
Vibrational Enthalpies of Solid Crystalline Materials


Abstract
:1. Introduction
1.1. Background
1.2. Motivation/Purpose
1.3. Status of the Field
1.4. Scope of the Paper/Summary of Paper
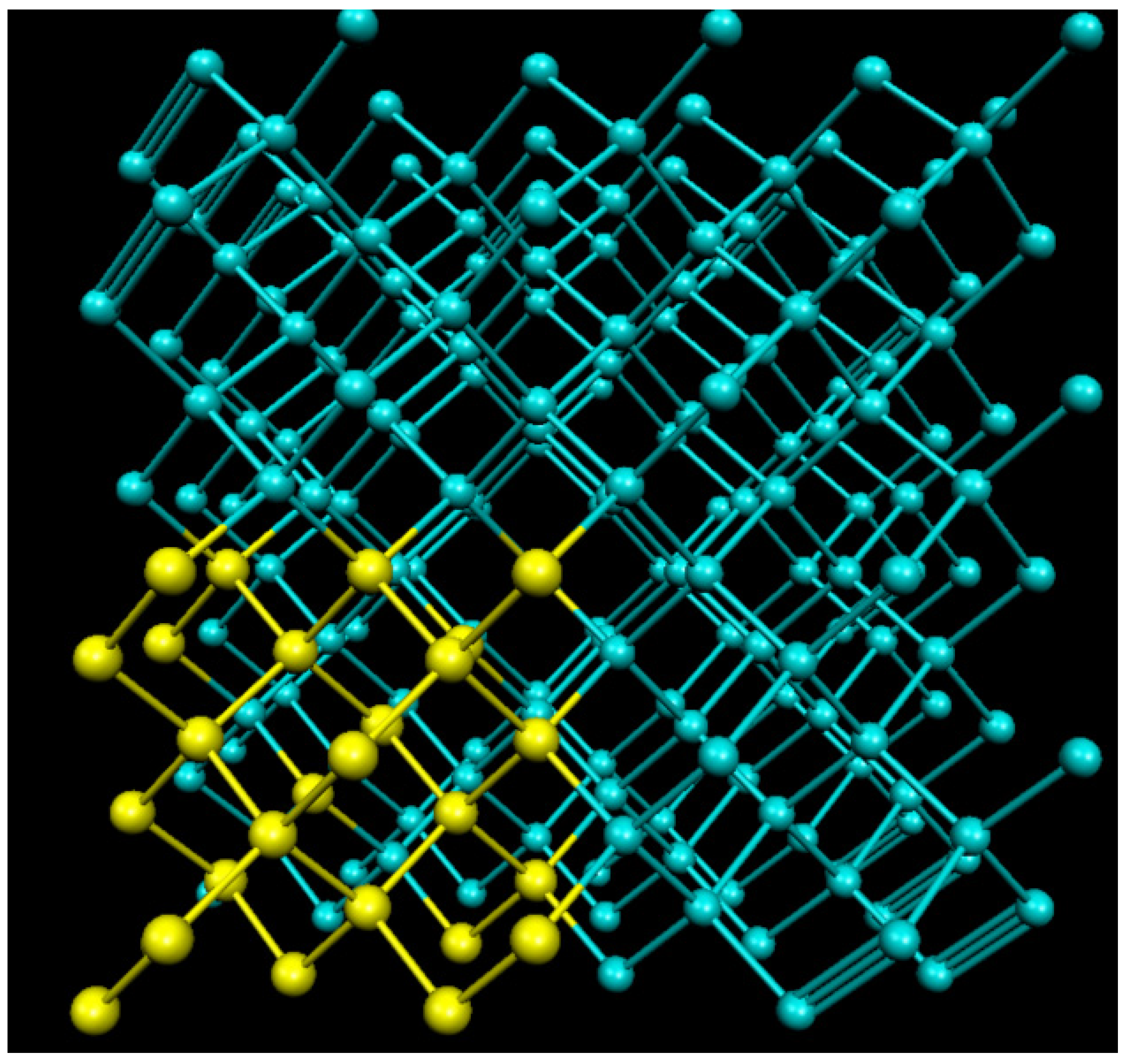
2. Methods
2.1. Density Functional Theory
2.2. Beyond Quasi-Harmonic Method
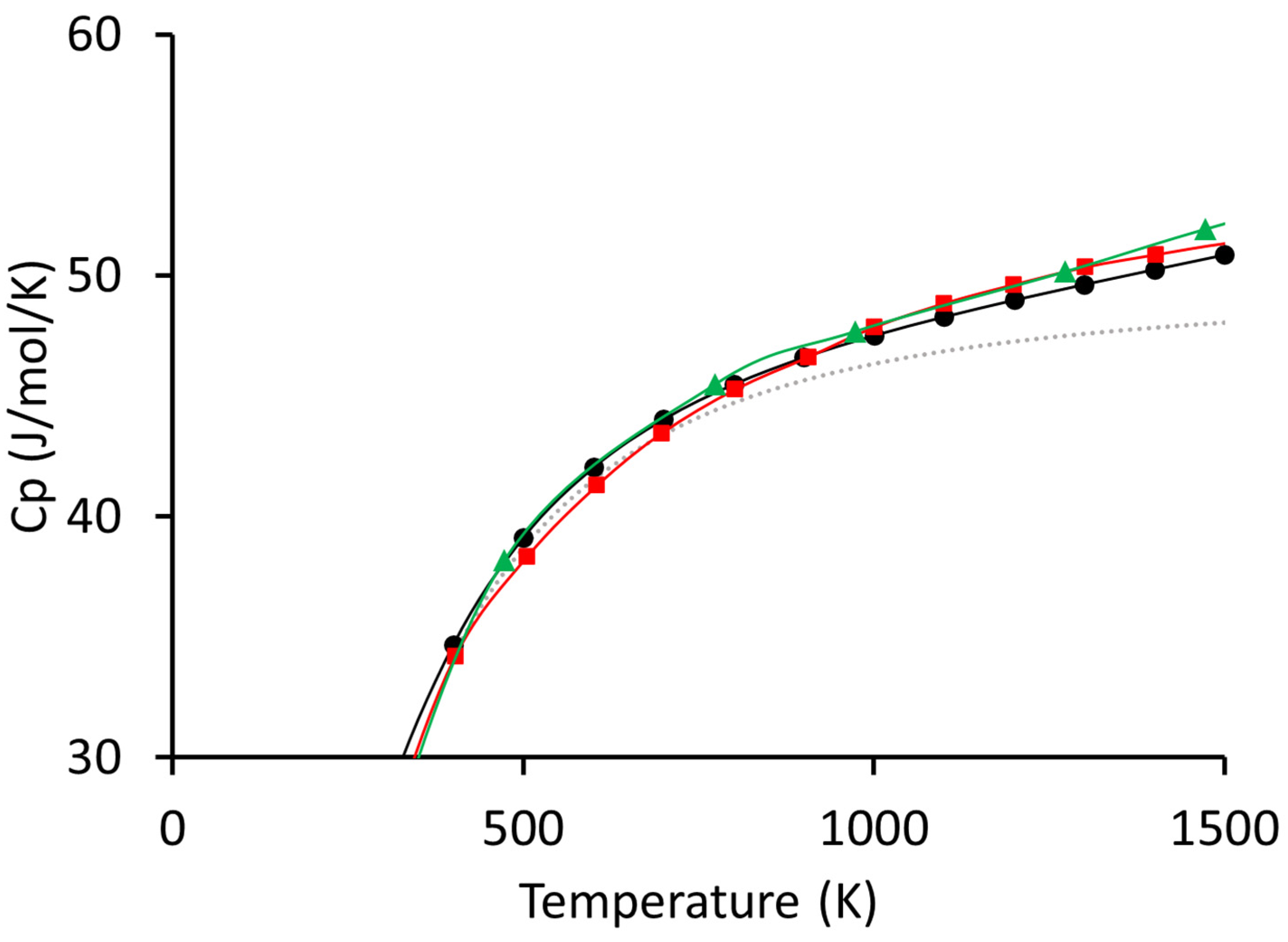
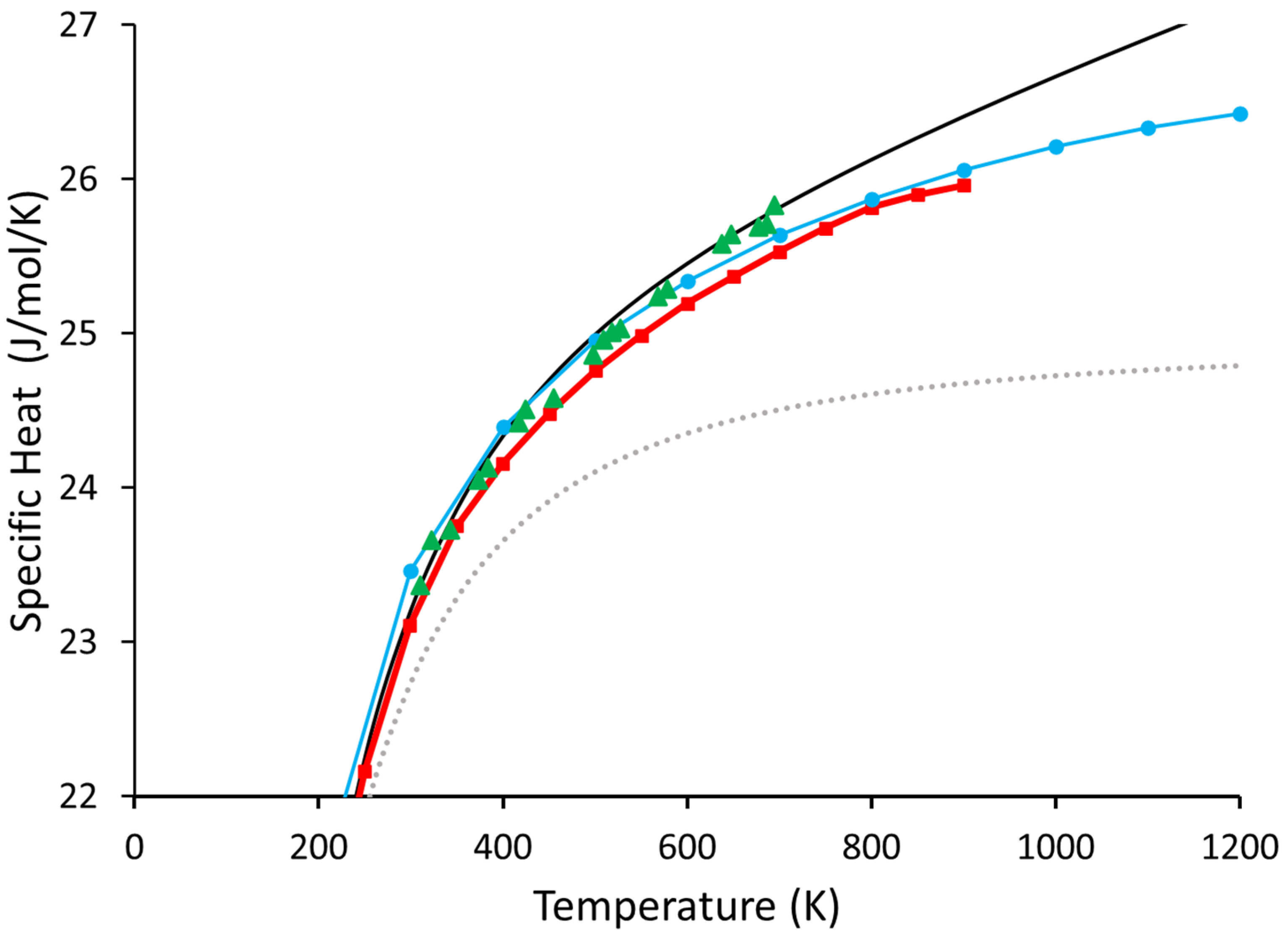
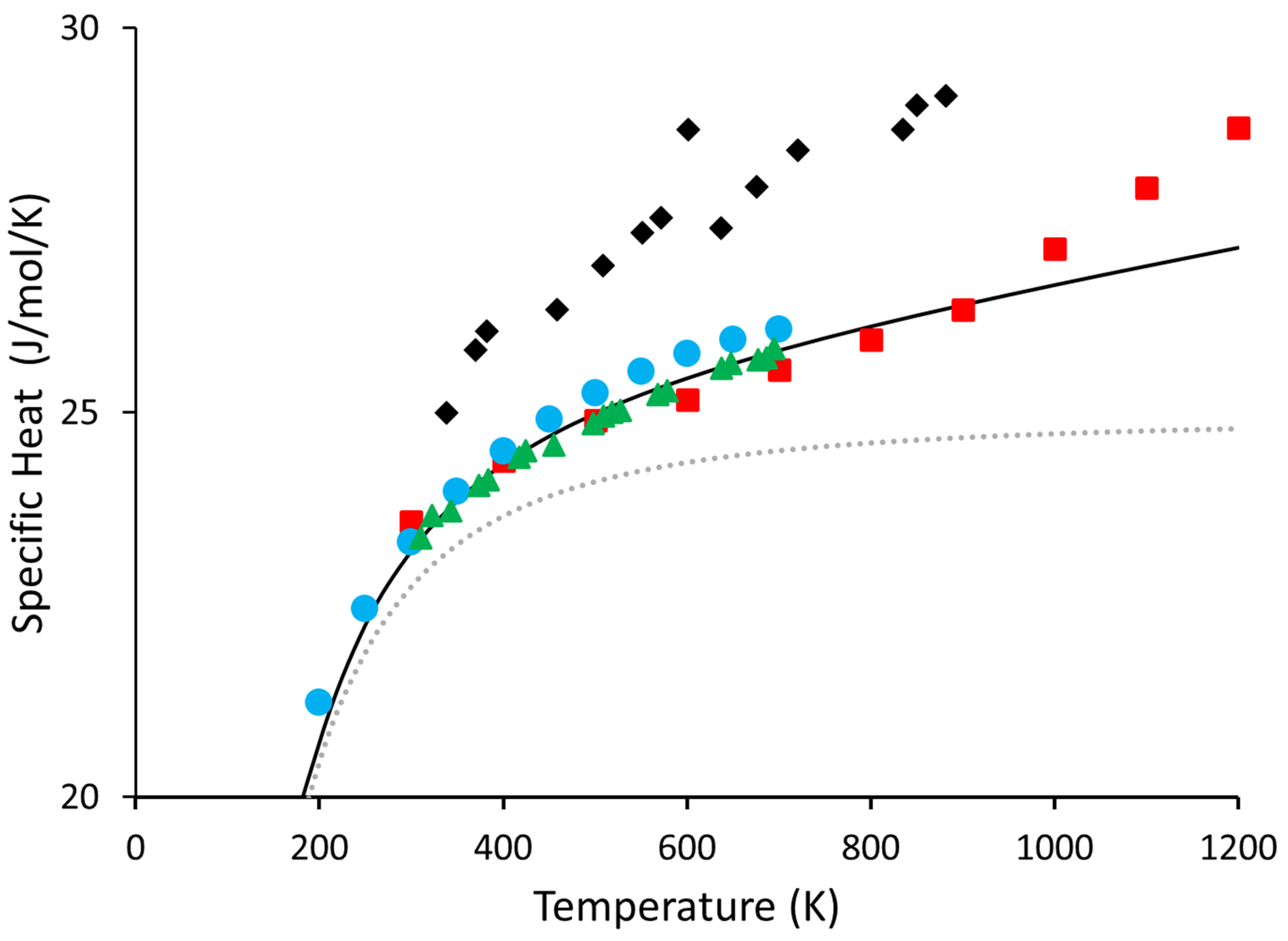
3. Results and Discussion
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Duff, A.I.; Davey, T.; Korbmacher, D.; Glensk, A.; Grabowski, B.; Neugebauer, J.; Finnis, M.W. Improved method of calculating ab initio high-temperature thermodynamic properties with application to ZrC. Phys. Rev. B 2015, 91, 214311. [Google Scholar] [CrossRef] [Green Version]
- Abdollahi, A. First-principle calculations of thermodynamic properties of ZrC and ZrN at high pressures and high temperatures. Phys. B Condens. Matter 2013, 410, 57–62. [Google Scholar] [CrossRef]
- Nelin, G.; Nilsson, G. Phonon anharmonicity of germanium in the temperature range 80–880 K. Phys. Rev. B 1974, 10, 612–620. [Google Scholar] [CrossRef]
- Leadbetter, A.J.; Settatree, G.R. Anharmonic effects in the thermodynamic properties of solids VI. Germanium: Heat capacity between 30 and 500 °C and analysis of data. J. Phys. C Solid State Phys. 1969, 2, 1105. [Google Scholar] [CrossRef]
- Estreicher, S.K.; Sanati, M.; West, D.; Ruymgaart, F. Thermodynamics of impurities in semiconductors. Phys. Rev. B 2004, 70, 125209. [Google Scholar] [CrossRef]
- Stanley, C.M. Specific heat at constant pressure from first principles: Contributions from fully anharmonic vibrations. Mater. Res. Express 2019, 6, 125924. [Google Scholar] [CrossRef]
- Khanzadeh, M.; Alahyarizadeh, G. A DFT study on pressure dependency of TiC and ZrC properties: Interconnecting elastic constants, thermodynamic, and mechanical properties. Ceram. Int. 2021, 47, 9990–10005. [Google Scholar] [CrossRef]
- Steurer, W. Quasicrystals: What do we know? What do we want to know? What can we know? Acta Cryst. 2018, A74, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gumber, S.; Kumar, M.; Gambhir, M.; Mohan, M.; Jha, P.K. Thermal and magnetic properties of cylindrical quantum dot with asymmetric confinement. Can. J. Phys. 2015, 93, 1264–1268. [Google Scholar]
- French, D.A.; Magee, M.; Furr, M.H.; Herzog, J.B. Plasmonic enhancement of photobrightening in CdSe quantum dots. J. Nanophotonics 2021, 15, 046005. [Google Scholar] [CrossRef]
- Grabowski, B.; Ikeda, Y.; Srinivasan, P.; Körmann, F.; Freysoldt, C.; Duff, A.I.; Shapeev, A.; Neugebauer, J. Ab initio vibrational free energies including anharmonicity for multicomponent alloys. Npj Comput. Mater. 2019, 5, 80. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Gupta, S.M.; Sharma, S.K. Carbon nanotubes: Synthesis, properties and engineering applications. Carbon Lett. 2019, 29, 419–447. [Google Scholar] [CrossRef]
- Hepplestone, S.P.; Ciavarella, A.M.; Janke, C.; Srivastava, G.P. Size and temperature dependence of the specific heat capacity of carbon nanotubes. Surf. Sci. 2006, 600, 3633–3636. [Google Scholar] [CrossRef]
- Cebe, P.; Partlow, B.P.; Kaplan, D.L.; Wurm, A.; Zhuravlev, E.; Schick, C. Fast Scanning Calorimetry of Silk Fibroin Protein: Sample Mass and Specific Heat Capacity Determination. In Fast Scanning Calorimetry; Schick, C., Mathot, V., Eds.; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Jiang, K.; Khan, F.; Thomas, J.; Desai, P.R.; Phani, A.; Das, S.; Thundat, T. Thermomechanical responses of microfluidic cantilever capture DNA melting and properties of DNA premelting states using picoliters of DNA solution. Appl. Phys. Lett. 2019, 114, 173703. [Google Scholar] [CrossRef]
- Ziman, J. Electrons and Phonons; Pergamon Press: Oxford, UK, 1958. [Google Scholar]
- Sagredo, F. Material Science Division; Private communication; Lawerence Berkeley National Lab: Berkeley, CA, USA, 2021. [Google Scholar]
- Soler, J.M.; Artacho, E.; Gale, J.D.; Garcia, A.; Junquera, J.; Ordejon, P.; Sanchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef] [Green Version]
- Emilio, A.; Anglada, E.; Diéguez, O.; Gale, J.D.; García, A.; Junquera, J.; Martin, R.M.; Ordejón, P.; Pruneda, J.M.; Sánchez-Portal, D.; et al. The SIESTA method; developments and applicability. J. Phys. Condens. Matter 2008, 20, 064208. [Google Scholar]
- García, A.; Papior, N.; Akhtar, A.; Artacho, E.; Blum, V.; Bosoni, E.; Brandimarte, P.; Brandbyge, M.; Cerdá, J.I.; Corsetti, F.; et al. Siesta: Recent developments and applications. J. Chem. Phys. 2020, 152, 204108. [Google Scholar] [CrossRef]
- Mu, F.; Cheng, Z.; Shi, J.; Shin, S.; Xu, B.; Shiomi, J.; Graham, S.; Suga, T. High Thermal Boundary Conductance across Bonded Heterogeneous GaN–SiC Interfaces. ACS Appl. Mater. Interfaces 2019, 11, 33428. [Google Scholar] [CrossRef]
- Cougo, B.; Morais, L.M.F.; Segond, G.; Riva, R.; Tran Duc, H. Influence of PWM Methods on Semiconductor Losses and Thermal Cycling of 15-kVA Three-Phase SiC Inverter for Aircraft Applications. Electronics 2020, 9, 620. [Google Scholar] [CrossRef] [Green Version]
- Ceperley, D.M.; Alder, B.J. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 1980, 45, 566. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048. [Google Scholar] [CrossRef] [Green Version]
- Kleinman, L.; Bylander, D.M. Efficacious form for model pseudopotentials. Phys. Rev. B 1982, 48, 1425. [Google Scholar] [CrossRef]
- Gibbons, T.M.; Bebek, M.B.; Kang, B.; Stanley, C.M.; Estreicher, S.K. Phonon-phonon interactions: First principles theory. J. Appl. Phys. 2015, 118, 085103. [Google Scholar] [CrossRef] [Green Version]
- Stanley, C.M.; Rader, B.K.; Laster, B.H.D.; Servati, M.; Estreicher, S.K. The Role of Interface Vibrational Modes in Thermal Boundary Resistance. Phys. Status Solidi A 2021, 218, 2100111. [Google Scholar] [CrossRef]
- Nakano, A.; Rino, J.P. Interation potential for Silicon Carbide: A Molecular Dynamics Study. JAP 2007, 101, 103515. [Google Scholar]
- CVD Technical Data Sheet. Available online: https://nanopdf.com/download/cvd-silicon-carbide-technical-data-sheet_pdf (accessed on 9 April 2022).
- Flubacher, P.; Leadbetter, A.J.; Morrison, J.A. The heat capacity of pure silicon and germanium 420 and properties of their vibrational frequency spectra. Philos. Mag. 1959, 4, 273–294. [Google Scholar] [CrossRef]
- Gerlich, D.; Abeles, B.; Miller, R.E. High-Temperature Specific Heats of Ge, Si, and Ge-Si Alloys. J. Appl. Phys. 1965, 36, 76–79. [Google Scholar] [CrossRef]
- Chen, H.S.; Turnbull, D. Specific heat and heat of crystallization of amorphous germanium. J. Appl. Phys. 1969, 40, 422. [Google Scholar] [CrossRef]
- Barin, I. Thermochemical Data of Pure Substances; VCH: Weinheim, Germany, 1995. [Google Scholar]
- Hedayat, A.; Khounsary, A.; Mashayek, F. Thermo-mechanical properties of silicon, germanium, diamond, beryllium and silicon carbide for high heat load X-ray optics applications. In Advances in X-ray/EUV Optics and Components VII; International Society for Optics and Photonics: Bellingham, WA, USA, 2012; p. 85020O. [Google Scholar]
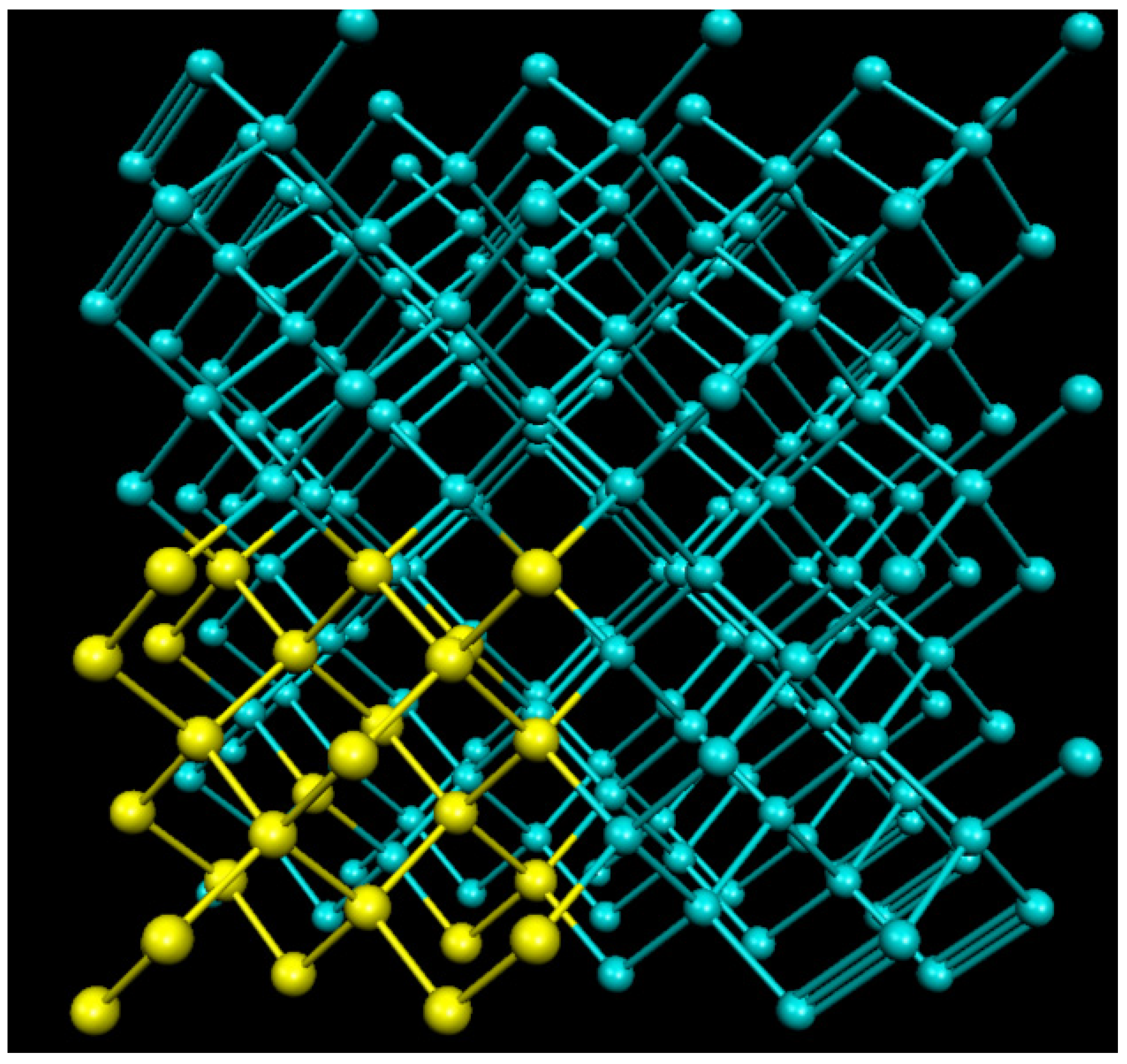
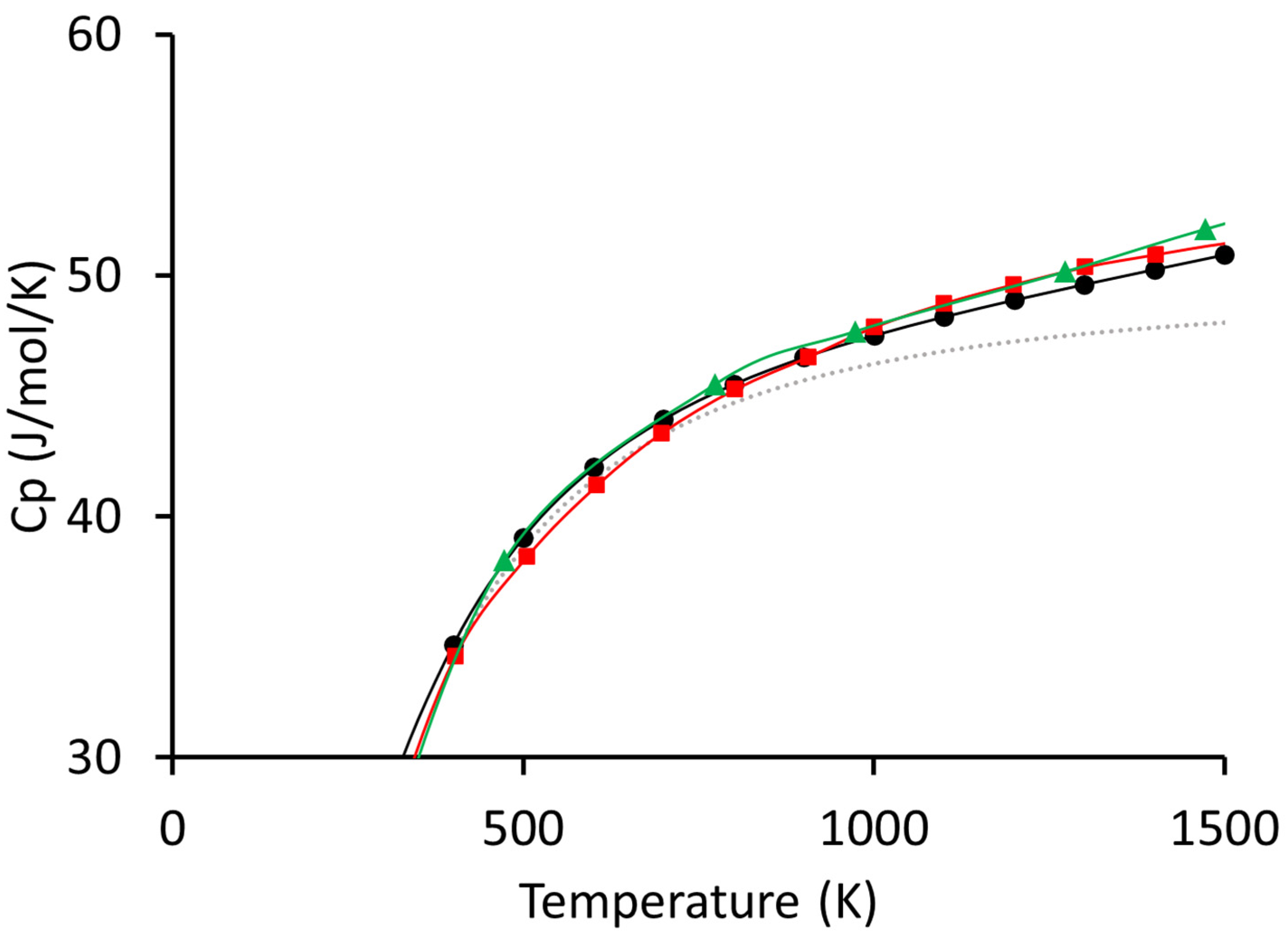
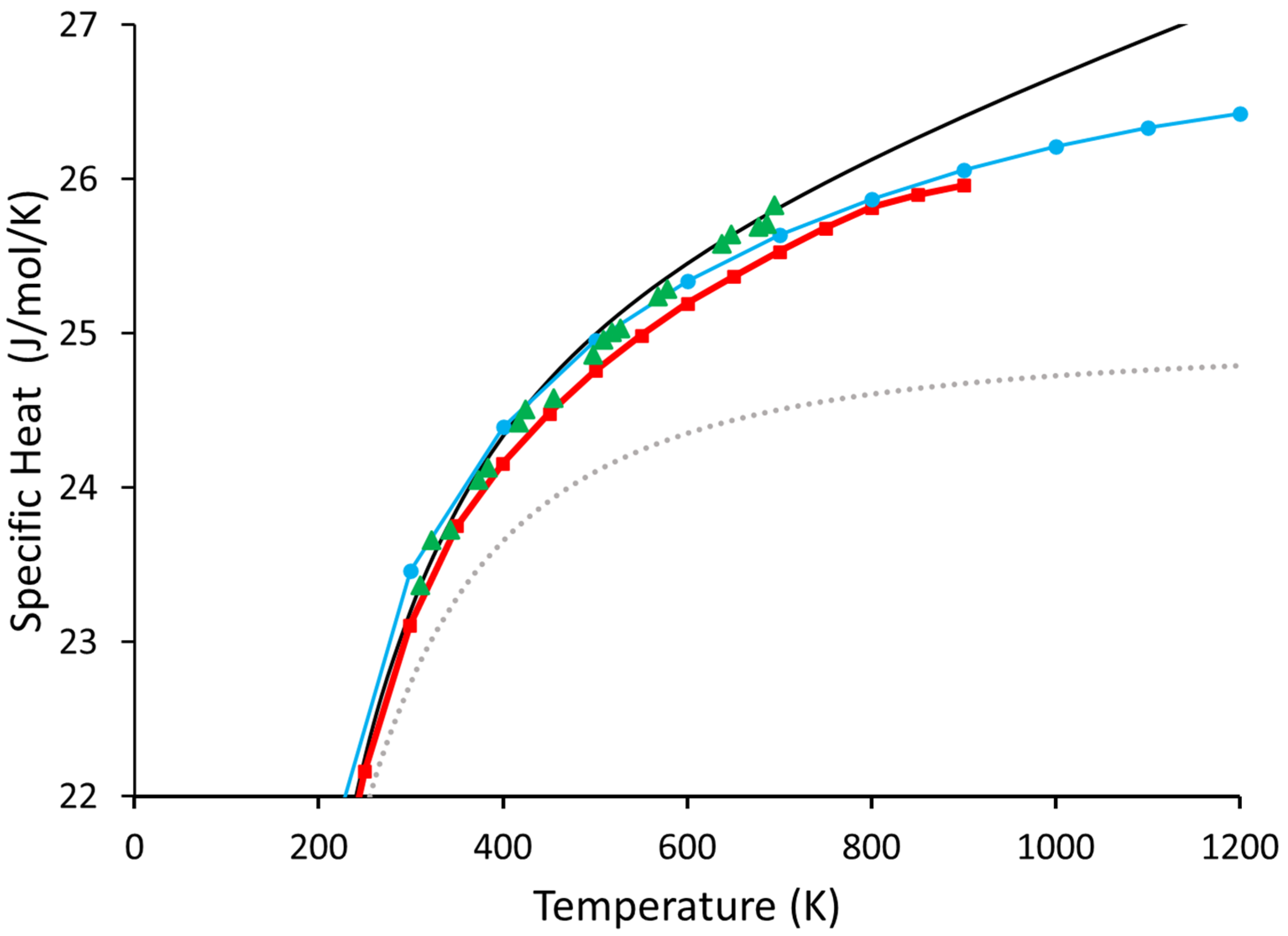
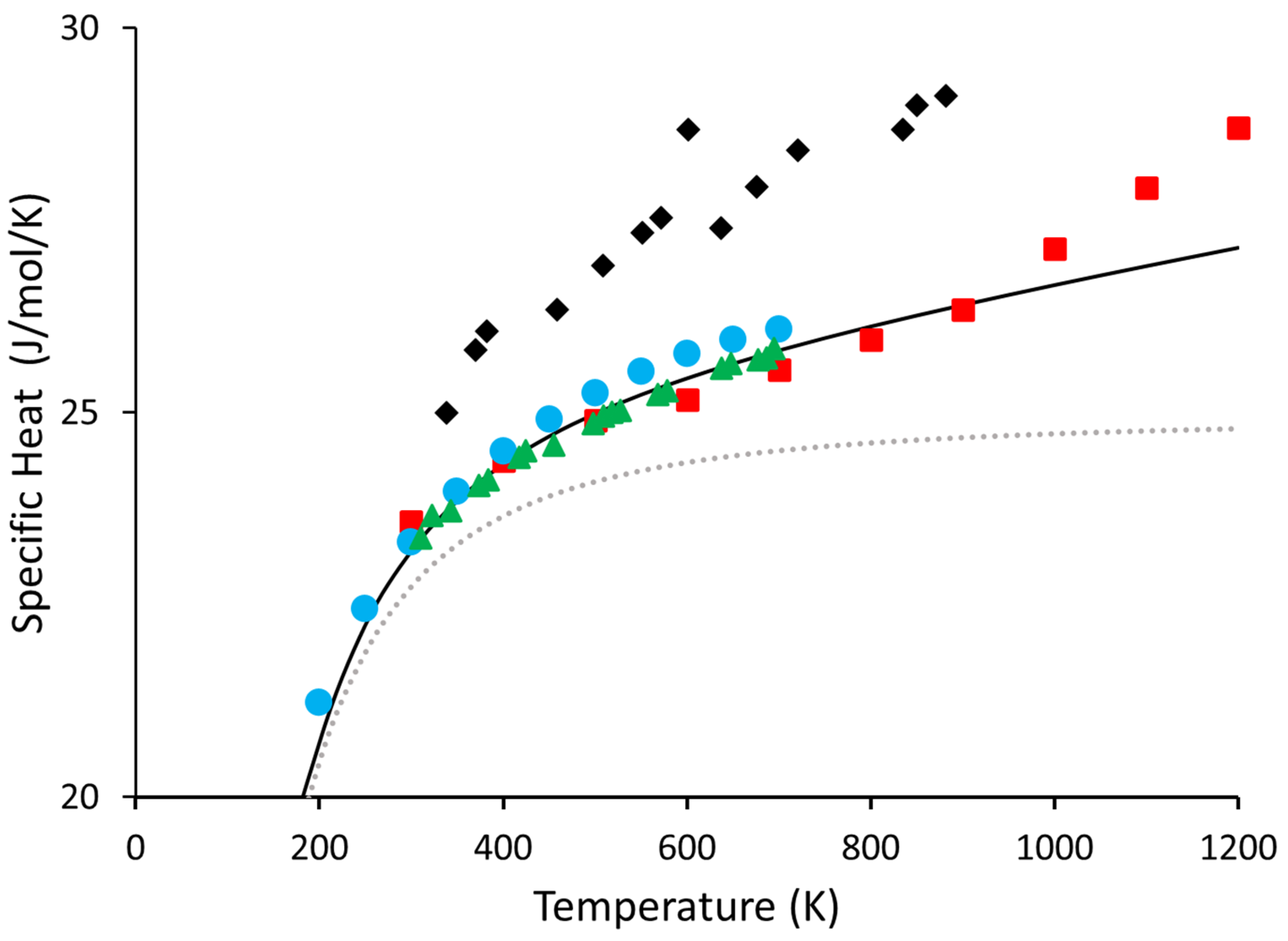

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | −2.812090475 ×10−12 |
| B | 5.690248937 × 10−9 |
| C | −4.285321740 × 10−6 |
| D | 1.344371869 × 10−3 |
| E | −6.020640219 × 10−2 |
| F | 6.053784794 × 10−1 |
| A | -- |
| B | −8.136069952 × 10−12 |
| C | 4.358400273 × 10−8 |
| D | −9.051433696 × 10−5 |
| E | 9.223260826 × 10−2 |
| F | 10.52098061 |
| A | -- |
| B | −1.238264848 × 10−11 |
| C | 4.420007261 × 10−8 |
| D | 5.902203596 × 10−5 |
| E | 3.773481439 × 10−2 |
| F | 16.13836107 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanley, C.M. Vibrational Enthalpies of Solid Crystalline Materials. Solids 2022, 3, 319-326. https://doi.org/10.3390/solids3020023
Stanley CM. Vibrational Enthalpies of Solid Crystalline Materials. Solids. 2022; 3(2):319-326. https://doi.org/10.3390/solids3020023
Chicago/Turabian StyleStanley, Christopher Martin. 2022. "Vibrational Enthalpies of Solid Crystalline Materials" Solids 3, no. 2: 319-326. https://doi.org/10.3390/solids3020023
APA StyleStanley, C. M. (2022). Vibrational Enthalpies of Solid Crystalline Materials. Solids, 3(2), 319-326. https://doi.org/10.3390/solids3020023