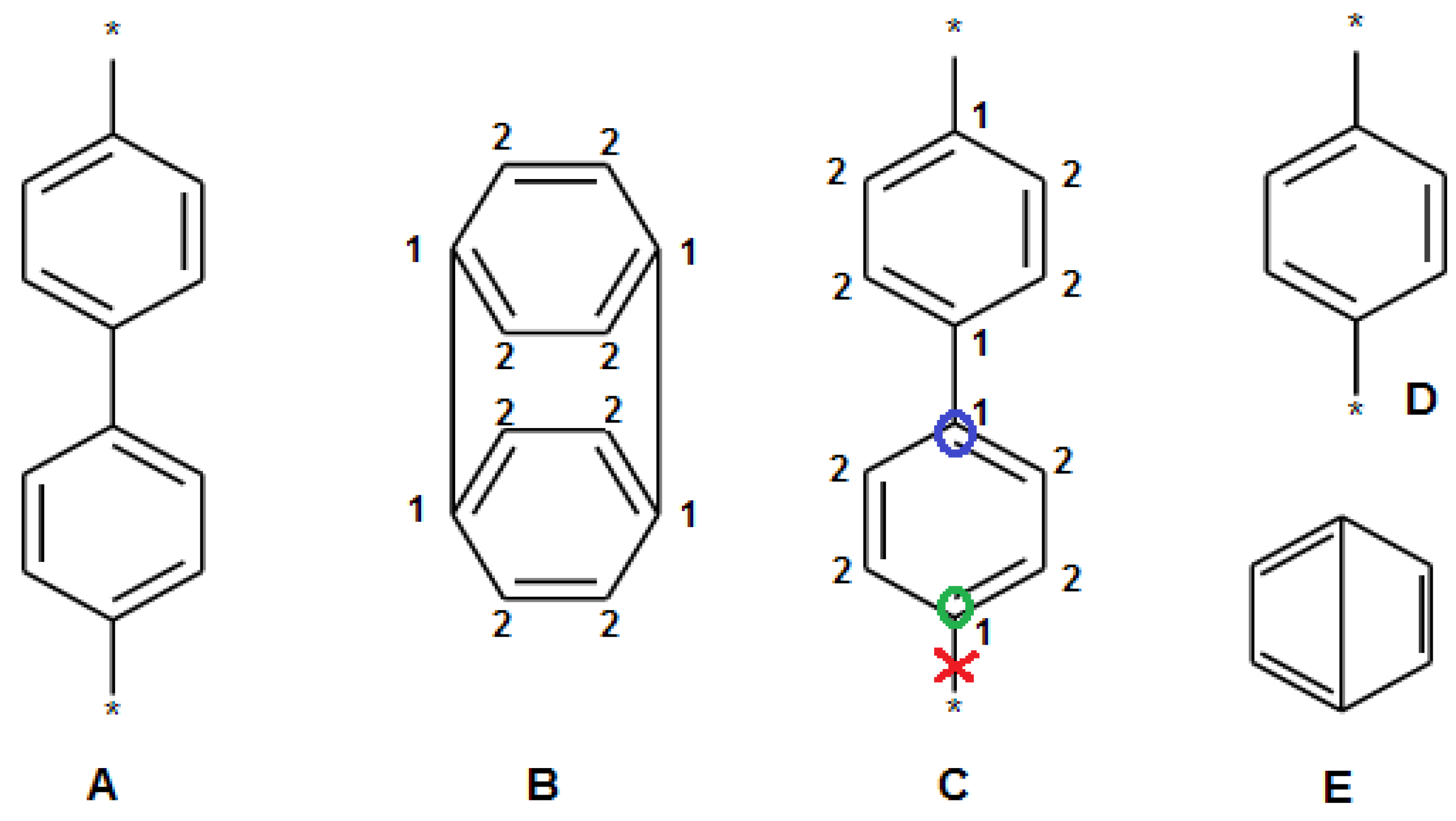
2.3. Filtration of Polymeric SRU Structures
- (1)
- Dummy atoms with an asterisk (*) to mark the SRU continuation;
- (2)
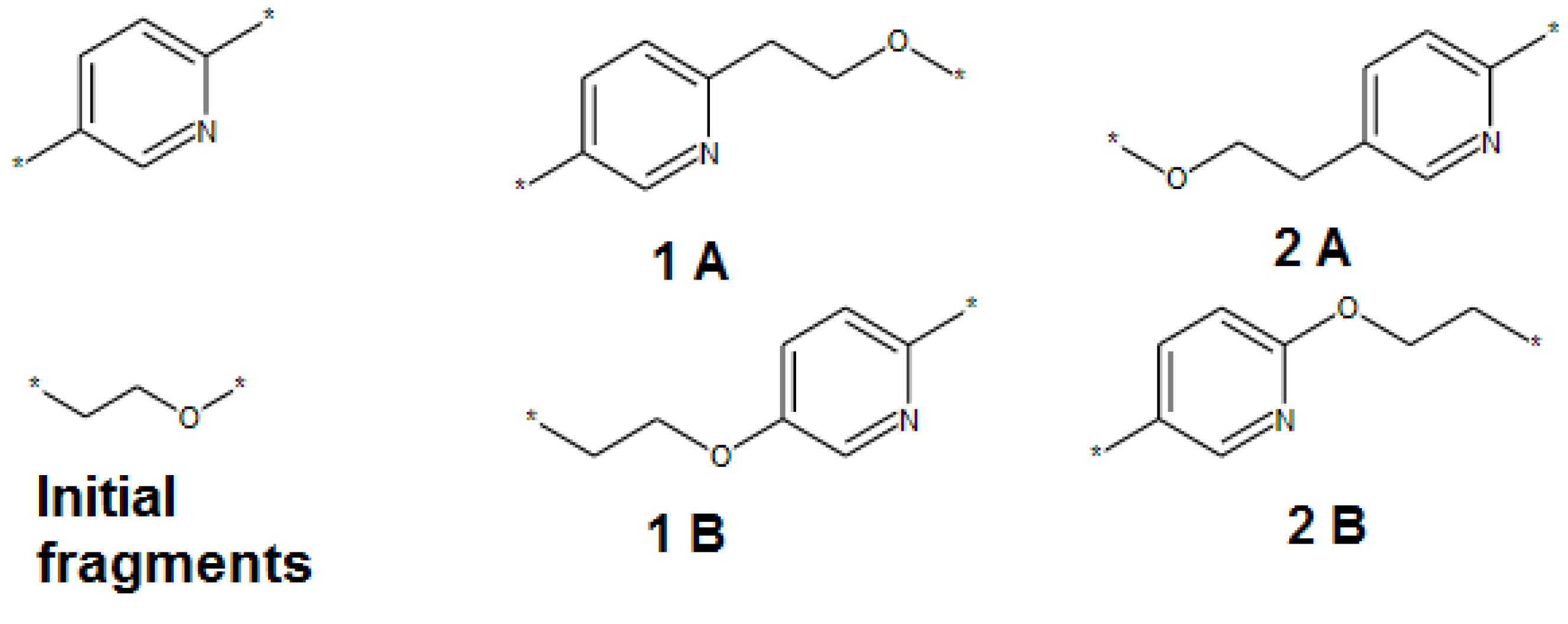
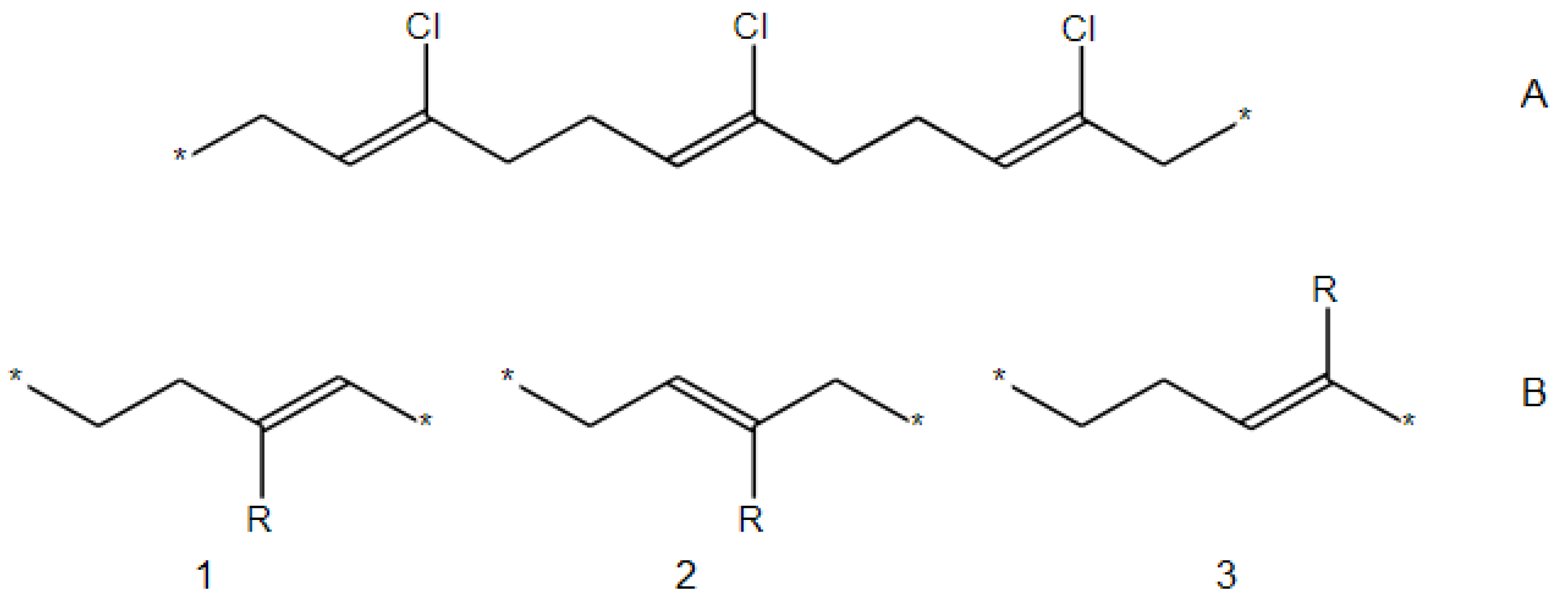
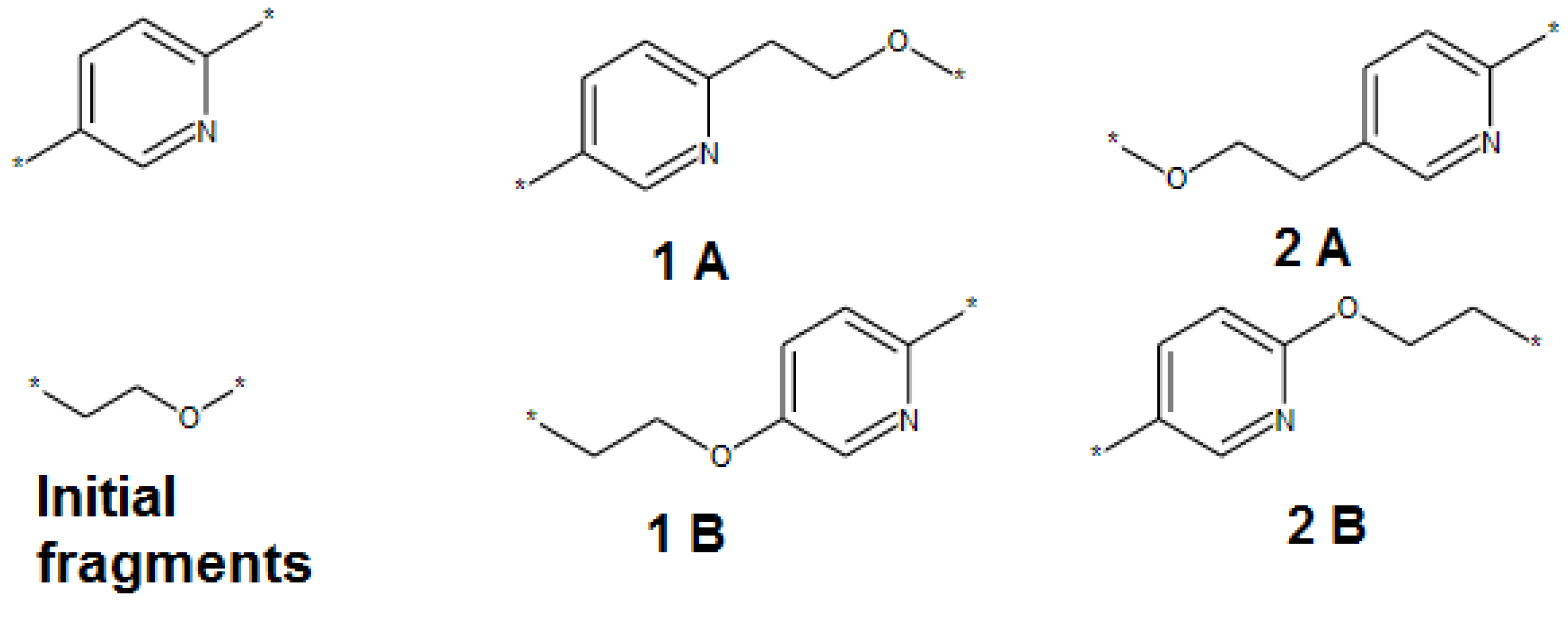
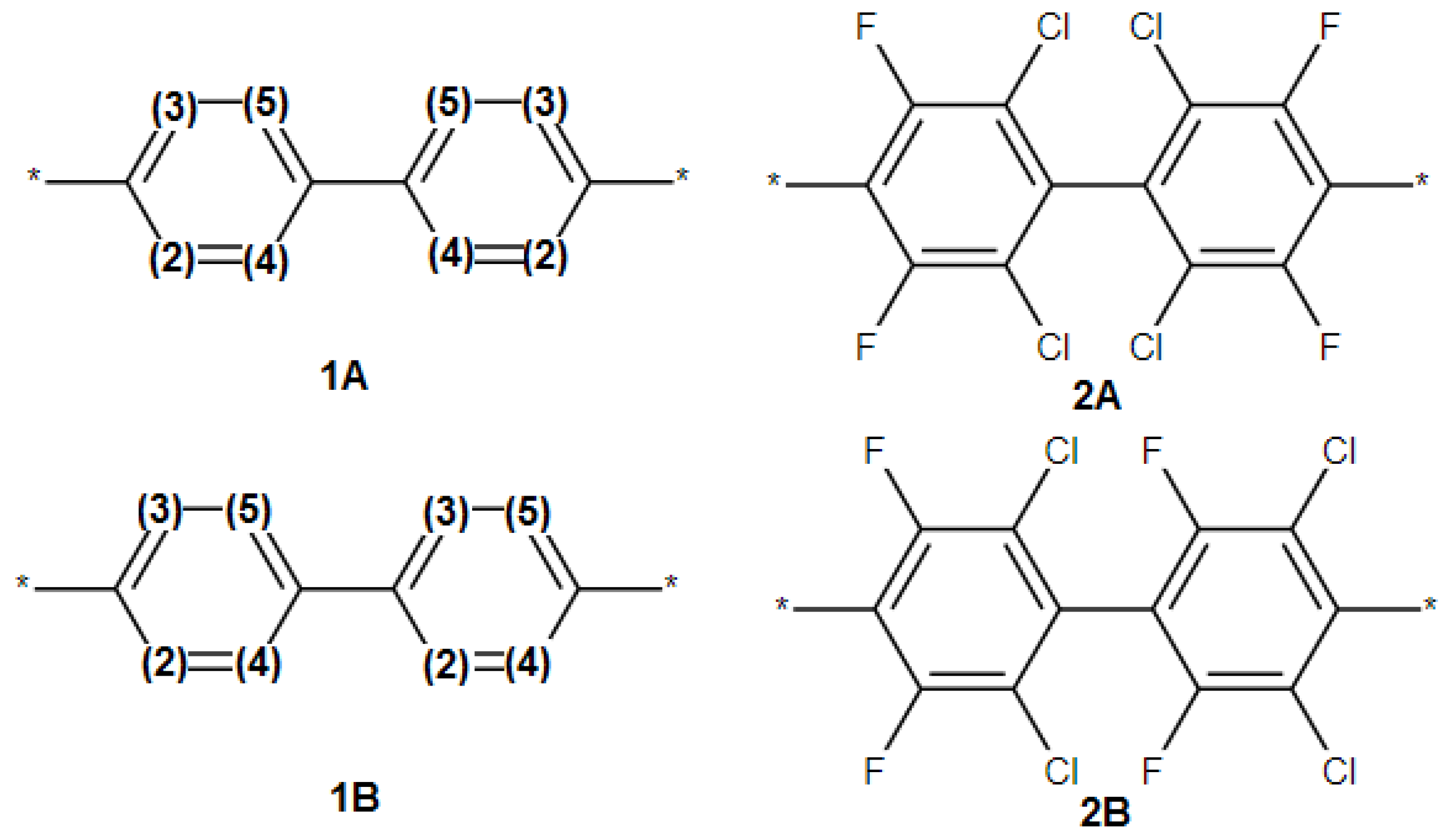
- The SRU may be represented by several equivalent chemical structures that are formally different (Figure 5A);
- (3)
- The polymer repeat unit may contain multiple SRUs, as shown in Figure 5B. For such chemical structures, the corresponding InChIkey must be identical to the InChIkey index generated for the backbone consisting of a single SRU.
2.4. Generation of New Polymer SRU Structures
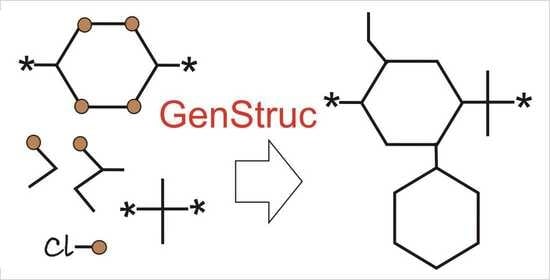
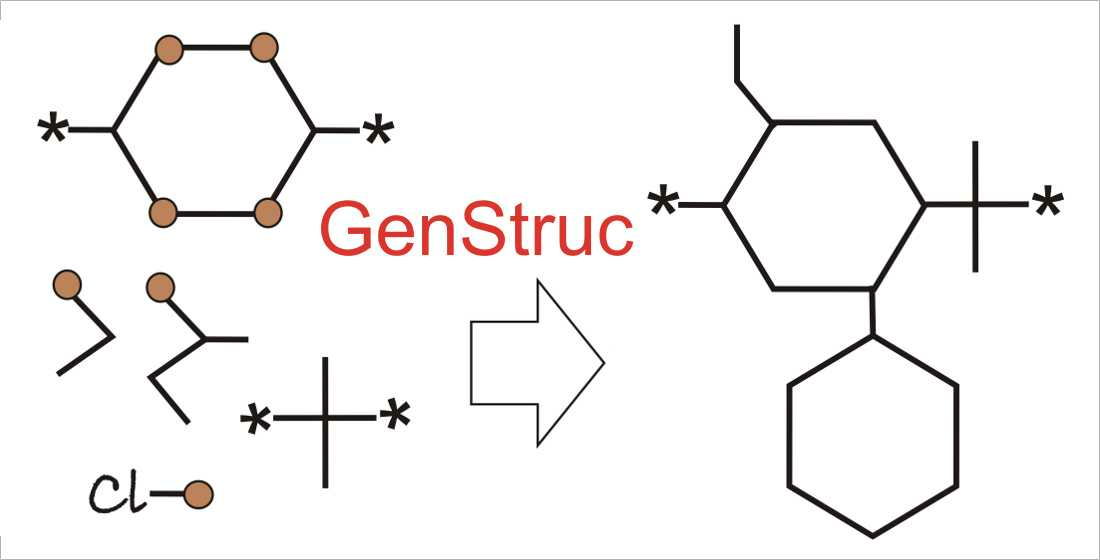
2.4.1. Polymer SRU Backbone Generation (Scaffold)
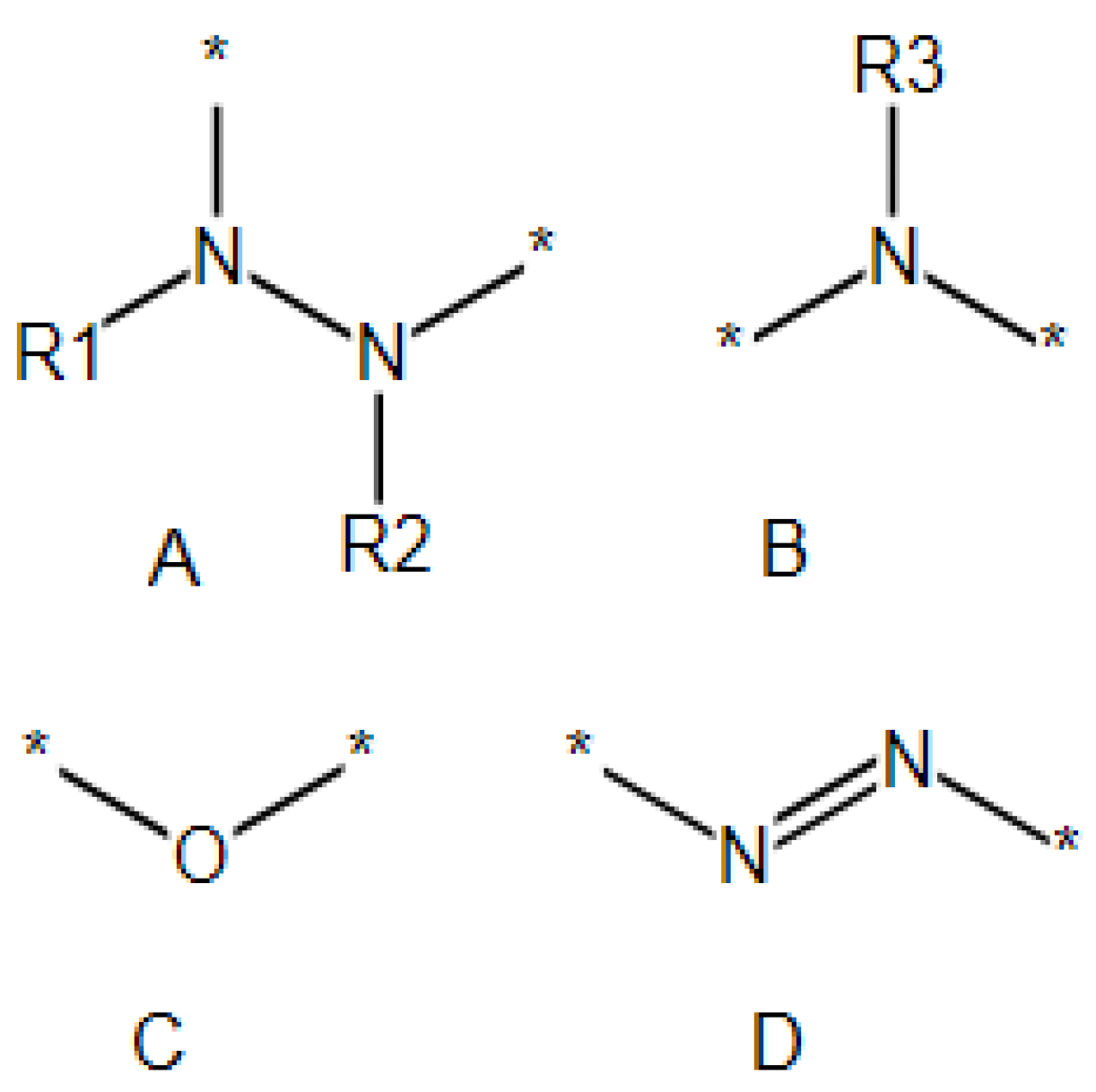
2.4.2. Adding Pendant Groups
2.5. The Program Description
2.5.1. GenStruc Program
- (1)
- Selection of parts of the databases from the backbone fragments and the pendant groups for the generation process;
- (2)
- Two generation methods: (a) enumeration of all available combinations of backbone fragments and pendant groups and (b) Monte Carlo algorithm;
- (3)
- In addition to the connecting points of the pendant groups extracted from the initial polymer set (Figure 1 point 1), hydrogen atoms attached to the backbone can also be used as additional connecting points;
- (4)
- One can specify the list of pendant groups for each atom of the backbone;
- (5)
- Setting the number of the fragments to generate the backbone of the polymer SRU. It is possible to use a variable number of fragments to generate the backbone as well as to filter the backbone by molecular weight;
- (6)
- Setting the weights of pendant groups when using the Monte Carlo algorithm. In this version of the program, the weights are the same for all connection points, but in the future it is planned to implement individual weights for each connection point;
- (7)
- Stopping the calculations when the specified number of chemical structures or the specified run time is reached;
- (8)
- The built-in filter blocks the formation of oxygen–oxygen and oxygen–nitrogen bonds in the backbone and halogen–nitrogen bonds when pendant groups are added. If such bonds are present, the structure is discarded and the program moves on to the next compound (the enumeration algorithm) or the new backbone fragments and pendant groups are reselected for SRU generation (the Monte Carlo algorithm).
2.5.2. PolyPred Program
- (1)
- Allowed chemical elements in the polymer composition are C, H, N, O, F, Si, S, Cl, and Br;
- (2)
- Two asterisk atoms are used to denote an SRU. The program does not handle carcass structures, grafted chains and block copolymers, or spatial polymers (where multiple asterisks must be used to denote an SRU);
- (3)
- Each asterisk must have a single bond to a single atom;
- (4)
- Polymers with isotopes are not processed; all isotope labels are removed before processing.
2.5.3. Program Generation Run
3. Results
3.1. The Design of “Hits”
3.2. Design of New Polymers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SRU | smallest polymer repeating unit |
| SRUDB | smallest polymer repeating unit database |
| PDB | pendant groups database |
| BDB | backbone database |
| BFDB | backbone fragments database |
| FDBs | fragments databases. Includes PDB, BDB, and BFDB |
References
- Patra, T.K. Data-Driven Methods for Accelerating Polymer Design. ACS Polym. Au 2022, 2, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Hiemenz, P.C.; Lodge, T.P. Polymer Chemistry, 2nd ed.; CRC: New York, NY, USA, 2007; pp. 1–575. [Google Scholar]
- Feldman, D. Polymer History. Des. Monomers Polym. 2008, 11, 1–15. [Google Scholar] [CrossRef]
- Mariello, M.; Kim, K.; Wu, K.; Lacour, S.P.; Leterrier, Y. Recent Advances in Encapsulation of Flexible Bioelectronic Implants: Materials, Technologies, and Characterization Methods. Adv. Mater. 2022, 34, e2201129. [Google Scholar] [CrossRef] [PubMed]
- Heath-Apostolopoulos, I.; Wilbraham, L.; Zwijnenburg, M.A. Computational high-through put screening of polymeric photocatalysts: Exploring the effect of composition, sequence isomerism and conformational degrees of freedom. Faraday Discuss. 2019, 215, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Ruipérez, F. High-Performance Quantum Chemical Calculations for Polymers. Int. Rev. Phys. Chem. 2019, 38, 343–403. [Google Scholar] [CrossRef]
- Shah, M.R.; Yadav, G.D. Prediction of sorption in polymers using quantum chemical calculations: Application to polymer membrane. J. Membr. Sci. 2013, 427, 108–117. [Google Scholar] [CrossRef]
- Gartner, T.E.; Jayaraman, A. Modeling and Simulations of Polymers: A Roadmap. Macromolecules 2019, 52, 755–786. [Google Scholar] [CrossRef]
- Khalatur, P.G. Molecular Dynamics Simulations in Polymer Science: Methods and Main Results. In Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Möller, M., Eds.; Elsevier: Dutch, The Netherlands, 2012; pp. 417–460. [Google Scholar] [CrossRef]
- Bhandari, S.; Lopez-Anido, R. Finite element analysis of thermoplastic polymer extrusion 3D printed material for mechanical property prediction. Addit. Manuf. 2018, 22, 187–196. [Google Scholar] [CrossRef]
- Bicerano, J. Prediction of Polymer Properties, 3rd ed.; Marcel Dekker Inc.: New York, NY, USA, 2002; pp. 1–746. [Google Scholar]
- Askadskii, A. Computational Materials Science of Polymers; Cambridge International Science Publishing: Cambridge, UK, 2003; pp. 1–650. [Google Scholar]
- Van Krevelen, D.W.; Te Nijenhuis, K. Properties of Polymers, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 1–1004. [Google Scholar]
- Polymer Genome. Available online: https://www.polymergenome.org/ (accessed on 23 November 2023).
- Park, J.; Shim, Y.; Lee, F.; Rammohan, A.; Goyal, S.; Shim, M.; Jeong, C.; Sin Kim, D. Prediction and Interpretation of Polymer Properties Using the Graph Convolutional Network. ACS Polym. Au 2022, 2, 213–222. [Google Scholar] [CrossRef]
- Guo, Z.X. Multiscale Materials Modelling Fundamentals and Applications. In Civil and Structural Engineering; Woodhead Publishing Series; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–293. [Google Scholar] [CrossRef]
- Zeng, Q.H.; Yu, A.B.; Lu, G.Q. Multiscale Modeling and Simulation of Polymer Nanocomposites. Prog. Polym. Sci. 2008, 33, 191–269. [Google Scholar] [CrossRef]
- Heiranian, M.; Du Chanois, R.M.; Ritt, C.L.; Violet, C.; Elimelech, M. Molecular simulations to elucidate transport phenomena in polymeric membranes. Environ. Sci. Technol. 2022, 56, 3313–3323. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.; Ostlund, N.S. Introduction to Advanced Electronic Structure Theory. In Modern Quantum Chemistry, Reprinted; Courier Corporation: Honolulu, HI, USA, 1996; pp. 1–480. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Stocker, S.; Csanyi, G.; Reuter, K.; Margraf, J.T. Machine Learning Chemical Reaction Space. Nat. Commun. 2020, 11, 5505. [Google Scholar] [CrossRef] [PubMed]
- National Research Council. Beyond the Molecular Frontier: Challenges for Chemistry and Chemical Engineering; The National Academies Press: Washington, DC, USA, 2003. [Google Scholar] [CrossRef]
- The Australian National University. Available online: https://www.anu.edu.au/news/all-news/researcher-sets-record-for-quantum-chemistry-calculation (accessed on 23 November 2023).
- Song, Y.; Xu, F.; Wei, M.J.; Wang, Y. Water Flow inside Polyamide Reverse Osmosis Membranes: A Non Equilibrium Molecular Dynamics Study. J. Phys. Chem. B 2017, 121, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wei, M.J.; Xu, F.; Wang, Y. Molecular Simulations of Water Transport Resistance in Polyamide RO Membranes: Interfacial and Interior Contributions. Engineering 2020, 6, 577–584. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, S.M.; Yang, B.Y.; Huo, J.; Zhang, X.P.; Bao, J.J.; Ruan, X.H.; He, G.H. Effect of Hydrogen-Bonding Interaction on the Arrangement and Dynamics of Water Confined in a Polyamide Membrane: A Molecular Dynamics Simulation. J. Phys. Chem. B 2018, 122, 4719–4728. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, S.L.; Huang, W.; Yu, C.Y.; Zhou, Y.F. MembrFactory: A Force Field and Composition Double Independent Universal Tool for Constructing Polyamide Reverse Osmosis Membranes. J. Comput. Chem. 2019, 40, 2432–2438. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, S.L.; Liu, L.F.; Huang, W.; Wang, Y.L.; Yu, C.Y.; Zhou, Y.F. Molecular Dynamics Simulation Studies of the Structure and Antifouling Performance of a Gradient Polyamide Membrane. Phys. Chem. 2019, 21, 19995–20002. [Google Scholar] [CrossRef]
- Field, M.J. A Practical Introduction to the Simulation of Molecular Systems, 2nd ed.; Cambridge University Press: Cambridge, UK, 1999; pp. 1–325. [Google Scholar]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef]
- Cheng, Y.; Guoqiang, L. The Rise of Machine Learning in Polymer Discovery. Adv. Intell. Syst. 2023, 5, 2200243. [Google Scholar] [CrossRef]
- Karelson, M. The Use of Topological Indices in QSAR and QSPR Modeling. In Advances in QSAR Modeling; Roy, K., Ed.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 57–88. [Google Scholar]
- Myint, K.-Z.; Wang, L.; Tong, Q.; Xie, X.-Q. Molecular Fingerprint-based Artificial Neural Networks QSAR for Ligand Biological Activity Predictions. Mol. Pharm. 2012, 9, 2912–2923. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C. The physicochemical approach to drug design and discovery (QSAR). Drug Dev. Res. 1981, 1, 267–309. [Google Scholar] [CrossRef]
- Ruimin, M.; Tengfei, L. PI1M: A Benchmark Database for Polymer Informatics. J. Chem. Inf. Model. 2020, 60, 4684–4690. [Google Scholar] [CrossRef]
- Suay-García, B.; Bueso-Bordils, J.I.; Falcó, A.; Antón-Fos, G.M.; Alemán-López, P.A. Virtual Combinatorial Chemistry and Pharmacological Screening: A Short Guide to Drug Design. Int. J. Mol. Sci. 2022, 23, 1620. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ru, Y.; Chen, Z.; Dong, C.; Dong, Y.; Liu, J. Accelerating the design and development of polymeric materials via deep learning: Current status and future challenges. APL Mach. Learn. 2023, 1, 021501. [Google Scholar] [CrossRef]
- Goodfellow, I.J.; Pouget-Abadie, J.; Mirza, M.; Bing, X.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, J. Generative Adversarial Networks. arXiv 2014, arXiv:1406.2661. [Google Scholar] [CrossRef]
- Sherstinsky, A. Fundamentals of recurrent neural network (RNNs) and longs hort-term memory (LSTM). arXiv 2018, arXiv:1808.03314. [Google Scholar] [CrossRef]
- Otsuka, S.; Kuwajima, I.; Hosoya, J.; Xu, Y.; Yamazaki, M. PoLyInfo: Polymer database for polymeric materials design. In Proceedings of the 2011 International Conference on Emerging Intelligent Data and Web Technologies, Tirana, Albania, 7–9 September 2011; pp. 22–29. [Google Scholar]
- Polymers: A Property Database. Available online: https://poly.chemnetbase.com/polymers/PolymerSearch.xhtml (accessed on 23 November 2023).
- Huan, T.; Mannodi-Kanakkithodi, A.; Kim, C.; Sharma, V.; Pilania, G.; Ramprasad, R. A polymer dataset for accelerated property prediction and design. Sci. Data 2016, 3, 160012. [Google Scholar] [CrossRef]
- Dalby, A.; Hourse, J.G.; Hounshell, W.D.; Gurchurst, A.K.I.; Grier, D.L.; Leland, B.A.; Laufer, J. Description of several chemical structure file formats used by computer programs developed at Molecular Design Limited. J. Chem. Inf. Comput. Sci. 1992, 32, 244–255. [Google Scholar] [CrossRef]
- Jin, W.; Barzilay, R.; Jaakkola, T.; Hierarchical Generation of Molecular Graphs Using Structural Motifs. arXiv 2020. Available online: https://arxiv.org/pdf/2002.03230 (accessed on 23 November 2023).
- Bonnici, V.; Giugno, R.; Pulvirenti, A.; Shasha, D.; Ferro, A. A subgraph isomorphism algorithm and its application to biochemical data. BMC Bioinform. 2013, 14, S13. [Google Scholar] [CrossRef]
- Yerin, A. InChI Encoding of Polymers Current Results and Further Tasks; InChITRUST: Bethesda, MD, USA, 2017; Available online: https://www.inchi-trust.org/wp/wp-content/uploads/2017/11/23.-InChI-Polymer-Yerin-201708.pdf (accessed on 23 November 2023).
- Inchi-Discuss Mailing List for InChI Facilities and Applications. Available online: https://sourceforge.net/p/inchi/mailman/inchi-discuss/?viewmonth=202301 (accessed on 23 November 2023).
- InChITRUST, Download Page. Available online: https://www.inchi-trust.org/download-latest-inchi-standard-software/ (accessed on 23 November 2023).
- Yu, M.; Shi, Y.; Jia, Q.; Wang, Q.; Luo, Z.-H.; Yan, F.; Zhou, Y.-N. Ring Repeating Unit: An Upgraded Structure Representation of Linear Condensation Polymers for Property Prediction. J. Chem. Inf. Model. 2023, 63, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- ISIS/Base. Available online: https://med.stanford.edu/content/dam/sm/htbc/documents/ISISBASE.pdf (accessed on 23 November 2023).
- Trepalin, S.V.; Yarkov, A.V. CheD—Chemical database compilation tool, Internet server and client for SQL servers. J. Chem. Inf. Comput. Sci. 2001, 41, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Akhukov, M.A.; Chorkov, V.A.; Gavrilov, A.A.; Guseva, D.V.; Khalatur, P.G.; Khokhlov, A.R.; Kniznik, A.A.; Komarov, P.V.; Okun, M.V.; Potapkin, B.V.; et al. MULTICOMP package for multilevel simulation of polymer nanocomposites. Comput. Mater. Sci. 2023, 216, 111832. [Google Scholar] [CrossRef]
- GitHub—Trepalin/KintechLab: Polymer Processing. Available online: https://github.com/trepalin/KintechLab (accessed on 23 November 2023).
- RUIMINMA1996. Available online: https://github.com/RUIMINMA1996/PI1M (accessed on 23 November 2023).
- Landrum, G. RDKit: Open-Source Cheminformatics Software. 2016. Available online: https://github.com/rdkit/rdkit/releases/tag/Release_2016_09_4 (accessed on 23 November 2023).
- Trepalin, S.V.; Gerasimenko, V.A.; Kozyukov, A.V.; Savchuk, N.P.; Ivaschenko, A.A. New Diversity Calculations Algorithms Used for Compound Selection. J. Chem. Inf. Comput. Sci. 2002, 42, 249–258. [Google Scholar] [CrossRef]
- Holliday, J.D.; Ranade, S.S.; Willett, P. A Fast Algorithm for Selecting Sets of Dissimilar Molecules from Large Chemical Databases. Quant. Struct.-Act. Relat. 1995, 14, 501–506. [Google Scholar] [CrossRef]
- Bremsler, W. HOSE-A Novel Substructure Code. Anal. Chim. Acta 1978, 103, 355–365. [Google Scholar] [CrossRef]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Lemonick, S. Exploring chemical space: Can AI take us where no human has gone before? Chem. Eng. News 2020, 98, 13. [Google Scholar]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef]
- Van der Maaten, L. Accelerating t-SNE using Tree-Based Algorithms. J. Mach. Learn. Res. 2014, 15, 1–21. [Google Scholar]
- Oftadeha, M.; Moshfegha, M.; Hadi Abdallahb, H. Optical Properties of Some Oligothiophene Derivatives: DFT Study. Phys. Chem. Res. 2016, 4, 35–46. [Google Scholar] [CrossRef]
- Graham, M.J. Development of High Refractive Index Poly(thiophene) for the Fabrication of All Organic 3-D Photonic Materials with a Complete Photonic Band Gap. Doctor Philosophy Dissertation, University of Akron, USA, December 2006. Available online: http://rave.ohiolink.edu/etdc/view?acc_num=akron1164049666 (accessed on 23 November 2023).
- Tran, H.D.; Kim, C.; Chen, L.; Chandrasekaran, A.; Batra, R.; Venkatram, S.; Kamal, D.; Lightstone, J.P.; Gurnani, R.; Shetty, P.; et al. Machine-learning predictions of polymer properties with Polymer Genome. J. Appl. Phys. 2020, 128, 171104. [Google Scholar] [CrossRef]
- Hamnett, A.; Hillman, A.R. An Ellipsometric Study of the Nucleation and Growth of Polythiophene Films. J. Electrochem. Soc. 1988, 135, 2517–2524. [Google Scholar] [CrossRef]
- Mannodi-Kanakkithodi, A.; Pilania, G.; Huan, T.D.; Lookman, T.; Ramprasad, R. Machine Learning Strategy for Accelerated Design of Polymer Dielectrics. Sci. Rep. 2016, 6, 20952. [Google Scholar] [CrossRef]
- Volgin, I.V.; Batyr, P.A.; Matseevich, A.V.; Dobrovskiy, A.Y.; Andreeva, M.V.; Nazarychev, V.M.; Larin, S.V.; Goikhman, M.Y.; Vizilter, Y.V.; Askadskii, A.A.; et al. Machine Learning with Enormous “Synthetic” Data Sets: Predicting Glass Transition Temperature of Polyimides Using Graph Convolutional Neural Networks. ACS Omega 2022, 7, 43678–43691. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Property Name | Unit of Measure |
|---|---|---|
| CL | specific heat capacity, liquid | J/g/K |
| CS | specific heat capacity, solid | J/g/K |
| COH1 | specific cohesion energy, Feudor | J/g |
| COH2 | specific cohesion energy, Van Krevelen | J/g |
| DELTA1 | delta solubility, Feudor | (J/cc)0.5 |
| DELTA2 | delta solubility, Van Krevelen | (J/cc)0.5 |
| RLL | specific refraction | cc/g |
| PLL | specific polarizability | cc/g |
| MU | dipole moment | Debye |
| MB | bulk modulus | MPa |
| STIFFNESS | molar stiffness | g0.25cm1.5/mole0.75 |
| EPSILON | dielectric constant | |
| N | refractive index | |
| VISFUNC | molar viscosity | gJ1/3mole−4/3 |
| EAFLOW | specific activation energy of viscous flow | kJ/g |
| O2PERM | permeability of oxygen | Barrers |
| N2PERM | permeability of nitrogen | Barrers |
| CO2PERM | permeability of carbon dioxide | Barrers |
| TDECOMP | decomposition temperature | K |
| SINF | brittle fracture stress at infinite mol weight | MPa |
| SIGMAF | brittle fracture stress at specified mol weight | MPa |
| SIGMAY | yield stress | MPa |
| Compound | Experimental Value | Bicerano [9] | Polymer Genome |
|---|---|---|---|
| polythiophene [*]c1ccc(s1)[*] | 1.4 [67], 3.36 [65] | 1.75 | 2.10 [14] |
| [*]c3ccc(Sc2ccc(Sc1ccc([*])cc1)cc2)cc3 | 1.75 | 1.68 | 1.72 [66] |
| [*]c3ccc(Sc2ccc(Sc1ccc([*])cc1)s2)cc3 | 1.75 | 1.71 | 1.77 [66] |
| [*]c3ccc(Sc2nnc(Sc1ccc([*])cc1)s2)cc3 | 1.75 | 1.71 | 1.71 [66] |
| [*]c5ccc(Sc4c1SCCSc1c(Sc2ccc([*])cc2)c3SCCSc34)cc5 | 1.77 | 1.76 | 1.80 [14] |
| Compound | Dielectric Constant | ||
|---|---|---|---|
| Figure 7g in Ref. [1] | Bicerano [11] | Polymer Genome [14] | |
| Hydroxylamines | |||
| -CO-NH-CO-NH-O-CH2-O-NH- | 4.69 | 6.88 | 5.0 |
| -NH-CO-NH-CO-O-NH-CO-O-CO- | 4.71 | 5.77 | 5.3 |
| -NH-O-NH-O-CH2-O-NH-CO-NH-CO- | 4.61 | 6.82 | 4.9 |
| -CO-O-CO-NH-CO-NH-CO-O-NH-CO-NH- | 4.78 | 6.33 | 5.3 |
| -NH-CO-O-CO-NH-O-CO-NH-CO-O-NH-CO- | 4.65 | 5.65 | 5.2 |
| Hydrazides | |||
| -NH-CO-NH- | 7.84 | 5.3 | |
| -NH-CO-NH-CO-NH- | 8.04 | 5.4 | |
| -NH-CO-NH-CO-NH-NH-CO- | 8.12 | 5.5 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knizhnik, A.A.; Komarov, P.V.; Potapkin, B.V.; Shirabaykin, D.B.; Sinitsa, A.S.; Trepalin, S.V. New Polymers In Silico Generation and Properties Prediction. Nanomanufacturing 2024, 4, 1-26. https://doi.org/10.3390/nanomanufacturing4010001
Knizhnik AA, Komarov PV, Potapkin BV, Shirabaykin DB, Sinitsa AS, Trepalin SV. New Polymers In Silico Generation and Properties Prediction. Nanomanufacturing. 2024; 4(1):1-26. https://doi.org/10.3390/nanomanufacturing4010001
Chicago/Turabian StyleKnizhnik, Andrey A., Pavel V. Komarov, Boris V. Potapkin, Denis B. Shirabaykin, Alexander S. Sinitsa, and Sergey V. Trepalin. 2024. "New Polymers In Silico Generation and Properties Prediction" Nanomanufacturing 4, no. 1: 1-26. https://doi.org/10.3390/nanomanufacturing4010001
APA StyleKnizhnik, A. A., Komarov, P. V., Potapkin, B. V., Shirabaykin, D. B., Sinitsa, A. S., & Trepalin, S. V. (2024). New Polymers In Silico Generation and Properties Prediction. Nanomanufacturing, 4(1), 1-26. https://doi.org/10.3390/nanomanufacturing4010001