Abstract
Hepatocellular carcinoma (HCC) is a leading cause of cancer-related mortality globally, with metabolic-dysfunction-associated steatohepatitis (MASH) and alcohol-related liver disease (ALD) emerging as major etiologies. This review explores the epidemiological trends, pathogenesis, and clinical management of HCC arising from MASH and ALD, highlighting both the shared and distinct mechanisms. MASH-HCC is driven by metabolic dysregulation, including obesity, insulin resistance, and lipotoxicity, with genetic polymorphisms such as PNPLA3 and TM6SF2 playing critical roles in disease progression. ALD-HCC, in contrast, is propelled by the toxic byproducts of ethanol metabolism, including acetaldehyde and reactive oxygen species, which induce chronic inflammation, and fibrosis. Both conditions also involve immune dysregulation, gut dysbiosis, and increased intestinal permeability, contributing to hepatic carcinogenesis. The review emphasizes that, while there is consensus regarding the screening of HCC in cirrhosis patients, there is lack of consensus on screening strategies for non-cirrhotic MASH patients who are also at risk for HCC. This underscores the importance of the early detection of cirrhosis using advanced diagnostic tools such as transient elastography and fibrosis scores. Current therapeutic approaches, ranging from surgical resection, liver transplantation, and locoregional therapies to systemic therapies like immune checkpoint inhibitors, are discussed, with an emphasis on the need for personalized treatment strategies. Finally, the review highlights future research priorities, including the development of novel biomarkers, exploration of the gut–liver axis, and deeper investigation of the interplay between genetic predisposition and environmental factors. By synthesizing these insights, the review aims to inform multidisciplinary approaches to reduce the global burden of MASH- and ALD-related HCC and improve patient outcomes.
Keywords:
hepatocellular carcinoma; HCC; liver cancer; MASLD; MASH; alcohol-related liver disease; ALD 1. Introduction
Primary liver carcinomas represent the sixth most common cancer worldwide and the third leading cause of cancer-related death worldwide, with an incidence of around 1 million cases annually [1,2]. Hepatocellular carcinoma accounts for approximately 90% of primary liver cancers, usually in the setting of chronic liver disease caused by hepatitis B virus (HBV), hepatitis C virus (HCV), alcohol-associated liver disease (ALD), and MASLD (metabolic-dysfunction-associated steatotic liver disease) [3,4]. MASLD and ALD are now classified under the overarching term Steatotic Liver Disease (SLD), which encompasses all the different etiologies of hepatic steatosis and is further classified into multiple subtypes [5,6]. Apart from MASLD and ALD, a new entity called MetALD has been defined to describe patients with features of MASLD who also have a significant intake of alcohol (140–350 g per week for women and 210–420 g per week for men) [5,6]. Additional sub-classes of SLD are ‘cryptogenic SLD’ and ‘specific etiology SLD’, which is further subdivided into ‘drug-induced liver injury’, monogenic diseases (e.g., lysosomal acid lipase deficiency, Wilson’s disease, etc.), and ‘miscellaneous diseases’ (e.g., celiac disease, malnutrition, chronic hepatitis C, etc.) [5,6]. In the present review, we will focus on MASLD and ALD. MASLD is defined as at least 5% hepatic steatosis with one or more conditions: T2DM, obesity, or metabolic dysregulation [7,8].
MASLD is defined as at least 5% hepatic steatosis with one or more conditions: T2DM, obesity, or metabolic dysregulation [7,8]. It is a spectrum of liver diseases, ranging from simple steatosis to its more aggressive form, metabolic-dysfunction-associated steatohepatitis (MASH), which is defined as the presence of inflammation and cellular injury (ballooning), with or without fibrosis. MASH can progress to cirrhosis and, ultimately, HCC, even in patients without advanced fibrosis. Alarmingly, MASH-HCC has become the fastest-growing etiology of HCC in the Western world, reflecting the global obesity epidemic and increased prevalence of metabolic disorders.
In contrast, ALD results from the hepatotoxic effects of chronic alcohol consumption, which triggers oxidative stress, immune dysregulation, and fibrogenic pathways. ALD-HCC remains a significant global issue, with alcohol use ranking among the leading modifiable risk factors for liver cancer. Although historically more prevalent in men, rising alcohol consumption among women has led to an increasing incidence of ALD-HCC in female populations, further exacerbating its public health impact.
While MASLD and ALD arise from distinct etiologies, they share critical pathogenic mechanisms, including oxidative stress, mitochondrial dysfunction, and chronic inflammation, that drive liver injury and promote carcinogenesis. However, differences in genetic predispositions, clinical presentations, and the impact of lifestyle factors demand individualized approaches to their management. This review explores the epidemiology, pathogenesis, and management of HCC in MASLD and ALD, emphasizing their shared and distinct features. By synthesizing the current evidence, we aim to provide a comprehensive understanding of these conditions, identify the gaps in our knowledge, and highlight potential avenues for further research.
2. Epidemiology
2.1. Overview of Epidemiological Trends of MASLD, MASH, and ALD HCC
It is estimated that more than one million individuals worldwide will be affected by liver cancer annually by 2025 [9]. Hepatocellular carcinoma is the most common form of liver cancer and accounts for around 90% of cases [10]. Although hepatocellular carcinoma (HCC) generally has a poor prognosis, with a 5-year survival rate below 20%, outcomes vary significantly based on the tumor stage. Patients with early-stage HCC can achieve a median survival of over 10 years, whereas those with advanced disease typically have a survival of only 1 to 3 years [11]. Studies comparing the overall survival of HCC patients caused by MASH have found no significant differences between the two groups [12].
MASLD has become the most prevalent chronic liver disease in history, affecting 1.66 billion individuals globally [13,14]. It is estimated that around 10% of all HCC cases are secondary to MASLD [15]. Data from the 2019 Global Burden of Disease (GBD) study estimated that the global prevalence of MASLD was 30.05% [16]. Western countries, with a high intake of high-fructose and processed foods, are experiencing rising rates of MASLD [17]. It is believed that approximately 20% of patients diagnosed with MASLD will progress to MASH [13], which has a global prevalence of 5.27% with significant geographic variations [16]. For example, Latin America has the highest prevalence of MASH at 7.11%, while Western Europe has the lowest prevalence at 4.02% [16]. Between 2010 and 2019, MASH was not only the fastest-growing etiology of liver cancer cases worldwide but also the leading cause of liver cancer deaths [18]. By 2030, it is projected that the incidence of MASH-HCC will increase by 130% in the United States [19]. This rise reflects a true epidemiologic trend rather than improved detection or changes in the diagnostic criteria [18].
Even as the MASLD prevalence evolves, ALD remains a highly significant global health problem, as it has been for millennia [20]. It was found that 25% of global deaths due to cirrhosis were associated with alcohol use. Alcohol use has been steadily rising globally, with alcohol consumption per capita in 2005 at 5.5 L, which is expected to increase to 7.6 L by 2030 [21,22]. A study by the USA Scientific Registry of Transplant Recipients from 2002 to 2016 found that 10.3% of the patients listed for a transplant had HCC secondary to ALD [23]. After MASH, alcohol use is the second-fastest-growing cause of liver cancer death [24]. ALD accounts for around 30% of HCC cases [25]. In 2019, alcohol was associated with an estimated 19% of deaths from liver cancer globally, with considerable regional variation [26,27]. Eastern Europe and parts of Asia have higher ALD-related HCC due to the elevated per capita alcohol consumption [24]. For example, the highest percentage of HCC mortality that was associated with alcohol was in Europe (35%), and the lowest percentage was in the Eastern Mediterranean region (10%) [24].
2.2. Sex Distribution in MASLD, ALD, and HCC
A meta-analysis of 22 studies found no difference in the prevalence of MASH between men and women who had biopsy-proven MASLD [28]. While it is believed that men generally have a higher prevalence of MASLD compared to women [29], studies have shown that post-menopausal women have a higher prevalence of MASLD compared to men [13,30]. This change in prevalence after menopause is hypothesized to be due to a protective effect that estrogen has during the pre-menopausal years. Regarding ALD, men have a higher rate of developing alcohol-related cirrhosis compared to women [31]. However, recent studies have shown a higher rate of increase in the prevalence of alcohol-related cirrhosis in women compared to men. In a retrospective study of privately insured persons in the US, between 2009 and 2015, there was a 50% increase in alcohol-related cirrhosis prevalence in women compared to a 30% increase in men [32]. This is postulated to be due to the increasing rate of alcohol use in women compared to men. A meta-analysis of six nationwide surveys in the US found that, between 2000 and 2016, the prevalence of alcohol use in women increased by approximately 0.6% per year, but no significant increase was found in men [33]. HCC has been well-known to disproportionately affect men more than women, with studies finding that men are two to four times more likely to develop liver cancer [34].
2.3. Risk Factors in MASH
Understanding the risk factors for the progression of MASH to cirrhosis is crucial, as they help identify individuals at a higher risk for developing advanced liver disease and guide targeted prevention and management strategies. The high prevalence of MASLD and its progression to MASH is strongly linked to chronic health conditions associated with metabolic syndrome (MetS): T2DM, HTN, HLD, heart disease, and obesity [35]. A meta-analysis of nine observational studies consisting of more than 1.5 million patients found that obese patients had a two-fold increase in HCC-associated mortality [4,36]. MASLD is characterized by an increase in hepatic lipid and triglyceride accumulation. Without early intervention, steatosis can progress to hepatocyte injury and, eventually, fibrosis, leading to liver cirrhosis and even higher chances of progression to HCC. The risk for developing HCC is increased 2.6-fold when obesity occurs with diabetes, hypertension, and hyperlipidemia [37]. Studies have found there to be a gender difference as well. A meta-analysis of 54 studies consisting of more than sixty-two thousand patients found that women had a 19% lower risk of MASLD but a 37% higher risk of advanced fibrosis than men [28]. In the US, around 30% of the adult population has MASLD, and 5% have progressed to MASH [38]. This percentage is vastly impacted by race in the US, with Hispanic individuals having the highest rates of MASLD, followed by White and Black individuals (21%, 12.5%, and 11.6%, respectively), which highlights the consideration of socioeconomic factors in patients [39]. Furthermore, genetic risk factors such as PNPLA3 variants are disproportionately prevalent in Hispanic populations, contributing to a higher MASLD-HCC incidence. Given the complex interplay of genetics, metabolic, and environmental factors in the progression of MASLD to MASH and cirrhosis, a multidisciplinary approach to early identification, risk stratification, and intervention is essential in order to mitigate the growing global burden of liver-related morbidity and mortality.
2.4. Risk Factors in ALD
Alcohol itself has been associated with an increased risk of cancer, and studies have found specific mechanisms that lead to the pathogenesis of HCC with alcohol use and ALD, discussed elsewhere in this article. Hence, the alarming increase in alcohol intake noted above increases the chances of ALD cirrhosis and progression to HCC. Females are more susceptible to ALD with lower volumes of alcohol, while males have a higher incidence of ALD-HCC [40]. Smoking and concomitant liver disease have also been shown to increase HCC with alcohol use. While the exact mechanism is unknown, it is believed that cigarette smoking and alcohol use have a synergetic carcinogenic effect [41]. ALD with a history of hepatitis B and C has also been shown to increase the risk of HCC [42]. The rising global alcohol consumption and its complex interplay with other risk factors underscore a critical need for increased awareness and targeted interventions to address these risk factors.
3. Pathogenesis
3.1. MASH-HCC
MASH is a multifactorial disease that involves a delicate relationship between numerous pathological mechanisms contributing to injury and fibrosis. Metabolic dysregulation (insulin resistance and lipotoxicity), oxidative stress, and increased pro-inflammatory cells are believed to be among the most vital contributors to chronic inflammation, which leads to hepatocyte damage and the potential progression to HCC [43,44,45,46].
3.2. Metabolic Dysregulation
A population-based case–control study using data from the Medicare database found that metabolic syndrome is associated with a 2.13-fold increase in HCC [47]. Interestingly, hypertension and hypercholesteremia did not show a correlation, while hyperglycemia and obesity increased HCC incidence, illustrating the key role glucose plays in hepatocarcinogenesis [48,49]. In T2DM patients, there is a 2-fold increase in the risk of HCC [50,51]. Physiologically, insulin promotes lipid synthesis and storage and reduces the formation of free fatty acids by inhibiting or suppressing lipolysis. In a state of insulin resistance, as in patients with diabetes, hyperinsulinemia and the subsequent hyperglycemia promote lipogenesis in hepatocytes.
Hepatic lipogenesis is modulated by two proteins—ChRBP (carbohydrate-response element-binding protein) and SREBP-1c (sterol regulatory element-binding protein 1c), both of which lead to overall lipid accumulation in hepatocytes [52]. To counteract the lipid-overloaded liver, adaptive changes in free fatty acid (FFA) metabolism must occur. It has been postulated that the total amount of triglycerides stored in hepatocytes is not the major determinant of lipotoxicity, and that specific lipid classes are agents of hepatocyte injury. Examples of such lipids are cholesterol, lysophosphatidylcholine, ceramides, and free fatty acids such as palmitic acid, which have been linked to the progression of MASH [53]. The increased lipids and FFAs in hepatocytes are believed to induce mitochondrial damage by producing mitochondrial reactive oxygen species (ROS). This can lead to protein and lipid peroxidation and impede β-oxidation, causing mitochondrial damage and, ultimately, resulting in cell death [54].
Previous studies have shown that saturated fatty acids induce liver damage by triggering pro-inflammatory macrophage activation through various signaling cascades, such as IKK/NF- kB and JAK/STAT3 [55]. While the specific biochemical pathways are beyond the scope of this article, pro-inflammatory cytokines such as TNF-α (tumor necrosis factor-alpha) and IL-6 (interleukin 6) are eventually produced, inducing a state of chronic inflammation in MASH. Furthermore, TNF-α has been shown to reprogram metabolic pathways by deactivating insulin signaling, thus enhancing lipolysis in adipocytes and delaying glucose clearance [43,44]. Mice studies have shown that the knockout of TNF-α protected them from insulin resistance and reduced serum fatty acid levels and oxidative stress [56,57,58,59,60]. Interestingly, studies by Nakagawa et al. [58] in 2014 showed that mice with TNF1 receptor knockout decreased the HCC development in obese mice. This complex interplay between biochemical pathways leads to chronic, low-grade inflammation, contributing to hepatocyte fibrosis and the eventual progression to HCC from MASH [61,62].
3.3. Oxidative Stress in Hepatocytes
Patients with MASH have higher levels of oxidative DNA damage than patients with other liver diseases [46,63]. As stated above, increased amounts of lipid and FFA lead to the production of mitochondrial reactive oxygen species due to mitochondrial damage. Increased lipid peroxidation and oxidative stress can further diminish mitochondrial function and affect the respiratory chain activity [64]. When lipotoxicity or oxidative damage occurs, an adaptive response is activated called the unfolded protein response (UPR), which is regulated by the endoplasmic reticulum (ER) [65]. It serves to bring homeostasis to the ER. However, during extended periods of ER stress, such as when hepatocytes are lipid-overloaded, the UPR is unable to restore homeostasis, and apoptosis occurs through a variety of proteins such as JNK (c-Jun N-terminal kinase), and CHOP (C/EBP homologous protein), as well as Bcl2 (B-cell lymphoma-2), PUMA (p53 up-regulated modulator of apoptosis), and DP5 (death protein 5) [66,67,68]. While ROS is usually beneficial to macrophages and is the first line against pathogens, DNA damage can occur as a bystander effect and likely plays a role in the progression of MASH to HCC and the associated proliferative response [69,70].
3.4. Gut Microbiome
The microbiome is a key modulator of metabolism and plays an essential role in immune system modulation, health maintenance, tolerance development, and the prevention of colonization by pathogens [71]. Studies have shown that the dysregulation of the normal gut microbiome, or dysbiosis, can contribute to chronic liver conditions such as MASH, ALD, and cirrhosis [72,73,74]. In MASH, the reduced butyrate-producing bacteria and elevated endotoxin-producing species create a pro-inflammatory state that promotes carcinogenesis. In ALD, alcohol disrupts the gut barrier integrity, allowing LPS to translocate to the liver and activate Kupffer cells. The gut microbiota affects the production of bacteria-derived metabolites like bile acid [75]. Recent studies have shown that regulating bile acid metabolism affects hepatic fibrogenesis and liver injury, particularly when there is an increased abundance of bacterial strains such as Lactobacilli and Bacteroides [76]. While the exact mechanism leading to liver injury is unknown, it is hypothesized that the tight junction within the intestinal epithelial barrier may play a role. Evidence suggests that dysfunctional gut permeability is present in patients with MASLD or MASH compared with healthy individuals, and the chronic inflammatory state may also be attributed to the epithelial barrier [77,78,79,80]. In mice studies, it was found that a compromised gut barrier led to the translocation of bacteria that could reach the liver through the portal vein and exacerbate liver inflammation and fibrosis [81]. While the exact mechanism that the gut microbiome plays in hepatic fibrosis is still under research, studies have shown a potential connection to fibrosis-related carcinogenesis in MASH-HCC [82,83].
3.5. Genetic Factors
Genetic predisposition significantly impacts the progression of MASH towards HCC. Several genetic polymorphisms have been associated with the progression of MASH fibrosis. The best known of these polymorphisms is PNPLA3 on chromosome 22 [84]. This variant leads to the impaired triglyceride mobilization of hepatic lipid droplets with increased hepatic lipid accumulation [84], eventually leading to increased oxidative stress. A European study found that PNPLA3 leads to a three-fold increase in HCC [71]. Furthermore, this increase is independent of other risk factors such as BMI, diabetes, and advanced fibrosis [85]. Another polymorphism is TM6SF2, located on chromosome 19. TM6SF2 causes the impaired secretion of very-low-density lipoproteins (VLDLs), thereby increasing hepatic lipid retention, leading to liver fibrosis and HCC [86,87]. On the other hand, variants in the HSD17B13 gene (17-beta hydroxysteroid dehydrogenase 13) located in chromosome 4 attenuate the risk of steatohepatitis and the progression to HCC [88,89]. Genomic mapping has been a valuable tool in identifying genetic variations linked to MASH-HCC. Research has found that the most frequent HCC drivers in MASH patients were mutations in TERT (telomerase reverse-transcriptase) promoter genes, CTNNB1 (catenin beta 1), TP53 single-nucleotide variants, and a TGF family activin receptor, ACVR2A, mutation [90]. While these polymorphisms and mutations are just a few associated with MASH and HCC, they highlight the complex pathogenesis involved in the progression to HCC. Incorporating genetic markers such as PNPLA3, TM6SF2, and HSD17B13 into polygenic risk models holds promise for the early stratification of HCC risk in MASLD and ALD patients. Future clinical tools may leverage these variants to personalize screening strategies and guide surveillance decisions.
3.6. Immune Response
Lobular inflammatory responses are believed to play an important role in hepatic fibrosis, cirrhosis, and HCC [91]. Both the innate and adaptive immune responses play a role in promoting hepatic inflammation in MASH. Kupffer cells, leukocytes, and natural killer cells all promote a pro-inflammatory state by releasing eicosanoids, nitric oxide, reactive oxygen species, and cytokines (e.g., TNF, IL-6, and chemokines) [53,92]. The continued liver injury sustained in MASH is believed to release damage-associated molecular patterns (DAMPs), which, in turn, causes the assembly of inflammatory corpuscles, activation of the inflammatory response, and further amplification of liver injury [93,94].
4. Pathogenesis
4.1. ALD-HCC
Alcohol promotes liver carcinogenesis through the formation of acetaldehyde and ROS, changes to the immune system, induction of chronic inflammation, and alterations to gene expression [95]. Chronic alcohol intake alters the architecture and compromises the functional capacity of the liver by triggering steatosis, steatohepatitis, and cirrhosis [95].
4.2. Alcohol Metabolism
Ethanol is metabolized into acetaldehyde by alcohol dehydrogenase (ADH) in the cytosol of hepatocytes. Acetaldehyde then enters the mitochondria, where it is oxidized to acetate by mitochondrial aldehyde dehydrogenase (ALDH) [91]. Acetaldehyde is a reactive and mutagenic compound that can promote DNA repair failure, lipid peroxidation, and mitochondrial damage by causing inflammation, extracellular matrix remolding, and promoting fibrogenesis, eventually leading to carcinogenesis [96,97]. While most alcohol is metabolized by ADH, CYP2E1 also metabolizes it, especially with excessive alcohol consumption [98]. The catalytic cycle of CYP2E1 produces ROS, contributing further to hepatic steatosis and DNA damage. Chronic alcohol exposure increases the levels of CYPE21, potentiating its damaging effects [99]. The enhanced alcohol metabolism in individuals with alcohol use disorder is due to this increase in CYP2E1 levels.
4.3. Oxidative Stress
Similar to the mechanism of cellular damage in MASLD, ROS generated as byproducts of alcohol metabolism in ALD cause cellular damage at the DNA level, impacting functions such as transcription and replication that can lead to ALD and hepatocarcinogenesis. Further, ROS induces the activation of cytokines and immune cells, leading to the upregulation of angiogenesis and metastatic processes [100]. In addition, ROS can lead to hepatic fibrosis via the activation of hepatic stellate cells through TGF-beta production. The ROS byproducts of CYP2E1 alcohol metabolism include H2O2, hydroxyl (OH−), and carbon-centered OH− [101]. In a healthy liver, antioxidants typically neutralize these ROS. However, chronic exposure to oxidative stress depletes the antioxidant system [99]. Downstream effects of oxidative stress include lipid peroxidation, which creates byproducts such as MDA (Malonaldehyde) and 4-HNE (trans-4-hydrox-2-nonenal) that have been known to cause mutations in p53 associated with HCC [102]. Further, the upregulation of VEGF (vascular endothelial growth factor) and MCP-1 (monocyte chemotactic protein-1) are also associated with tumor angiogenesis and metastasis [98]. Chronic alcohol consumption leads to structural and functional abnormalities in the hepatic mitochondria: it increases their size, impairs the integrity of the mitochondrial DNA (mtDNA), reduces hepatic ATP levels, and impairs the biosynthesis of mitochondrial proteins [99]. As noted above, chronic alcohol metabolism increases the levels of mitochondrial CYP2E1, which, in parallel with the associated mitochondrial dysfunction, further intensifies the generation and accumulation of ROS in hepatic mitochondria [98].
4.4. Role of the Immune System and Gut Microbiome
Like MASH, there is a complex interplay between the gut microbiota and the immune system with ALD. Alcohol consumption can disrupt the intestinal barrier, predisposing one to gut dysbiosis and leading to an influx of endotoxins and inflammatory cytokines in the liver [103,104,105]. With chronic alcohol use, there is an increased translocation of bacteria-derived lipopolysaccharide (LPS) from the gut to the liver, leading to the activation of the innate immune system by Kupffer cells [106]. Downstream molecular mechanisms such as the IKK/NF-Kb pathway lead to the release of pro-inflammatory cytokines such as TNF-alpha and IL-1β, contributing to hepatocyte injury [107]. Kupffer cells also impact anti-inflammatory cytokines such as IL-10, which have downstream effects on the signaling protein STAT3. Under normal conditions, STAT3 has a hepatoprotective effect but it is linked to an oncogenic effect in HCC [108]. NK cells are naturally part of the innate immune system and play a protective role against infection, but they also have a role in cancer surveillance [109]. Studies have shown that not only can alcohol consumption impair NK cell function, leading to hepatocyte injury, but their quantity is also reduced in HCC [103,104]. Regarding the gut microbiota, studies have found a difference in the gut microflora in patients with HCC compared to health controls. Among patients with early-stage HCC, the levels of butyrate-producing bacterial families (protective to the intestinal barrier) decreased, whereas Klebsiella and Haemophilus species, which produce LPS, increased [110,111]. As mentioned previously, LPS activates pro-inflammatory cytokines, leading to hepatocyte injury [112]. While further research is needed to elucidate the impact of the gut microbiota on HCC, current studies have shown that the intestinal microbiome plays a significant role in ALD and ALD-associated carcinogenesis to HCC.
4.5. Genetic Variations
Growing evidence shows that genetic factors play a role in developing HCC in ALD. A genome-wide association study was performed on individuals of European descent, which found that the rs738409 variant of the PNPLA3 gene increased the risk of developing cirrhosis after alcohol consumption [113]. This gene variant has also been linked to MASH-HCC, as described previously. In MASH, PNPLA3 impacts intrahepatic triglyceride accumulation, but its role in ALD remains unclear. Further, a meta-analysis of five studies has found that the GG phenotype, compared to the CC phenotype, has a higher prevalence among patients with alcohol-associated cirrhosis with HCC [114]. Other genome-wide association studies have found that MBOAT7 (membrane-bound O-acyltransferase domain-containing protein 7) and TM6SF2 are genetic factors that can increase the risk of alcohol-associated cirrhosis [115]. However, FAF2 (Fas-Associated Factor 2), which plays a role in lipid droplet regulation, is protective against alcohol-associated cirrhosis [115]. Researchers have also found the rs708113 [T] minor allele of the WNT3A-WNT9A gene on chromosome 1 is associated with a lower risk of developing alcohol-associated HCC [116]. The human telomerase reverse-transcriptase (TERT) gene on chromosome 5 is crucial in the proliferation of multiple cancers. It encodes a ribonuclease telomerase enzyme that prevents the shortening of telomeres. The rs2736098*A allele and rs2736100*T allele are believed to be associated with telomere shortening and an increased risk of hepatocellular carcinoma [117]. Genetic polymorphisms in alcohol dehydrogenase (ADH), specifically ADH1C*1, have also been linked to carcinogenesis due to its ability to generate ROS through acetaldehyde [118]. Studies have shown the vital interplay between genetics and the development of ALD-HCC, highlighting the need for personalized approaches to risk assessment and management in affected individuals.
5. Clinical Management
5.1. Surveillance
The preferred imaging modality for HCC surveillance remains an abdominal ultrasound [119,120]. Studies have shown that an abdominal ultrasound with alpha-fetoprotein (AFP) levels provides a better diagnostic odds ratio than an ultrasound alone [119]. Hence, the AASLD guidance recommends both an ultrasound and AFP should be used for HCC surveillance. Surveillance should occur at 6-month intervals indefinitely in patients with liver cirrhosis of any etiology, including MASH and ALD [119]. While MRI is the most accurate modality for detecting and characterizing HCC, with a sensitivity ranging from 47% to 95% for lesions smaller than 2 cm, it is typically used as a diagnostic test rather than a primary screening tool due to its cost and limited availability [121]. Abbreviated MRI (AMRI) has emerged as a potential alternative for HCC surveillance. AMRI involves a reduced number of sequences, which shortens the acquisition time and improves cost-effectiveness while maintaining a high sensitivity and specificity for early-stage HCC detection [122]. Currently, the AASLD recommends against the use of MRI for routine HCC screening due to radiology service capacity, patient acceptance, and cost-effectiveness [119].
Despite the widespread global prevalence of MASLD and ALD, current guidelines do not recommend screening in non-cirrhotic MASH due to the lower absolute risk, lack of validated risk models, and cost-effectiveness concerns [123]. In contrast, ALD-HCC typically arises in cirrhotic patients, who are already included in standard surveillance protocols. Additionally, ultrasound performs poorly in obese individuals, a common feature in MASLD, further limiting effective screening. Emerging tools, such as FIB-4, elastography, and genetic risk scores, are promising but not yet endorsed by guidelines [124,125]. Hence, there is a need to develop biomarkers or risk stratification models which will help us identify MASLD and ALD patients with a high-enough risk of HCC to warrant surveillance.
5.2. Diagnosis
Currently, there is no difference in the diagnosis of MASH/ALD-associated HCC compared to other etiologies. The diagnosis of HCC is primarily based on imaging without the need for pathologic confirmation. However, imaging can be used for diagnosis only in patients with cirrhosis (including ALD-cirrhosis and MASH-cirrhosis), chronic HBV, or a history of HCC. The preferred imaging modality is contrast-enhanced multiphase CT or MRI of the abdomen. With both modalities, a finding of arterial phase hyperenhancement (APHE) and washout on the portal venous or delayed phases is considered a radiological hallmark of HCC with a high predictive value and sensitivity for lesions greater than 1 cm in size in patients with chronic hepatitis B, cirrhosis, or a history of HCC [126,127]. Evaluating non-invasive imagining is based on the Liver Imaging Reporting and Data System (LI-RADS) criteria ranging from LR-1 (benign) to LR-5 (HCC) [128,129]. Figure 1 outlines the LI-RADS criteria in further detail.
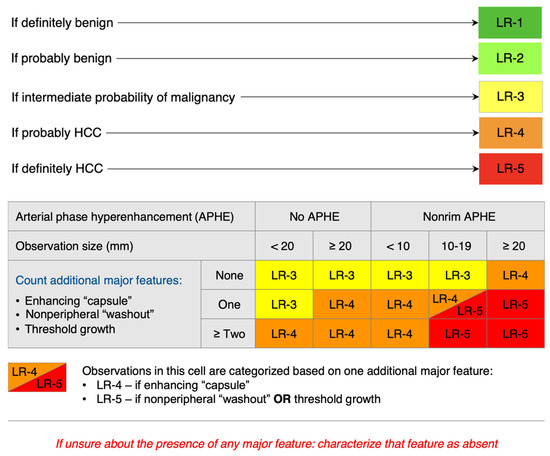
Figure 1.
CT/MRI LI-RADS diagnostic algorithm [121,128].
A histological diagnosis is required if cirrhosis, HBV, or a history of HCC is not present, as the LI-RADS criteria do not apply to this population. One caveat to this general rule is that the LI-RADS criteria also do not apply to patients with cirrhosis from vascular etiologies (e.g., cardiac cirrhosis), and the diagnosis in these patients should also be made with histopathology. Diagnosis should be based on the International Consensus Group for Hepatocellular Neoplasia using histological and immunohistochemical analyses [130]. The exact histological findings are beyond the scope of this article. A negative liver biopsy does not exclude the diagnosis of HCC in inconclusive cases; a second biopsy is recommended, especially in cases of high clinical suspicion. Previously, AFP (alpha-fetoprotein) levels greater than 400 ng/mL were regarded as a diagnostic criterion, but, due to its inadequate accuracy, current guidelines recommend against using AFP for diagnosis [131,132].
5.3. Staging
All patients with HCC should undergo cancer staging with contrast-enhanced multiphase CT or MRI of the abdomen to assess the extent of malignancy. The TNM staging system is not typically used for HCC. The most universally accepted staging system for HCC is the Barcelona Clinic Liver Cancer (BCLC) staging system (Figure 2) [133]. The BCLC classifies tumors from stage 0 to stage D, with D being the terminal stage based on end-stage liver dysfunction, irrespective of tumor burden. Patients beyond Stage A should undergo noncontract CT of the chest to evaluate for metastatic disease. At the initial evaluation, the tumor staging, burden, degree of liver dysfunction, and Eastern Cooperative Oncology Group (ECOG) performance status (PS) should be documented for baseline measurements as this helps delineate appropriate treatment regimens for patients [119].
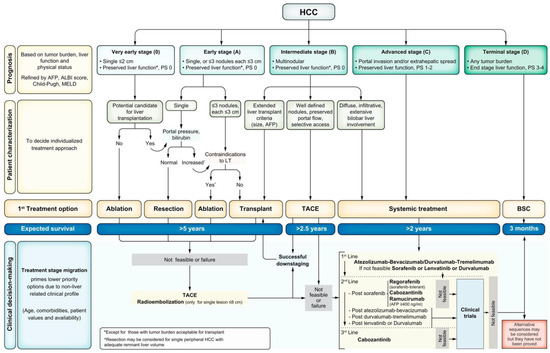
Figure 2.
BCLC staging [133].
5.4. Treatment
The treatment of HCC can be broadly divided into surgical, locoregional, and systemic therapies. A multidisciplinary approach to care is critical. A multidisciplinary team should be established not only to opine on the initial diagnosis and decide on the initial treatment plan but also to follow the patient over time to adjust the management plan as the tumor burden and the patient’s condition change.
5.5. Surgical Resection
In patients with HCC without cirrhosis, the treatment of choice is surgical resection for localized tumors [119]. In those with cirrhosis, surgical resection should be considered the treatment of choice for patients with a limited tumor burden and an amenable anatomic location in well-compensated cirrhosis without clinically significant portal hypertension and an adequate future liver remnant [134]. Surgical resection can be accomplished with minimally invasive surgery with laparoscopic or robotic approaches. The 5-year survival rate in these patients exceeds 70% [119]. The Child–Turcotte–Pugh (CTP) score has been used to assess patients for surgical candidacy. Typically, patients in CTP Class A can be considered for surgery since they usually have an appropriate liver reserve and are amenable to surgical resection. The recurrence of HCC after resection remains a significant obstacle, with a recurrence rate of as high as 70% at five years [119]. The risk of recurrence is the highest in the first year post-resection, which fortifies the importance of continued surveillance with cross-sectional imaging in addition to AFP [119].
5.6. Liver Transplantation
In patients with early-stage HCC who do not qualify for resection due to underlying liver disease, liver transplantation (LT) should be considered. Doyle et al., in 2012, reported a 69.1% 10-year survival rate in HCC patients who underwent LT [135]. Importantly, LT has a lower recurrence rate compared to patients undergoing liver resection or ablation. A meta-analysis of 18,421 patients found that the risk of recurrence after liver resection is threefold that of LT [136]. The same analysis found that the mortality rate after liver resection is about 50% higher than LT [135]. To optimize the outcomes after LT, the Milan Criteria (a single tumor with a diameter ≤ 5 cm, or ≤3 tumors with none exceeding 3 cm in diameter) have been the gold standard for patient selection [119]. Patients who are beyond the Milan criteria can be “downstaged”, and then considered for liver transplantation. In these patients who do not initially qualify for an LT due tumor burden, and in those who do qualify but must wait for allograft availability, bridge therapy to downstage and control the tumor size is performed using locoregional therapies (LRTs) as noted below. Understanding the etiologic background of HCC has important clinical implications. In MASH-HCC, metabolic comorbidities such as obesity and cardiovascular disease can complicate surgical or transplant eligibility. In contrast, ALD-HCC patients often face challenges related to alcohol relapse risk, malnutrition, and psychosocial evaluation. Tailoring surveillance, therapeutic selection, and perioperative planning to etiology can improve outcomes. Socioeconomic and clinical barriers influence transplant eligibility. ALD patients may face stigma and sobriety-related delays, while MASH patients often have comorbidities (e.g., obesity and cardiovascular disease) that increase the surgical risk. Furthermore, race, insurance status, and geographic location impact both transplant access and post-transplant outcomes [137].
5.7. Surgical Risk
The management of patients with MASLD-HCC and ALD-HCC may differ in surgical risk evaluation. Studies have found that patients with MASLD-HCC have a higher rate of post-surgical complications compared to patients with HCC from other etiologies [138,139]. It is estimated that the 90-day mortality in this subgroup is 11%, with a major complication rate of 31% after surgical resection [140]. Chronic conditions such as T2DM, HTN, and HLD are increased in this subgroup, and studies have shown these conditions have a negative post-operative effect [141]. Interestingly, it was found that the degree of steatosis also affected post-operative outcomes—patients with a higher degree of steatosis were found to have increased morbidity and mortality [140]. Regarding the surgical risk in ALD-HCC, studies have found that patients with ALD have more adverse effects and a higher in-hospital mortality that is estimated to be 2.6-fold higher compared to patients without ALD [142]. One potential reason for this is the degree of malnutrition in this cohort, which is known to increase the post-operative risk significantly [143,144]. While no direct comparison of surgical risk between MASH and ALD-HCC has been made, peri-operative risk management is imperative, and further research is needed in order to identify areas for improvement.
5.8. Locoregional Therapy
Locoregional therapies are imaging-guided, tumor-specific procedures used in about half of the patients with HCC [145]. The mainstay modalities in these scenarios include radiofrequency ablation (RFA), where thermal damage is delivered to the tumors using electromagnetic energy microwave ablation (MWA), which also relies on thermal damage but differs in its energy delivery methods [146,147,148]; TACE (transarterial chemoembolization), which embolizes the distal branches of the hepatic artery supplying the tumor using embolic particles like gelatin sponges, or synthetic polymer beads, and chemotherapeutic agents like doxorubicin, cisplatin, or irinotecan [146,149]; TARE (transarterial radioembolization), which delivers a high dose of targeted radiation to the tumors utilizing Y90 (yttrium-90)-impregnated microspheres [146,150]; and SBRT (stereotactic body radiotherapy), which delivers high doses of radiation with a high precision to a targeted tumor in a small number of fractions, typically between 1 to 5 sessions [151].
5.9. Systemic Therapy
Systemic therapy is reserved for patients with unresectable HCC who are not candidates for locoregional therapy. Systemic treatment for MASH-HCC and ALD-HCC is the same. Medications used in systemic treatment can be divided into the following categories: immune checkpoint inhibitors (ICIs), multi-target tyrosine kinase inhibitors (MTKIs), and monoclonal antiangiogenic antibodies (anti-VEGF antibodies). ICIs include PD1 inhibitors (programmed death 1) like pembrolizumab and nivolumab, inhibitors of the ligand of PD1 (PD-L1) like durvalumab and atezolizumab, or CTLA4 inhibitors (cytotoxic T lymphocyte-associated protein 4) like tremelimumab and ipilimumab [119]. Examples of MTKIs are sorafenib, lenvatinib, cabozantinib, and regorafenib. The monoclonal antiangiogenic antibodies comprise ramucirumab and bevacizumab, and they can be used in combination with ICIs or as a monotherapy [119]. Intravenous ICI treatment regimens are recommended as a first-line therapy since they have a higher efficacy and are better tolerated than MTKIs. However, they are associated with immune-related adverse effects. Since ICIs enhance T-cell responses, they are not recommended in post-transplant patients due to a high risk of graft loss, as multiple cases of severe rejection after LT have been reported [152]. MTKIs are oral agents but have a poorer tolerance profile than the ICIs. Prominent MTKI adverse side effects include hand–foot skin reaction, diarrhea, hypertension, proteinuria, gastrointestinal side effects, the inhibition of wound healing, hemorrhage, and thromboembolism [119]. Notably, the immunotherapy response may differ by etiology [153]. MASH-HCC appears less responsive to ICIs due to an immunosuppressive microenvironment dominated by dysfunctional T cells. In contrast, ALD-HCC may exhibit a more inflamed immune profile that enhances ICI efficacy, although direct comparative trials remain limited. While the specific details of each of these therapies are beyond the scope of this article, the correct systemic treatment should be chosen in a multidisciplinary manner as each of these medications has specific indications and side effect profiles. Clinical trials often underrepresent patients with significant metabolic comorbidities or advanced liver dysfunction, which are common in MASH-HCC. This limits the generalizability and underscores the need for etiology-specific trial stratification and broader inclusion criteria. Therapy needs to be tailored to each patient (Table 1).

Table 1.
Comparison between MASH-HCC and ALD-HCC.
6. Conclusions
The global rise in hepatocellular carcinoma associated with MASH and ALD represents a critical challenge that demands urgent, multifaceted intervention. While MASH-HCC is projected to become the predominant etiology of liver cancer in the Western world by 2030, ALD-HCC continues to account for a significant proportion of cases worldwide, driven by high and rising alcohol consumption. These trends emphasize the need for early detection and treatment strategies tailored to the unique characteristics of these conditions.
MASH-HCC highlights the pivotal role of metabolic dysregulation, with obesity, type 2 diabetes, and other components of metabolic syndrome serving as major drivers of hepatic inflammation and carcinogenesis. Genetic polymorphisms, including PNPLA3, TM6SF2, and HSD17B13, offer opportunities for personalized risk stratification and targeted therapies.
In contrast, the pathogenesis of ALD-HCC is driven by alcohol metabolism and its toxic byproducts, such as acetaldehyde and reactive oxygen species (ROS), which lead to direct hepatocyte injury, immune dysregulation, and fibrogenesis. Furthermore, genetic insights, such as variations in ADH, TERT, and PNPLA3, should guide individualized care plans for those at heightened risk.
Both MASH-HCC and ALD-HCC share common pathways, such as oxidative stress, mitochondrial dysfunction, and the role of the gut–liver axis. Dysbiosis and increased intestinal permeability exacerbate hepatic inflammation and fibrosis, making the microbiome a promising area for therapeutic exploration. Research into microbiome modulation, such as probiotics, prebiotics, and fecal microbiota transplantation, could offer novel strategies to mitigate disease progression in both etiologies. The mitigation of MASLD and ALD will undoubtedly have a corresponding dampening effect on the disease burden of HCC related to these conditions.
Therapeutic advancements are vital to improving outcomes in HCC. Early-stage disease management should focus on curative options, such as surgical resection, liver transplantation, and ablation. However, for patients with advanced disease, locoregional therapies (e.g., TACE, TARE, and ablation) and systemic treatments (e.g., immune checkpoint inhibitors, tyrosine kinase inhibitors, and antiangiogenic agents) provide valuable pathways for disease control.
Future research should prioritize validated screening algorithms for non-cirrhotic MASLD, the integration of genetic and microbiome-based risk stratification tools, and etiology-specific therapeutic trials. The clinical trial design must also account for patient heterogeneity in liver function and metabolic status to improve external validity. Equally important is the need to address healthcare disparities that influence the outcomes in HCC, particularly in underserved populations disproportionately affected by MASH and ALD. By embracing a multidisciplinary and personalized approach to prevention, diagnosis, and treatment, we can reduce the burden of these diseases, improve survival rates, and enhance the quality of life for patients worldwide.
Author Contributions
Conceptualization, A.B.; methodology, A.B., S.P., F.K. and D.F.; investigation, A.B., S.P., F.K. and D.F.; resources, A.B., S.P., F.K. and D.F.; data curation, A.B., S.P., F.K. and D.F.; writing—original draft presentation, A.B., S.P., F.K. and D.F.; writing—review and editing, A.B., S.P., F.K. and D.F.; visualization, A.B., S.P., F.K. and D.F.; supervision, A.B.; project administration, A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data were created in this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Rinella, M.E.; Sanyal, A.J.; Harrison, S.A.; Brunt, E.M.; Goodman, Z.; Cohen, D.E.; Loomba, R. From NAFLD to MAFLD: Implications of a Premature Change in Terminology. Hepatology 2021, 73, 1194–1198. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023, 78, 1966–1986. [Google Scholar] [CrossRef] [PubMed]
- Boccatonda, A.; Andreetto, L.; D’Ardes, D.; Cocco, G.; Rossi, I.; Vicari, S.; Schiavone, C.; Cipollone, F.; Guagnano, M.T. From NAFLD to MAFLD: Definition, Pathophysiological Basis and Cardiovascular Implications. Biomedicines 2023, 11, 883. [Google Scholar] [CrossRef]
- Llovet, J.M.; Willoughby, C.E.; Singal, A.G.; Greten, T.F.; Heikenwälder, M.; El-Serag, H.B.; Finn, R.S.; Friedman, S.L. Nonalcoholic steatohepatitis-related hepatocellular carcinoma: Pathogenesis and treatment. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 487–503. [Google Scholar] [CrossRef]
- Cancer Today. Available online: https://gco.iarc.who.int/today/ (accessed on 25 September 2024).
- Ioannou, G.N.; Splan, M.F.; Weiss, N.S.; McDonald, G.B.; Beretta, L.; Lee, S.P. Incidence and Predictors of Hepatocellular Carcinoma in Patients with Cirrhosis. Clin. Gastroenterol. Hepatol. 2007, 5, 938–945.e4. [Google Scholar] [CrossRef]
- Kanneganti, M.; Singal, A.G. Diagnosis and management of indeterminate liver nodules in patients with cirrhosis. Clin. Liver Dis. 2023, 22, 181. [Google Scholar] [CrossRef]
- Holzner, M.L.; Florman, S.; Schwartz, M.E.; Tabrizian, P. Outcomes of liver transplantation for nonalcoholic steatohepatitis-associated hepatocellular carcinoma. HPB 2022, 24, 470–477. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Lazarus, J.V.; Colombo, M.; Cortez-Pinto, H.; Huang, T.T.-K.; Miller, V.; Ninburg, M.; Schattenberg, J.M.; Seim, L.; Wong, V.W.S.; Zelber-Sagi, S. NAFLD—sounding the alarm on a silent epidemic. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 377–379. [Google Scholar] [CrossRef]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335. [Google Scholar] [CrossRef]
- Yu, S.; Li, C.; Ji, G.; Zhang, L. The Contribution of Dietary Fructose to Non-alcoholic Fatty Liver Disease. Front. Pharmacol. 2021, 12, 783393. [Google Scholar] [CrossRef]
- Huang, D.Q.; Singal, A.G.; Kono, Y.; Tan, D.J.H.; El-Serag, H.B.; Loomba, R. Changing global epidemiology of liver cancer from 2010 to 2019: NASH is the fastest growing cause of liver cancer. Cell Metab. 2022, 34, 969–977.e2. [Google Scholar] [CrossRef]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef]
- Seitz, H.K.; Neuman, M.G. The History of Alcoholic Liver Disease: From an Unrecognized Disease to One of the Most Frequent Diseases in Hepatology. J. Clin. Med. 2021, 10, 858. [Google Scholar] [CrossRef]
- Global Status Report on Alcohol and Health. 2018. Available online: https://www.who.int/publications/i/item/9789241565639 (accessed on 12 March 2025).
- Manthey, J.; Shield, K.D.; Rylett, M.; Hasan, O.S.M.; Probst, C.; Rehm, J. Global alcohol exposure between 1990 and 2017 and forecasts until 2030: A modelling study. Lancet 2019, 393, 2493–2502. [Google Scholar] [CrossRef]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 2019, 17, 748–755.e3. [Google Scholar] [CrossRef]
- Huang, D.Q.; Mathurin, P.; Cortez-Pinto, H.; Loomba, R. Global epidemiology of alcohol-associated cirrhosis and HCC: Trends, projections and risk factors. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 37–49. [Google Scholar] [CrossRef]
- Ganne-Carrié, N.; Nahon, P. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J. Hepatol. 2019, 70, 284–293. [Google Scholar] [CrossRef]
- GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Castro Narro, G.E.; Díaz, L.A.; Ortega, E.K.; Bautista Garín, M.F.; Reyes, E.C.; Martinez Delfin, P.S.; Arab, J.P.; Bataller, R. Alcohol-related liver disease: A global perspective. Ann. Hepatol. 2024, 29, 101499. [Google Scholar] [CrossRef]
- Balakrishnan, M.; Patel, P.; Dunn-Valadez, S.; Dao, C.; Khan, V.; Ali, H.; El-Serag, L.; Hernaez, R.; Sisson, A.; Thrift, A.P.; et al. Women Have a Lower Risk of Nonalcoholic Fatty Liver Disease but a Higher Risk of Progression vs Men: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2021, 19, 61–71.e15. [Google Scholar] [CrossRef]
- Burra, P.; Bizzaro, D.; Gonta, A.; Shalaby, S.; Gambato, M.; Morelli, M.C.; Trapani, S.; Floreani, A.; Marra, F.; Brunetto, M.R.; et al. Clinical impact of sexual dimorphism in non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH). Liver Int. 2021, 41, 1713–1733. [Google Scholar] [CrossRef]
- Eng, P.C.; Forlano, R.; Tan, T.; Manousou, P.; Dhillo, W.S.; Izzi-Engbeaya, C. Non-alcoholic fatty liver disease in women—Current knowledge and emerging concepts. JHEP Rep. 2023, 5, 100835. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457. [Google Scholar] [CrossRef]
- Carlini, L.E.; Fernandez, A.C.; Mellinger, J.L. Sex and gender in alcohol use disorder and alcohol-associated liver disease in the United States: A narrative review. Hepatology 2024. Available online: https://journals.lww.com/hep/fulltext/9900/sex_and_gender_in_alcohol_use_disorder_and.864.aspx (accessed on 12 March 2025). [CrossRef]
- Grucza, R.A.; Sher, K.J.; Kerr, W.C.; Krauss, M.J.; Lui, C.K.; McDowell, Y.E.; Hartz, S.; Virdi, G.; Bierut, L.J. Trends in Adult Alcohol Use and Binge Drinking in the Early 21st Century United States: A Meta-Analysis of Six National Survey Series. Alcohol. Clin. Exp. Res. 2018, 42, 1939–1950. [Google Scholar] [CrossRef]
- Wu, E.M.; Wong, L.L.; Hernandez, B.Y.; Ji, J.-F.; Jia, W.; Kwee, S.A.; Kalathil, S. Gender differences in hepatocellular cancer: Disparities in nonalcoholic fatty liver disease/steatohepatitis and liver transplantation. Hepatoma Res. 2018, 4, 66. [Google Scholar] [CrossRef]
- Paschos, P.; Paletas, K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia 2009, 13, 9–19. [Google Scholar]
- Gupta, A.; Das, A.; Majumder, K.; Arora, N.; Mayo, H.G.; Singh, P.P.; Beg, M.S.; Singh, S. Obesity is Independently Associated with Increased Risk of Hepatocellular Cancer–related Mortality: A Systematic Review and Meta-Analysis. Am. J. Clin. Oncol. 2018, 41, 874. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients with Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e2. [Google Scholar] [CrossRef]
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef]
- Schneider, A.L.C.; Lazo, M.; Selvin, E.; Clark, J.M. Racial Differences in Nonalcoholic Fatty Liver Disease in the U.S. Population. Obesity 2014, 22, 292–299. [Google Scholar] [CrossRef]
- Becker, U.; Deis, A.; Sørensen, T.I.; Grønbaek, M.; Borch-Johnsen, K.; Müller, C.F.; Schnohr, P.; Jensen, G. Prediction of risk of liver disease by alcohol intake, sex, and age: A prospective population study. Hepatology 1996, 23, 1025–1029. [Google Scholar] [CrossRef]
- Marti-Aguado, D.; Clemente-Sanchez, A.; Bataller, R. Cigarette smoking and liver diseases. J. Hepatol. 2022, 77, 191–205. [Google Scholar] [CrossRef]
- Donato, F.; Tagger, A.; Gelatti, U.; Parrinello, G.; Boffetta, P.; Albertini, A.; Decarli, A.; Trevisi, P.; Ribero, M.L.; Martelli, C.; et al. Alcohol and Hepatocellular Carcinoma: The Effect of Lifetime Intake and Hepatitis Virus Infections in Men and Women. Am. J. Epidemiol. 2002, 155, 323–331. [Google Scholar] [CrossRef]
- Zhang, H.H.; Halbleib, M.; Ahmad, F.; Manganiello, V.C.; Greenberg, A.S. Tumor necrosis factor-alpha stimulates lipolysis in differentiated human adipocytes through activation of extracellular signal-related kinase and elevation of intracellular cAMP. Diabetes 2002, 51, 2929–2935. [Google Scholar] [CrossRef]
- del Aguila, L.F.; Claffey, K.P.; Kirwan, J.P. TNF-alpha impairs insulin signaling and insulin stimulation of glucose uptake in C2C12 muscle cells. Am. J. Physiol. 1999, 276, E849–E855. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Miyanishi, K.; Kobune, M.; Kawano, Y.; Hoki, T.; Kubo, T.; Hayashi, T.; Sato, T.; Sato, Y.; Takimoto, R.; et al. Increased hepatic oxidative DNA damage in patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. J. Gastroenterol. 2013, 48, 1249–1258. [Google Scholar] [CrossRef]
- Welzel, T.M.; Graubard, B.I.; Zeuzem, S.; El-Serag, H.B.; Davila, J.A.; McGlynn, K.A. Metabolic syndrome increases the risk of primary liver cancer in the United States: A population-based case-control study. Hepatology 2011, 54, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Turati, F.; Talamini, R.; Pelucchi, C.; Polesel, J.; Franceschi, S.; Crispo, A.; Izzo, F.; La Vecchia, C.; Boffetta, P.; Montella, M. Metabolic syndrome and hepatocellular carcinoma risk. Br. J. Cancer 2013, 108, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Kurahashi, N.; Iwasaki, M.; Tanaka, Y.; Mizokami, M.; Noda, M.; Tsugane, S.; Japan Public Health Center-Based Prospective Study Group. Metabolic factors and subsequent risk of hepatocellular carcinoma by hepatitis virus infection status: A large-scale population-based cohort study of Japanese men and women (JPHC Study Cohort II). Cancer Causes Control 2009, 20, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Ajmera, V.; Cepin, S.; Tesfai, K.; Hofflich, H.; Cadman, K.; Lopez, S.; Madamba, E.; Bettencourt, R.; Richards, L.; Behling, C.; et al. A prospective study on the prevalence of NAFLD, advanced fibrosis, cirrhosis and hepatocellular carcinoma in people with type 2 diabetes. J. Hepatol. 2023, 78, 471–478. [Google Scholar] [CrossRef]
- Siegel, A.B.; Zhu, A.X. Metabolic syndrome and hepatocellular carcinoma: Two growing epidemics with a potential link. Cancer 2009, 115, 5651–5661. [Google Scholar] [CrossRef]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Barbuio, R.; Milanski, M.; Bertolo, M.B.; Saad, M.J.; Velloso, L.A. Infliximab reverses steatosis and improves insulin signal transduction in liver of rats fed a high-fat diet. J. Endocrinol. 2007, 194, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- Palisso, G.; Gambardella, A.; Tagliamonte, M.R.; Saccomanno, F.; Salvatore, T.; Gualdiero, P.; D’Onofrio, M.V.; Howard, B.V. Available online: https://academic.oup.com/jcem/article-abstract/81/12/4244/2650453?redirectedFrom=fulltext&login=false (accessed on 12 March 2025).
- Koo, S.-Y.; Park, E.-J.; Lee, C.-W. Immunological distinctions between nonalcoholic steatohepatitis and hepatocellular carcinoma. Exp. Mol. Med. 2020, 52, 1209–1219. [Google Scholar] [CrossRef]
- Torre, P.; Motta, B.M.; Sciorio, R.; Masarone, M.; Persico, M. Inflammation and Fibrogenesis in MAFLD: Role of the Hepatic Immune System. Front. Med. 2021, 8, 781567. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.-A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef]
- Henkel, A.; Green, R.M. The unfolded protein response in fatty liver disease. Semin. Liver Dis. 2013, 33, 321–329. [Google Scholar] [CrossRef]
- Flessa, C.-M.; Kyrou, I.; Nasiri-Ansari, N.; Kaltsas, G.; Papavassiliou, A.G.; Kassi, E.; Randeva, H.S. Endoplasmic Reticulum Stress and Autophagy in the Pathogenesis of Non-alcoholic Fatty Liver Disease (NAFLD): Current Evidence and Perspectives. Curr. Obes. Rep. 2021, 10, 134–161. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Huang, S.; Berger, E.; Liu, L.; Gross, N.; Heinzmann, F.; Ringelhan, M.; Connor, T.O.; Stadler, M.; Meister, M.; et al. Kupffer Cell-Derived Tnf Triggers Cholangiocellular Tumorigenesis through JNK due to Chronic Mitochondrial Dysfunction and ROS. Cancer Cell 2017, 31, 771–789.e6. [Google Scholar] [CrossRef] [PubMed]
- Canli, Ö.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Löwer, M.; Sahin, U.; et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 2017, 32, 869–883.e5. [Google Scholar] [CrossRef] [PubMed]
- Leyh, C.; Coombes, J.D.; Schmidt, H.H.; Canbay, A.; Manka, P.P.; Best, J. MASLD-Related HCC—Update on Pathogenesis and Current Treatment Options. J. Pers. Med. 2024, 14, 370. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.A.; Paik, Y.-H.; Schnabl, B. Role of Gut Microbiota in Liver Disease. J. Clin. Gastroenterol. 2015, 49 (Suppl. S1), S25–S27. [Google Scholar] [CrossRef]
- Etienne-Mesmin, L.; Chassaing, B.; Gewirtz, A.T. Tryptophan: A gut microbiota-derived metabolites regulating inflammation. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 7–9. [Google Scholar] [CrossRef]
- Konturek, P.C.; Harsch, I.A.; Konturek, K.; Schink, M.; Konturek, T.; Neurath, M.F.; Zopf, Y. Gut–Liver Axis: How Do Gut Bacteria Influence the Liver? Med. Sci. 2018, 6, 79. [Google Scholar] [CrossRef]
- Chavez-Talavera, O. 2017. Available online: https://www.gastrojournal.org/article/S0016-5085(17)30157-9/fulltext?referrer=https%3A%2F%2Fpubmed.ncbi.nlm.nih.gov%2F (accessed on 12 March 2025).
- Sydor, S.; Best, J.; Messerschmidt, I.; Manka, P.; Vilchez-Vargas, R.; Brodesser, S.; Lucas, C.; Wegehaupt, A.; Wenning, C.; Aßmuth, S.; et al. Altered Microbiota Diversity and Bile Acid Signaling in Cirrhotic and Noncirrhotic NASH-HCC. Clin. Transl. Gastroenterol. 2020, 11, e00131. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Volynets, V.; Küper, M.A.; Strahl, S.; Maier, I.B.; Spruss, A.; Wagnerberger, S.; Königsrainer, A.; Bischoff, S.C.; Bergheim, I. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig. Dis. Sci. 2012, 57, 1932–1941. [Google Scholar] [CrossRef]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 222–232. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302. [Google Scholar] [CrossRef]
- Bechmann, L.P.; Kocabayoglu, P.; Sowa, J.-P.; Sydor, S.; Best, J.; Schlattjan, M.; Beilfuss, A.; Schmitt, J.; Hannivoort, R.A.; Kilicarslan, A.; et al. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology 2013, 57, 1394–1406. [Google Scholar] [CrossRef]
- Caussy, C.; Tripathi, A.; Humphrey, G.; Bassirian, S.; Singh, S.; Faulkner, C.; Bettencourt, R.; Rizo, E.; Richards, L.; Xu, Z.Z.; et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 2019, 10, 1406. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Patman, G.L.; Leathart, J.B.S.; Piguet, A.-C.; Burt, A.D.; Dufour, J.-F.; Day, C.P.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef]
- Singh, S.; Anshita, D.; Ravichandiran, V. MCP-1: Function, regulation, and involvement in disease. Int. Immunopharmacol. 2021, 101 Pt B, 107598. [Google Scholar] [CrossRef]
- Stickel, F.; Buch, S.; Nischalke, H.D.; Weiss, K.H.; Gotthardt, D.; Fischer, J.; Rosendahl, J.; Marot, A.; Elamly, M.; Casper, M.; et al. Genetic variants in PNPLA3 and TM6SF2 predispose to the development of hepatocellular carcinoma in individuals with alcohol-related cirrhosis. Am. J. Gastroenterol. 2018, 113, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Trépo, E.; Nahon, P.; Cao, Q.; Moreno, C.; Letouzé, E.; Imbeaud, S.; Gustot, T.; Deviere, J.; Debette, S.; et al. PNPLA3 and TM6SF2 variants as risk factors of hepatocellular carcinoma across various etiologies and severity of underlying liver diseases. Int. J. Cancer 2019, 144, 533–544. [Google Scholar] [CrossRef]
- Gellert-Kristensen, H.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Stender, S. High Risk of Fatty Liver Disease Amplifies the Alanine Transaminase-Lowering Effect of a HSD17B13 Variant. Hepatology 2020, 71, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Pinyol, R.; Torrecilla, S.; Wang, H.; Montironi, C.; Piqué-Gili, M.; Torres-Martin, M.; Wei-Qiang, L.; Willoughby, C.E.; Ramadori, P.; Andreu-Oller, C.; et al. Molecular characterisation of hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 865–878. [Google Scholar] [CrossRef]
- Niemelä, O.; Parkkila, S.; Pasanen, M.; Iimuro, Y.; Bradford, B.; Thurman, R.G. Early alcoholic liver injury: Formation of protein adducts with acetaldehyde and lipid peroxidation products, and expression of CYP2E1 and CYP3A. Alcohol. Clin. Exp. Res. 1998, 22, 2118–2124. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef]
- Zhang, W.-J.; Chen, S.-J.; Zhou, S.-C.; Wu, S.-Z.; Wang, H. Inflammasomes and Fibrosis. Front. Immunol. 2021, 12, 643149. [Google Scholar] [CrossRef]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef]
- Cederbaum, A.I. CYP2E1—Biochemical and toxicological aspects and role in alcohol-induced liver injury. Mt. Sinai J. Med. N. Y. 2006, 73, 657–672. [Google Scholar]
- Mello, T.; Ceni, E.; Surrenti, C.; Galli, A. Alcohol induced hepatic fibrosis: Role of acetaldehyde. Mol. Asp. Med. 2008, 29, 17–21. [Google Scholar] [CrossRef]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef]
- Tan, H.K.; Yates, E.; Lilly, K.; Dhanda, A.D. Oxidative stress in alcohol-related liver disease. World J. Hepatol. 2020, 12, 332–349. [Google Scholar] [CrossRef]
- Hagymási, K.; Blázovics, A.; Lengyel, G.; Kocsis, I.; Fehér, J. Oxidative damage in alcoholic liver disease. Eur. J. Gastroenterol. Hepatol. 2001, 13, 49–53. [Google Scholar] [CrossRef]
- Hu, W.; Feng, Z.; Eveleigh, J.; Iyer, G.; Pan, J.; Amin, S.; Chung, F.-L.; Tang, M.-S. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 2002, 23, 1781–1789. [Google Scholar] [CrossRef]
- Wang, F.; Yang, J.-L.; Yu, K.; Xu, M.; Xu, Y.; Chen, L.; Lu, Y.; Fang, H.; Wang, X.; Hu, Z.; et al. Activation of the NF-κB pathway as a mechanism of alcohol enhanced progression and metastasis of human hepatocellular carcinoma. Mol. Cancer 2015, 14, 10. [Google Scholar] [CrossRef]
- Bajaj, J.S. Alcohol, liver disease and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 235–246. [Google Scholar] [CrossRef]
- Dubinkina, V.B.; Tyakht, A.V.; Odintsova, V.Y.; Yarygin, K.S.; Kovarsky, B.A.; Pavlenko, A.V.; Ischenko, D.S.; Popenko, A.S.; Alexeev, D.G.; Taraskina, A.Y.; et al. Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome 2017, 5, 141. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Stärkel, P.; Windey, K.; Tremaroli, V.; Bäckhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Hara, M.; Higaki, Y.; Ichiba, M.; Horita, M.; Mizuta, T.; Eguchi, Y.; Yasutake, T.; Ozaki, I.; Yamamoto, K.; et al. Influence of alcohol consumption and gene polymorphisms of ADH2 and ALDH2 on hepatocellular carcinoma in a Japanese population. Int. J. Cancer 2006, 118, 1501–1507. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.-Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef]
- Lee, C.; Cheung, S.T. STAT3: An Emerging Therapeutic Target for Hepatocellular Carcinoma. Cancers 2019, 11, 1646. [Google Scholar] [CrossRef]
- Tian, Z.; Chen, Y.; Gao, B. Natural killer cells in liver disease. Hepatology 2013, 57, 1654–1662. [Google Scholar] [CrossRef]
- Dapito, D.H.; Mencin, A.; Gwak, G.-Y.; Pradere, J.-P.; Jang, M.-K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Greten, T.F. Gut microbiome in HCC—Mechanisms, diagnosis and therapy. J. Hepatol. 2020, 72, 230–238. [Google Scholar] [CrossRef]
- Ren, Z.; Li, A.; Jiang, J.; Zhou, L.; Yu, Z.; Lu, H.; Xie, H.; Chen, X.; Shao, L.; Zhang, R.; et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2019, 68, 1014–1023. [Google Scholar] [CrossRef]
- Buch, S.; Stickel, F.; Trépo, E.; Way, M.; Herrmann, A.; Nischalke, H.D.; Brosch, M.; Rosendahl, J.; Berg, T.; Ridinger, M.; et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef]
- Salameh, H.; Raff, E.; Erwin, A.; Seth, D.; Nischalke, H.D.; Falleti, E.; Burza, M.A.; Leathert, J.; Romeo, S.; Molinaro, A.; et al. PNPLA3 Gene Polymorphism Is Associated with Predisposition to and Severity of Alcoholic Liver Disease. Am. J. Gastroenterol. 2015, 110, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Schwantes-An, T.-H.; Darlay, R.; Mathurin, P.; Masson, S.; Liangpunsakul, S.; Mueller, S.; Aithal, G.P.; Eyer, F.; Gleeson, D.; Thompson, A.; et al. Genome-wide Association Study and Meta-analysis on Alcohol-Associated Liver Cirrhosis Identifies Genetic Risk Factors. Hepatology 2021, 73, 1920–1931. [Google Scholar] [CrossRef]
- Trépo, E.; Caruso, S.; Yang, J.; Imbeaud, S.; Couchy, G.; Bayard, Q.; Letouzé, E.; Ganne-Carrié, N.; Moreno, C.; Oussalah, A.; et al. Common genetic variation in alcohol-related hepatocellular carcinoma: A case-control genome-wide association study. Lancet Oncol. 2022, 23, 161–171. [Google Scholar] [CrossRef]
- Seif Eldin, W.R.; Saad, E.A.; Monier, A.; Elshazli, R.M. Association of TERT (rs2736098 and rs2736100) genetic variants with elevated risk of hepatocellular carcinoma: A retrospective case–control study. Sci. Rep. 2023, 13, 18382. [Google Scholar] [CrossRef]
- Homann, N.; Stickel, F.; König, I.R.; Jacobs, A.; Junghanns, K.; Benesova, M.; Schuppan, D.; Himsel, S.; Zuber-Jerger, I.; Hellerbrand, C.; et al. Alcohol dehydrogenase 1C*1 allele is a genetic marker for alcohol-associated cancer in heavy drinkers. Int. J. Cancer 2006, 118, 1998–2002. [Google Scholar] [CrossRef]
- Singal, A.G.; Llovet, J.M.; Yarchoan, M.; Mehta, N.; Heimbach, J.K.; Dawson, L.A.; Jou, J.H.; Kulik, L.M.; Agopian, V.G.; Marrero, J.A.; et al. AASLD Practice Guidance on prevention, diagnosis, and treatment of hepatocellular carcinoma. Hepatology 2023, 78, 1922–1965. [Google Scholar] [CrossRef]
- Galle, P.R.; Forner, A.; Llovet, J.M.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.-L.; Schirmacher, P.; Vilgrain, V. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]
- ACR Appropriateness Criteria® Chronic Liver Disease—ClinicalKey. Available online: https://www.clinicalkey.com/#!/content/playContent/1-s2.0-S1546144020301113?returnurl=https:%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS1546144020301113%3Fshowall%3Dtrue&referrer=https:%2F%2Fpubmed.ncbi.nlm.nih.gov%2F (accessed on 3 December 2024).
- Gupta, P.; Soundararajan, R.; Patel, A.; Kumar-M, P.; Sharma, V.; Kalra, N. Abbreviated MRI for hepatocellular carcinoma screening: A systematic review and meta-analysis. J. Hepatol. 2021, 75, 108–119. [Google Scholar] [CrossRef]
- Singal, A.G.; El-Serag, H.B. Rational HCC screening approaches for patients with NAFLD. J. Hepatol. 2022, 76, 195–201. [Google Scholar] [CrossRef]
- Kjaergaard, M.; Lindvig, K.P.; Thorhauge, K.H.; Andersen, P.; Hansen, J.K.; Kastrup, N.; Jensen, J.M.; Hansen, C.D.; Johansen, S.; Israelsen, M.; et al. Using the ELF test, FIB-4 and NAFLD fibrosis score to screen the population for liver disease. J. Hepatol. 2023, 79, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, R.; Kc, S.; Thapa, P.; Pokharel, A.; Karki, N.; Jaishi, B. Estimation of Liver Fat by FibroScan in Patients with Nonalcoholic Fatty Liver Disease. Cureus 2021, 13, e16414. [Google Scholar] [CrossRef] [PubMed]
- Bolondi, L.; Gaiani, S.; Celli, N.; Golfieri, R.; Grigioni, W.F.; Leoni, S.; Venturi, A.M.; Piscaglia, F. Characterization of small nodules in cirrhosis by assessment of vascularity: The problem of hypovascular hepatocellular carcinoma. Hepatology 2005, 42, 27–34. [Google Scholar] [CrossRef]
- Matsui, O.; Kobayashi, S.; Sanada, J.; Kouda, W.; Ryu, Y.; Kozaka, K.; Kitao, A.; Nakamura, K.; Gabata, T. Hepatocelluar nodules in liver cirrhosis: Hemodynamic evaluation (angiography-assisted CT) with special reference to multi-step hepatocarcinogenesis. Abdom. Imaging 2011, 36, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.G.; Bruix, J.; Sherman, M.; Sirlin, C.B. LI-RADS (Liver Imaging Reporting and Data System): Summary, discussion, and consensus of the LI-RADS Management Working Group and future directions. Hepatology 2015, 61, 1056–1065. [Google Scholar] [CrossRef]
- Tang, A.; Bashir, M.R.; Corwin, M.T.; Cruite, I.; Dietrich, C.F.; Do, R.K.G.; Ehman, E.C.; Fowler, K.J.; Hussain, H.K.; Jha, R.C.; et al. Evidence Supporting LI-RADS Major Features for CT- and MR Imaging-based Diagnosis of Hepatocellular Carcinoma: A Systematic Review. Radiology 2018, 286, 29–48. [Google Scholar] [CrossRef]
- International Consensus Group for Hepatocellular Neoplasia the International Consensus Group for Hepatocellular Neoplasia. Pathologic diagnosis of early hepatocellular carcinoma: A report of the international consensus group for hepatocellular neoplasia. Hepatology 2009, 49, 658–664. [Google Scholar] [CrossRef] [PubMed]
- European Association for The Study of The Liver; European Organisation for Research and Treatment of Cancer. EASL-EORTC clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943. [Google Scholar] [CrossRef]
- Heimbach, J.K.; Kulik, L.M.; Finn, R.S.; Sirlin, C.B.; Abecassis, M.M.; Roberts, L.R.; Zhu, A.X.; Murad, M.H.; Marrero, J.A. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018, 67, 358–380. [Google Scholar] [CrossRef]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fàbrega, J.; Burrel, M.; Garcia-Criado, Á.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC strategy for prognosis prediction and treatment recommendation: The 2022 update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef]
- Guglielmi, A.; Ruzzenente, A.; Conci, S.; Valdegamberi, A.; Iacono, C. How much remnant is enough in liver resection? Dig. Surg. 2012, 29, 6–17. [Google Scholar] [CrossRef]
- Doyle, M.B.M.; Vachharajani, N.; Maynard, E.; Shenoy, S.; Anderson, C.; Wellen, J.R.; Lowell, J.A.; Chapman, W.C. Liver transplantation for hepatocellular carcinoma: Long-term results suggest excellent outcomes. J. Am. Coll. Surg. 2012, 215, 19–28; discussion 28–30. [Google Scholar] [CrossRef]
- Koh, J.H.; Tan, D.J.H.; Ong, Y.; Lim, W.H.; Ng, C.H.; Tay, P.W.L.; Yong, J.N.; Muthiah, M.D.; Tan, E.X.; Pang, N.Q.; et al. Liver resection versus liver transplantation for hepatocellular carcinoma within Milan criteria: A meta-analysis of 18,421 patients. Hepatobiliary Surg. Nutr. 2022, 11, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.C.; Yu, R.L.; Alqahtani, S.; Tamim, H.; Saberi, B.; Bonder, A. Racial, ethnic, and socioeconomic disparities impact post-liver transplant survival in patients with hepatocellular carcinoma. Ann. Hepatol. 2023, 28, 101127. [Google Scholar] [CrossRef] [PubMed]
- Xin Koh, Y.; Jin Tan, H.; Xin Liew, Y.; Syn, N.; Yao Teo, J.; Yee Lee, S.; Goh, B.K.P.; Goh, G.B.B.; Yip Chan, C. Liver Resection for Nonalcoholic Fatty Liver Disease-Associated Hepatocellular Carcinoma. J. Am. Coll. Surg. 2019, 229, 467–478e1. [Google Scholar] [CrossRef] [PubMed]
- Wakai, T.; Shirai, Y.; Sakata, J.; Korita, P.V.; Ajioka, Y.; Hatakeyama, K. Surgical outcomes for hepatocellular carcinoma in nonalcoholic fatty liver disease. J. Gastrointest. Surg. 2011, 15, 1450–1458. [Google Scholar] [CrossRef]
- Cauchy, F.; Zalinski, S.; Dokmak, S.; Fuks, D.; Farges, O.; Castera, L.; Paradis, V.; Belghiti, J. Surgical treatment of hepatocellular carcinoma associated with the metabolic syndrome. Br. J. Surg. 2013, 100, 113–121. [Google Scholar] [CrossRef]
- Balzan, S.; Nagarajan, G.; Farges, O.; Galleano, C.Z.; Dokmak, S.; Paugam, C.; Belghiti, J. Safety of liver resections in obese and overweight patients. World J. Surg. 2010, 34, 2960–2968. [Google Scholar] [CrossRef]
- Wu, H.-Y.; Chang, C.-C.; Yeh, C.-C.; Chen, M.-Y.; Cherng, Y.-G.; Chen, T.-L.; Liao, C.-C. Adverse outcomes after non-hepatic surgeries in patients with alcoholic liver diseases: A propensity-score matched study. BMC Gastroenterol. 2022, 22, 475. [Google Scholar] [CrossRef]
- McClain, C.J.; Barve, S.S.; Barve, A.; Marsano, L. Alcoholic liver disease and malnutrition. Alcohol. Clin. Exp. Res. 2011, 35, 815–820. [Google Scholar] [CrossRef]
- Ford, K.L.; Prado, C.M.; Weimann, A.; Schuetz, P.; Lobo, D.N. Unresolved issues in perioperative nutrition: A narrative review. Clin. Nutr. 2022, 41, 1578–1590. [Google Scholar] [CrossRef]
- Llovet, J.M.; De Baere, T.; Kulik, L.; Haber, P.K.; Greten, T.F.; Meyer, T.; Lencioni, R. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 293–313. [Google Scholar] [CrossRef]
- Mazzaferro, V.; Regalia, E.; Doci, R.; Andreola, S.; Pulvirenti, A.; Bozzetti, F.; Montalto, F.; Ammatuna, M.; Morabito, A.; Gennari, L. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N. Engl. J. Med. 1996, 334, 693–699. [Google Scholar] [CrossRef]
- Lencioni, R.; Crocetti, L. Radiofrequency ablation of liver cancer. Tech. Vasc. Interv. Radiol. 2007, 10, 38–46. [Google Scholar] [CrossRef]
- Gao, Q.; Anwar, I.J.; Abraham, N.; Barbas, A.S. Liver Transplantation for Hepatocellular Carcinoma after Downstaging or Bridging Therapy with Immune Checkpoint Inhibitors. Cancers 2021, 13, 6307. [Google Scholar] [CrossRef] [PubMed]
- Glassberg, M.B.; Ghosh, S.; Clymer, J.W.; Qadeer, R.A.; Ferko, N.C.; Sadeghirad, B.; Wright, G.W.; Amaral, J.F. Microwave ablation compared with radiofrequency ablation for treatment of hepatocellular carcinoma and liver metastases: A systematic review and meta-analysis. OncoTargets Ther. 2019, 12, 6407–6438. [Google Scholar] [CrossRef]
- Imai, N.; Ishigami, M.; Ishizu, Y.; Kuzuya, T.; Honda, T.; Hayashi, K.; Hirooka, Y.; Goto, H. Transarterial chemoembolization for hepatocellular carcinoma: A review of techniques. World J. Hepatol. 2014, 6, 844–850. [Google Scholar] [CrossRef]
- Kallini, J.R.; Gabr, A.; Salem, R.; Lewandowski, R.J. Transarterial Radioembolization with Yttrium-90 for the Treatment of Hepatocellular Carcinoma. Adv. Ther. 2016, 33, 699–714. [Google Scholar] [CrossRef]
- Qin, S.; Chen, M.; Cheng, A.-L.; Kaseb, A.O.; Kudo, M.; Lee, H.C.; Yopp, A.C.; Zhou, J.; Wang, L.; Wen, X.; et al. Atezolizumab plus bevacizumab versus active surveillance in patients with resected or ablated high-risk hepatocellular carcinoma (IMbrave050): A randomised, open-label, multicentre, phase 3 trial. Lancet 2023, 402, 1835–1847. [Google Scholar] [CrossRef]
- Ganne-Carrié, N.; Nahon, P. Differences between hepatocellular carcinoma caused by alcohol and other aetiologies. J. Hepatol. 2025, 82, 909–917. [Google Scholar] [CrossRef]
- Why Is Nutrition So Important in Alcohol Associated Liver Disease?|AASLD. Available online: https://www.aasld.org/liver-fellow-network/core-series/why-series/why-nutrition-so-important-alcohol-associated-liver (accessed on 14 August 2022).
- Kim, H.; Lee, D.S.; An, T.H.; Park, H.-J.; Kim, W.K.; Bae, K.-H.; Oh, K.-J. Metabolic Spectrum of Liver Failure in Type 2 Diabetes and Obesity: From NAFLD to NASH to HCC. Int. J. Mol. Sci. 2021, 22, 4495. [Google Scholar] [CrossRef]
- Kraglund, F.; Birn-Rydder, R.; Sørensen, M.D.; Abazi, R.; Villadsen, G.E.; Jepsen, P. Alcohol use and hepatocellular carcinoma risk in patients with alcohol-related cirrhosis. Scand. J. Gastroenterol. 2023, 58, 1321–1327. [Google Scholar] [CrossRef]
- Wattacheril, J.J.; Abdelmalek, M.F.; Lim, J.K.; Sanyal, A.J. AGA Clinical Practice Update on the Role of Noninvasive Biomarkers in the Evaluation and Management of Nonalcoholic Fatty Liver Disease: Expert Review. Gastroenterology 2023, 165, 1080–1088. [Google Scholar] [CrossRef]
- Jophlin, L.L.; Singal, A.K.; Bataller, R.; Wong, R.J.; Sauer, B.G.; Terrault, N.A.; Shah, V.H. ACG Clinical Guideline: Alcohol-Associated Liver Disease. Am. J. Gastroenterol. 2024, 119, 30–54. [Google Scholar] [CrossRef] [PubMed]
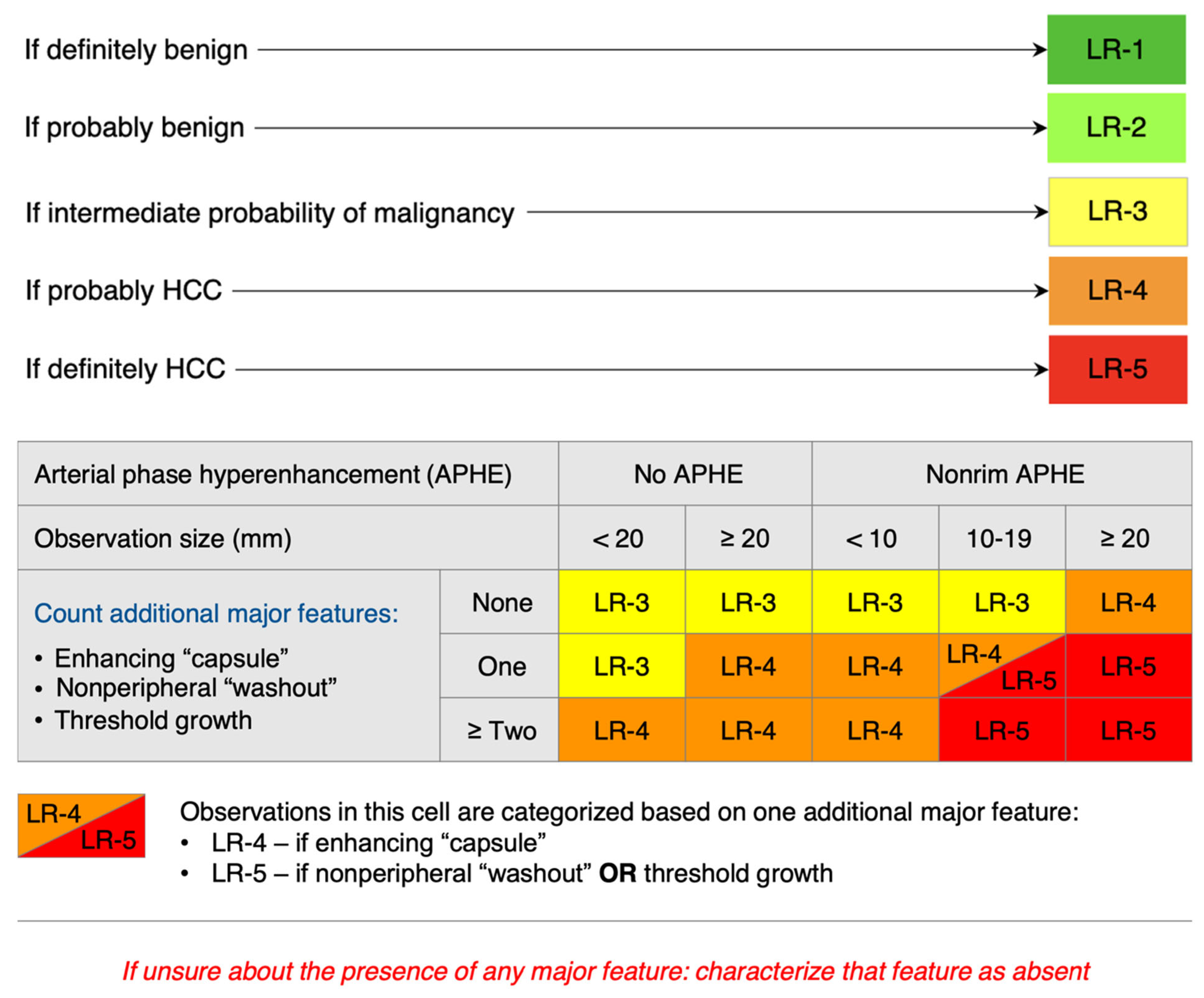
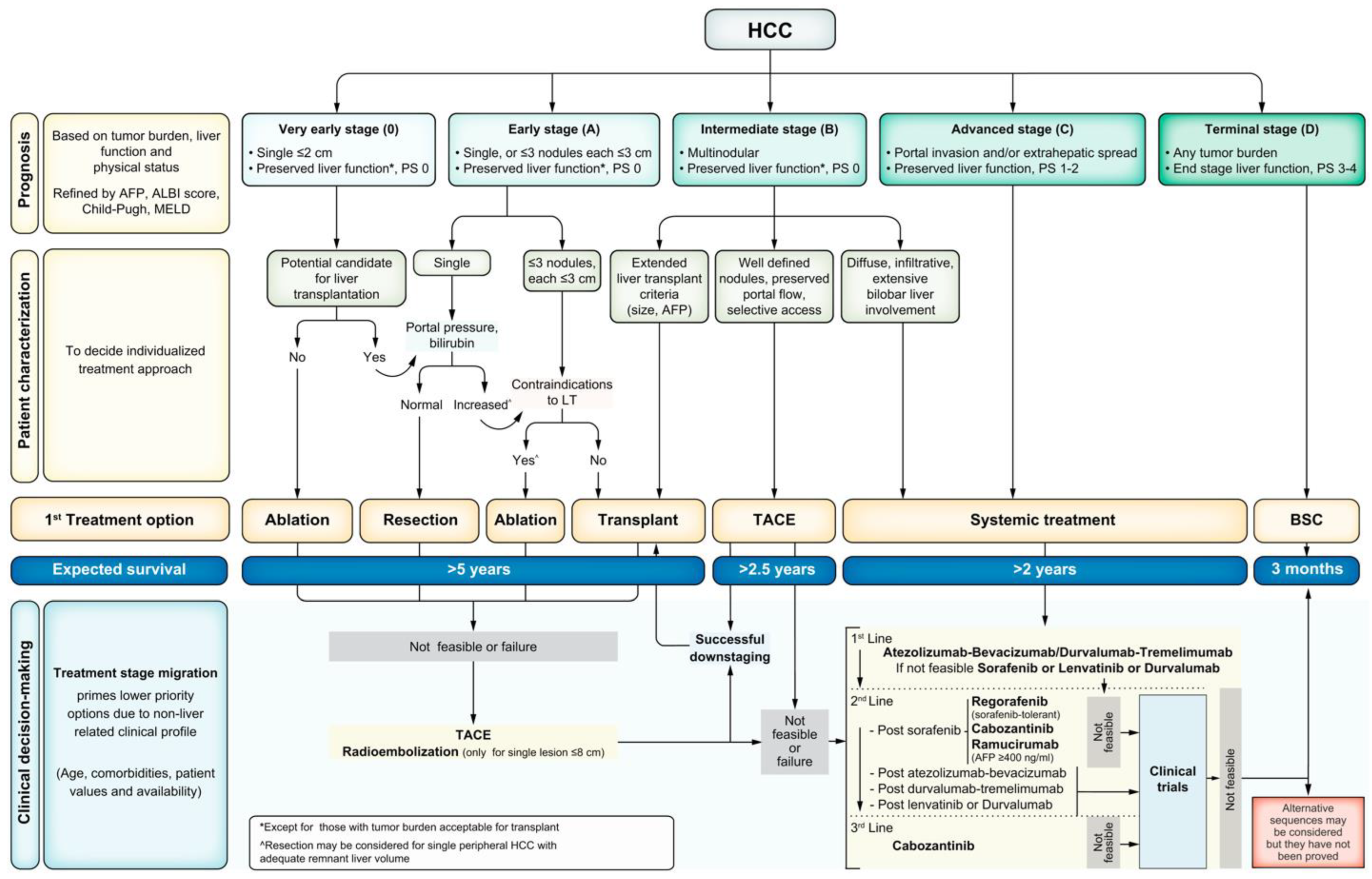
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).