
Protein Ligases: Nature’s Gift for Protein/Peptide Synthesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
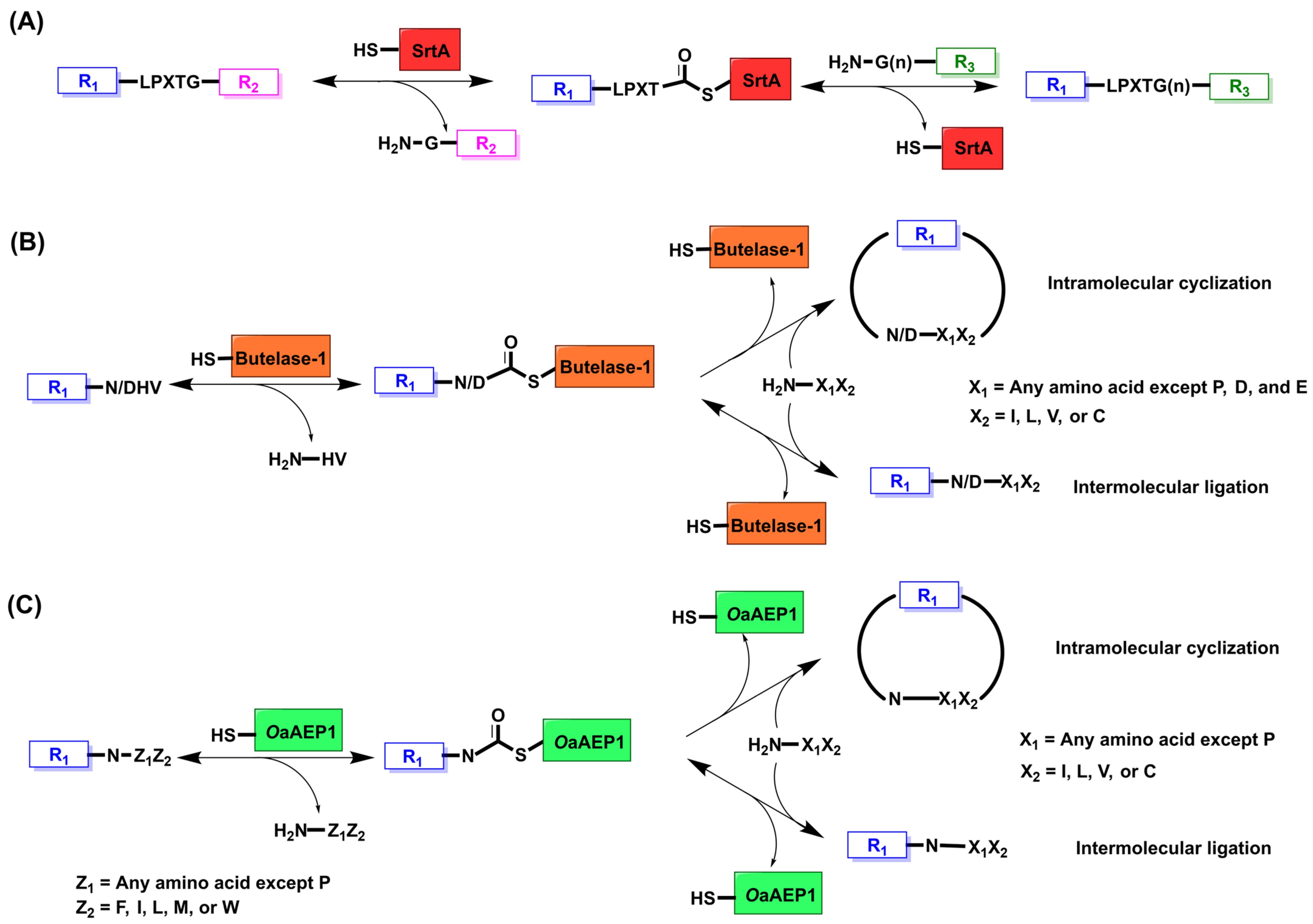
2. Sortase A
2.1. Mechanism and Engineering
2.2. Applications
2.2.1. Histone Modification In Vitro and In Cellulo
2.2.2. Protein Engineering Through “Sortagging”
2.2.3. Bacterial Cell Wall Engineering
3. Butelase
3.1. Mechanism and Engineering
3.2. Applications
4. OaAEP1
4.1. Mechanism and Engineering
4.2. Applications
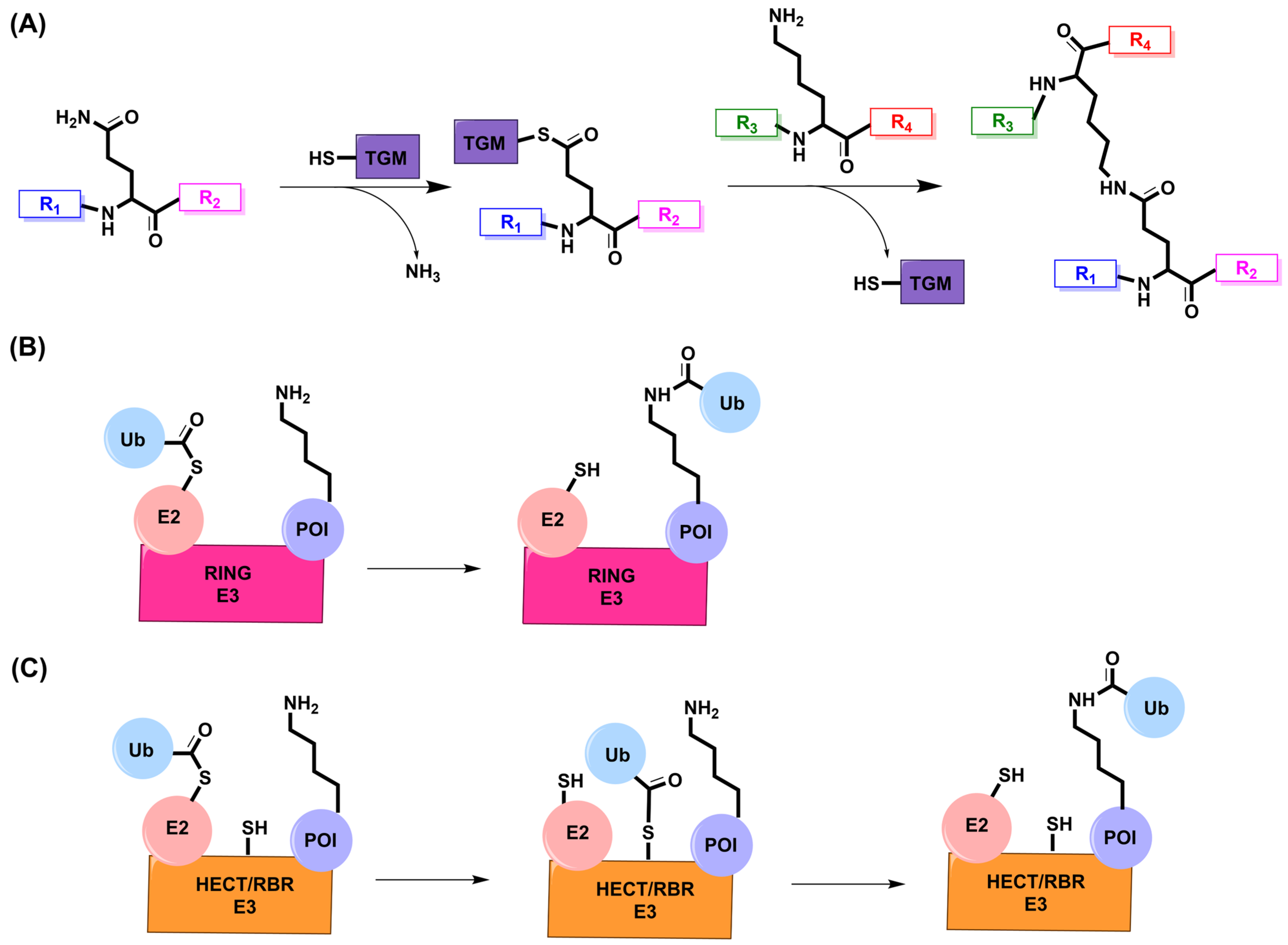
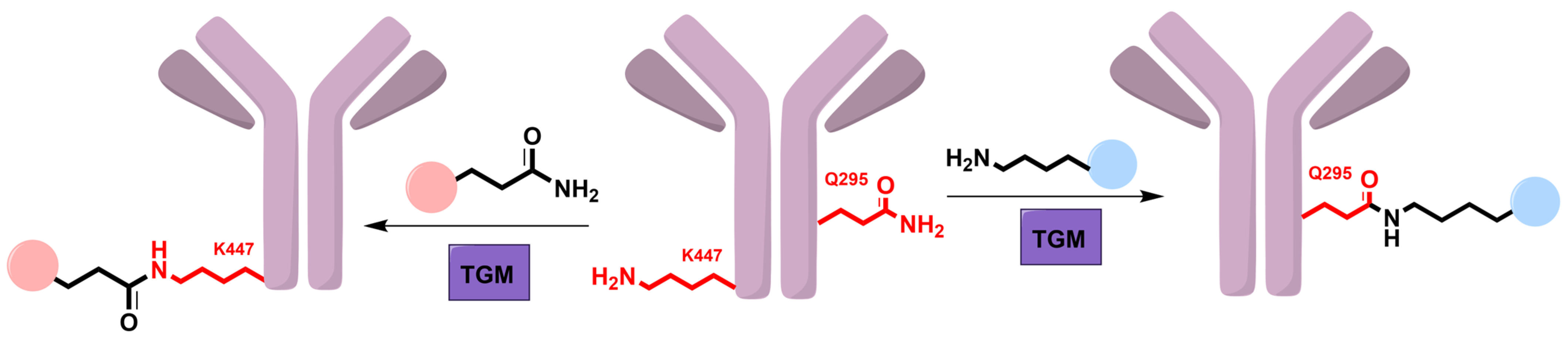
5. Transglutaminase
5.1. Mechanism and Engineering
5.2. Applications
6. E3 Ligase
6.1. Mechanism
6.2. Applications
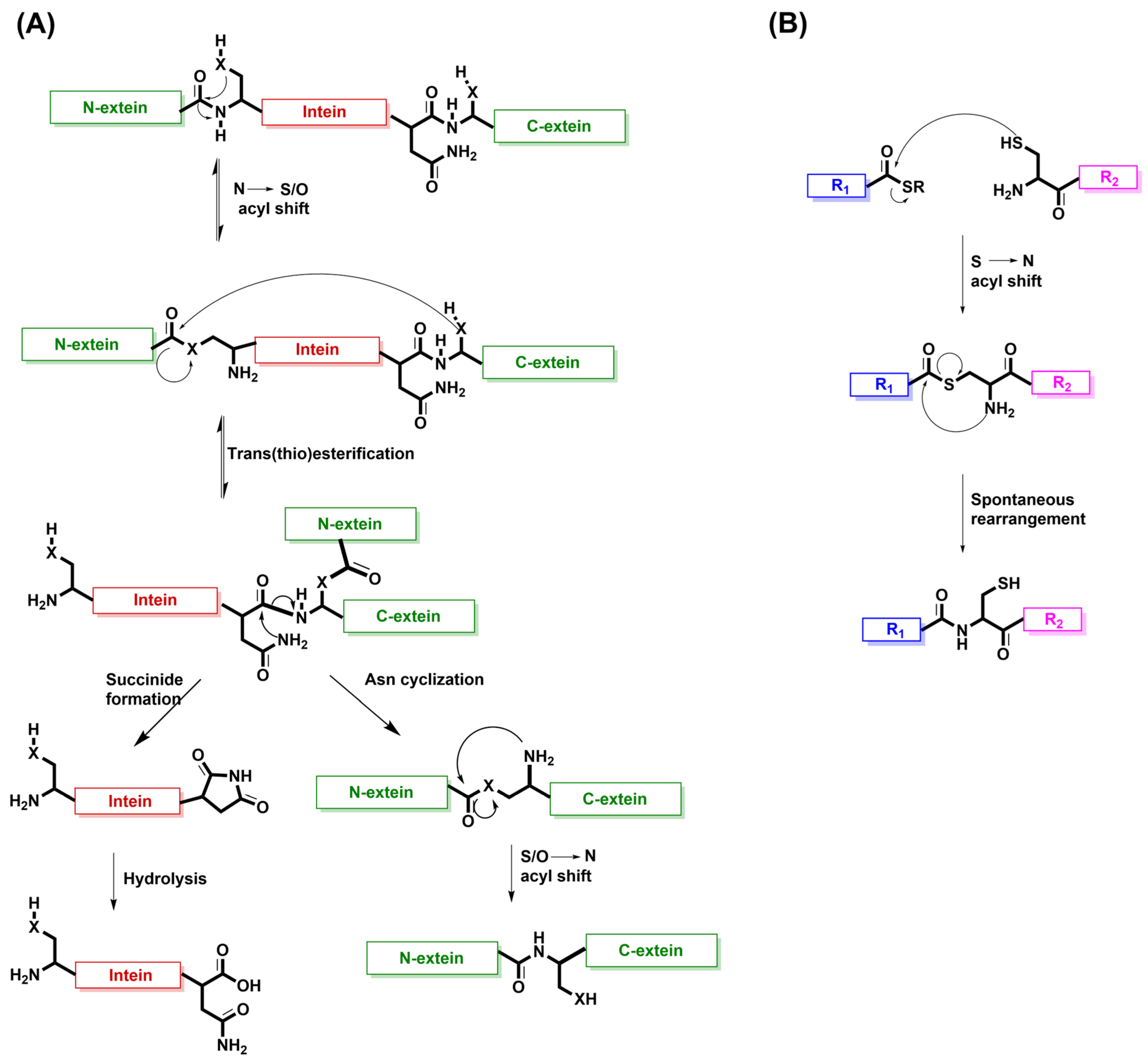
7. Intein Splicing as the Inspiration for Native Chemical Ligation (NCL)
8. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Listov, D.; Goverde, C.A.; Correia, B.E.; Fleishman, S.J. Opportunities and Challenges in Design and Optimization of Protein Function. Nat. Rev. Mol. Cell Biol. 2024, 25, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Jiang, W.; Chen, Z.; Huang, Y.; Mao, J.; Zheng, W.; Hu, Y.; Shi, J. Advance in Peptide-Based Drug Development: Delivery Platforms, Therapeutics and Vaccines. Signal Transduct. Target. Ther. 2025, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, S.B.; Samanta, D. Engineering Protein-Based Therapeutics through Structural and Chemical Design. Nat. Commun. 2023, 14, 2411. [Google Scholar] [CrossRef] [PubMed]
- Johnson, I.S. Human Insulin from Recombinant DNA Technology. Science 1983, 219, 632–637. [Google Scholar] [CrossRef]
- Schellenberger, V.; Wang, C.; Geething, N.C.; Spink, B.J.; Campbell, A.; To, W.; Scholle, M.D.; Yin, Y.; Yao, Y.; Bogin, O.; et al. A Recombinant Polypeptide Extends the in Vivo Half-Life of Peptides and Proteins in a Tunable Manner. Nat. Biotechnol. 2009, 27, 1186–1190. [Google Scholar] [CrossRef]
- Tokmakov, A.A.; Kurotani, A.; Takagi, T.; Toyama, M.; Shirouzu, M.; Fukami, Y.; Yokoyama, S. Multiple Post-Translational Modifications Affect Heterologous Protein Synthesis. J. Biol. Chem. 2012, 287, 27106–27116. [Google Scholar] [CrossRef]
- Berrade, L.; Camarero, J.A. Expressed Protein Ligation: A Resourceful Tool to Study Protein Structure and Function. Cell. Mol. Life Sci. 2009, 66, 3909–3922. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Sayers, J.; Premdjee, B.; Payne, R.J. Rapid and Efficient Protein Synthesis through Expansion of the Native Chemical Ligation Concept. Nat. Rev. Chem. 2018, 2, 0122. [Google Scholar] [CrossRef]
- Bondalapati, S.; Jbara, M.; Brik, A. Expanding the Chemical Toolbox for the Synthesis of Large and Uniquely Modified Proteins. Nat. Chem. 2016, 8, 407–418. [Google Scholar] [CrossRef]
- Jacobitz, A.W.; Kattke, M.D.; Wereszczynski, J.; Clubb, R.T. Sortase Transpeptidases: Structural Biology and Catalytic Mechanism. Adv. Protein Chem. Struct. Biol. 2017, 109, 223–264. [Google Scholar] [CrossRef]
- Spirig, T.; Weiner, E.M.; Clubb, R.T. Sortase Enzymes in Gram-Positive Bacteria. Mol. Microbiol. 2011, 82, 1044–1059. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J. Surface Proteins of Staphylococcus Aureus. Microbiol. Spectr. 2019, 7, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Amacher, J.F.; Antos, J.M. Sortases: Structure, Mechanism, and Implications for Protein Engineering. Trends Biochem. Sci. 2024, 49, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, G.; Ton-That, H.; Schneewind, O. Staphylococcus Aureus Sortase, an Enzyme That Anchors Surface Proteins to the Cell Wall. Science 1999, 285, 760–763. [Google Scholar] [CrossRef]
- Bradshaw, W.J.; Davies, A.H.; Chambers, C.J.; Roberts, A.K.; Shone, C.C.; Acharya, K.R. Molecular Features of the Sortase Enzyme Family. FEBS J. 2015, 282, 2097–2114. [Google Scholar] [CrossRef]
- Antos, J.M.; Truttmann, M.C.; Ploegh, H.L. Recent Advances in Sortase-Catalyzed Ligation Methodology. Curr. Opin. Struct. Biol. 2016, 38, 111–118. [Google Scholar] [CrossRef]
- Pihl, R.; Zheng, Q.; David, Y. Nature-Inspired Protein Ligation and Its Applications. Nat. Rev. Chem. 2023, 7, 234–255. [Google Scholar] [CrossRef]
- Chen, I.; Dorr, B.M.; Liu, D.R. A General Strategy for the Evolution of Bond-Forming Enzymes Using Yeast Display. Proc. Natl. Acad. Sci. USA 2011, 108, 11399–11404. [Google Scholar] [CrossRef]
- Hirakawa, H.; Ishikawa, S.; Nagamune, T. Ca2+-Independent Sortase-A Exhibits High Selective Protein Ligation Activity in the Cytoplasm of Escherichia coli. Biotechnol. J. 2015, 10, 1487–1492. [Google Scholar] [CrossRef]
- Wuethrich, I.; Peeters, J.G.C.; Blom, A.E.M.; Theile, C.S.; Li, Z.; Spooner, E.; Ploegh, H.L.; Guimaraes, C.P. Site-Specific Chemoenzymatic Labeling of Aerolysin Enables the Identification of New Aerolysin Receptors. PLoS ONE 2014, 9, e109883. [Google Scholar] [CrossRef]
- Piotukh, K.; Geltinger, B.; Heinrich, N.; Gerth, F.; Beyermann, M.; Freund, C.; Schwarzer, D. Directed Evolution of Sortase A Mutants with Altered Substrate Selectivity Profiles. J. Am. Chem. Soc. 2011, 133, 17536–17539. [Google Scholar] [CrossRef] [PubMed]
- Whedon, S.D.; Lee, K.; Wang, Z.A.; Zahn, E.; Lu, C.; Yapa Abeywardana, M.; Fairall, L.; Nam, E.; DuBois-Coyne, S.; De Ioannes, P.; et al. Circular Engineered Sortase for Interrogating Histone H3 in Chromatin. J. Am. Chem. Soc. 2024, 146, 33914–33927. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Gao, Y.; Liu, X.; Xiao, Y.; Wu, M. A General Method to Edit Histone H3 Modifications on Chromatin Via Sortase-Mediated Metathesis. Angew. Chem. Int. Ed. 2022, 61, e202209945. [Google Scholar] [CrossRef] [PubMed]
- Dorr, B.M.; Ham, H.O.; An, C.; Chaikof, E.L.; Liu, D.R. Reprogramming the Specificity of Sortase Enzymes. Proc. Natl. Acad. Sci. USA 2014, 111, 13343–13348. [Google Scholar] [CrossRef]
- Zhulenkovs, D.; Jaudzems, K.; Zajakina, A.; Leonchiks, A. Enzymatic Activity of Circular Sortase A under Denaturing Conditions: An Advanced Tool for Protein Ligation. Biochem. Eng. J. 2014, 82, 200–209. [Google Scholar] [CrossRef]
- Li, Z.; Tong, Z.; Gong, Q.; Ai, H.; Peng, S.; Chen, C.; Chu, G.-C.; Li, J.-B. The Expedient, CAET-Assisted Synthesis of Dual-Monoubiquitinated Histone H3 Enables Evaluation of Its Interaction with DNMT1. Chem. Sci. 2023, 14, 5681–5688. [Google Scholar] [CrossRef]
- Li, W.; Cao, P.; Xu, P.; Sun, F.; Wang, C.; Zhang, J.; Dong, S.; Wilson, J.R.; Xu, D.; Fan, H.; et al. Rapid Reconstitution of Ubiquitinated Nucleosome Using a Non-Denatured Histone Octamer Ubiquitylation Approach. Cell Biosci. 2024, 14, 81. [Google Scholar] [CrossRef]
- Wang, Z.A.; Whedon, S.D.; Wu, M.; Wang, S.; Brown, E.A.; Anmangandla, A.; Regan, L.; Lee, K.; Du, J.; Hong, J.Y.; et al. H2B Deacylation Selectivity: Exploring Chromatin’s Dark Matter with an Engineered Sortase. J. Am. Chem. Soc. 2022, 144, 3360–3364. [Google Scholar] [CrossRef]
- Xiao, Y.; Zou, K.; Yang, J.; Wu, M. Deciphering Histone H4 Lysine Acetylation and Methylation via Sortase-Mediated Semisynthesis. Cell Rep. Phys. Sci. 2023, 4, 101638. [Google Scholar] [CrossRef]
- Zou, Z.; Ji, Y.; Schwaneberg, U. Empowering Site-Specific Bioconjugations In Vitro and In Vivo: Advances in Sortase Engineering and Sortase-Mediated Ligation. Angew. Chem. Int. Ed. 2024, 63, e202310910. [Google Scholar] [CrossRef]
- Witte, M.D.; Cragnolini, J.J.; Dougan, S.K.; Yoder, N.C.; Popp, M.W.; Ploegh, H.L. Preparation of Unnatural N-to-N and C-to-C Protein Fusions. Proc. Natl. Acad. Sci. USA 2012, 109, 11993–11998. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Saxon, E.; Bertozzi, C.R. Cell Surface Engineering by a Modified Staudinger Reaction. Science 2000, 287, 2007–2010. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Liang, C.; Yu, X.-H.; Ma, X.; Qu, Y.; Zhuang, W.-R.; Li, W.; Nie, W.; Ren, Y.; Lei, Y.; et al. Chemistry-Enabled Intercellular Enzymatic Labeling for Monitoring the Immune Effects of Cytotoxic T Lymphocytes In Vivo. Anal. Chem. 2024, 96, 13996–14003. [Google Scholar] [CrossRef]
- Ito, H.; Seishima, M. Regulation of the Induction and Function of Cytotoxic T Lymphocytes by Natural Killer T Cell. J. Biomed. Biotechnol. 2010, 2010, 641757. [Google Scholar] [CrossRef]
- Nelson, J.W.; Chamessian, A.G.; McEnaney, P.J.; Murelli, R.P.; Kazmierczak, B.I.; Spiegel, D.A. Correction to A Biosynthetic Strategy for Re-Engineering the Staphylococcus Aureus Cell Wall with Non-Native Small Molecules. ACS Chem. Biol. 2011, 6, 971. [Google Scholar] [CrossRef]
- Jiang, F.; Cai, C.; Wang, X.; Han, S. A Dual Biomarker-Targeting Probe Enables Signal-on Surface Labeling of Staphylococcus aureus. Bioorg. Med. Chem. Lett. 2023, 93, 129428. [Google Scholar] [CrossRef]
- Self, W.H.; Wunderink, R.G.; Williams, D.J.; Zhu, Y.; Anderson, E.J.; Balk, R.A.; Fakhran, S.S.; Chappell, J.D.; Casimir, G.; Courtney, D.M.; et al. Staphylococcus aureus Community-Acquired Pneumonia: Prevalence, Clinical Characteristics, and Outcomes. Clin. Infect. Dis. 2016, 63, 300–309. [Google Scholar] [CrossRef]
- Silversides, J.A.; Lappin, E.; Ferguson, A.J. Staphylococcal Toxic Shock Syndrome: Mechanisms and Management. Curr. Infect. Dis. Rep. 2010, 12, 392–400. [Google Scholar] [CrossRef]
- Nguyen, G.K.T.; Kam, A.; Loo, S.; Jansson, A.E.; Pan, L.X.; Tam, J.P. Butelase 1: A Versatile Ligase for Peptide and Protein Macrocyclization. J. Am. Chem. Soc. 2015, 137, 15398–15401. [Google Scholar] [CrossRef]
- Nguyen, G.K.T.; Wang, S.; Qiu, Y.; Hemu, X.; Lian, Y.; Tam, J.P. Butelase 1 Is an Asx-Specific Ligase Enabling Peptide Macrocyclization and Synthesis. Nat. Chem. Biol. 2014, 10, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Hemu, X.; Zhang, X.; Bi, X.; Liu, C.-F.; Tam, J.P. Butelase 1-Mediated Ligation of Peptides and Proteins. In Enzyme-Mediated Ligation Methods; Nuijens, T., Schmidt, M., Eds.; Springer: New York, NY, USA, 2019; pp. 83–109. [Google Scholar] [CrossRef]
- Morgan, H.E.; Turnbull, W.B.; Webb, M.E. Challenges in the Use of Sortase and Other Peptide Ligases for Site-Specific Protein Modification. Chem. Soc. Rev. 2022, 51, 4121–4145. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.K.T.; Cao, Y.; Wang, W.; Liu, C.F.; Tam, J.P. Site-Specific N-Terminal Labeling of Peptides and Proteins Using Butelase 1 and Thiodepsipeptide. Angew. Chem. Int. Ed. 2015, 54, 15694–15698. [Google Scholar] [CrossRef] [PubMed]
- Hemu, X.; Zhang, X.; Nguyen, G.K.T.; To, J.; Serra, A.; Loo, S.; Sze, S.K.; Liu, C.-F.; Tam, J.P. Characterization and Application of Natural and Recombinant Butelase-1 to Improve Industrial Enzymes by End-to-End Circularization. RSC Adv. 2021, 11, 23105–23112. [Google Scholar] [CrossRef]
- Zhao, J.; Fan, R.; Jia, F.; Huang, Y.; Huang, Z.; Hou, Y.; Hu, S.-Q. Enzymatic Properties of Recombinant Ligase Butelase-1 and Its Application in Cyclizing Food-Derived Angiotensin I-Converting Enzyme Inhibitory Peptides. J. Agric. Food Chem. 2021, 69, 5976–5985. [Google Scholar] [CrossRef]
- Nguyen, G.K.T.; Qiu, Y.; Cao, Y.; Hemu, X.; Liu, C.-F.; Tam, J.P. Butelase-Mediated Cyclization and Ligation of Peptides and Proteins. Nat. Protoc. 2016, 11, 1977–1988. [Google Scholar] [CrossRef]
- Zhao, J.; Song, W.; Huang, Z.; Yuan, X.; Huang, Y.; Hou, Y.; Liu, K.; Jin, P.; Hu, S.-Q. “Top-down” Overexpression Optimization of Butelase-1 in Escherichia coli and Its Application in Anti-Tumor Peptides. Int. J. Biol. Macromol. 2024, 276, 133933. [Google Scholar] [CrossRef]
- Zhao, J.; Ge, G.; Huang, Y.; Hou, Y.; Hu, S.-Q. Study on Activation Mechanism and Cleavage Sites of Recombinant Butelase-1 Zymogen Derived from Clitoria ternatea. Biochimie 2022, 199, 12–22. [Google Scholar] [CrossRef]
- Singh, A.K.; Antonenko, A.; Kocyła, A.; Krężel, A. An Efficient and Easily Obtainable Butelase Variant for Chemoenzymatic Ligation and Modification of Peptides and Proteins. Microb. Cell Fact. 2024, 23, 325. [Google Scholar] [CrossRef]
- Cui, Y.; Han, D.; Bai, X.; Shi, W. Development and Applications of Enzymatic Peptide and Protein Ligation. J. Pept. Sci. 2025, 31, e3657. [Google Scholar] [CrossRef]
- Cao, Y.; Nguyen, G.K.T.; Chuah, S.; Tam, J.P.; Liu, C.-F. Butelase-Mediated Ligation as an Efficient Bioconjugation Method for the Synthesis of Peptide Dendrimers. Bioconjug. Chem. 2016, 27, 2592–2596. [Google Scholar] [CrossRef] [PubMed]
- Galvez, A.F.; Chen, N.; Macasieb, J.; de Lumen, B.O. Chemopreventive Property of a Soybean Peptide (Lunasin) That Binds to Deacetylated Histones and Inhibits Acetylation1. Cancer Res. 2001, 61, 7473–7478. [Google Scholar] [PubMed]
- Zhao, J.; Ge, G.; Huang, Y.; Hou, Y.; Hu, S.-Q. Butelase 1-Mediated Enzymatic Cyclization of Antimicrobial Peptides: Improvements on Stability and Bioactivity. J. Agric. Food Chem. 2022, 70, 15869–15878. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.S.; Durek, T.; Kaas, Q.; Poth, A.G.; Gilding, E.K.; Conlan, B.F.; Saska, I.; Daly, N.L.; van der Weerden, N.L.; Craik, D.J.; et al. Efficient Backbone Cyclization of Linear Peptides by a Recombinant Asparaginyl Endopeptidase. Nat. Commun. 2015, 6, 10199. [Google Scholar] [CrossRef]
- Tang, T.M.S.; Cardella, D.; Lander, A.J.; Li, X.; Escudero, J.S.; Tsai, Y.-H.; Luk, L.Y.P. Use of an Asparaginyl Endopeptidase for Chemo-Enzymatic Peptide and Protein Labeling. Chem. Sci. 2020, 11, 5881–5888. [Google Scholar] [CrossRef]
- Yang, R.; Wong, Y.H.; Nguyen, G.K.T.; Tam, J.P.; Lescar, J.; Wu, B. Engineering a Catalytically Efficient Recombinant Protein Ligase. J. Am. Chem. Soc. 2017, 139, 5351–5358. [Google Scholar] [CrossRef]
- Rehm, F.B.H.; Tyler, T.J.; Xie, J.; Yap, K.; Durek, T.; Craik, D.J. Asparaginyl Ligases: New Enzymes for the Protein Engineer’s Toolbox. ChemBioChem 2021, 22, 2079–2086. [Google Scholar] [CrossRef]
- Rehm, F.B.H.; Harmand, T.J.; Yap, K.; Durek, T.; Craik, D.J.; Ploegh, H.L. Site-Specific Sequential Protein Labeling Catalyzed by a Single Recombinant Ligase. J. Am. Chem. Soc. 2019, 141, 17388–17393. [Google Scholar] [CrossRef]
- Rehm, F.B.H.; Tyler, T.J.; de Veer, S.J.; Craik, D.J.; Durek, T. Enzymatic C-to-C Protein Ligation. Angew. Chem. Int. Ed. 2022, 61, e202116672. [Google Scholar] [CrossRef]
- Rehm, F.B.H.; Tyler, T.J.; Yap, K.; Durek, T.; Craik, D.J. Improved Asparaginyl-Ligase-Catalyzed Transpeptidation via Selective Nucleophile Quenching. Angew. Chem. Int. Ed. 2021, 60, 4004–4008. [Google Scholar] [CrossRef]
- Tang, J.; Hao, M.; Liu, J.; Chen, Y.; Wufuer, G.; Zhu, J.; Zhang, X.; Zheng, T.; Fang, M.; Zhang, S.; et al. Author Correction: Design of a Recombinant Asparaginyl Ligase for Site-Specific Modification Using Efficient Recognition and Nucleophile Motifs. Commun. Chem. 2024, 7, 121. [Google Scholar] [CrossRef] [PubMed]
- Simon Tang, T.M.; Mason, J.M. Intracellular Application of an Asparaginyl Endopeptidase for Producing Recombinant Head-to-Tail Cyclic Proteins. JACS Au 2023, 3, 3290–3296. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.-C.; Zhang, Y.-N.; Zhang, H.; Chen, Y.; Cui, Z.-H.; Zhu, W.-J.; Fang, G.-M. Asparaginyl Endopeptidase-Mediated Peptide Cyclization for Phage Display. Org. Lett. 2024, 26, 2601–2605. [Google Scholar] [CrossRef] [PubMed]
- Fottner, M.; Heimgärtner, J.; Gantz, M.; Mühlhofer, R.; Nast-Kolb, T.; Lang, K. Site-Specific Protein Labeling and Generation of Defined Ubiquitin-Protein Conjugates Using an Asparaginyl Endopeptidase. J. Am. Chem. Soc. 2022, 144, 13118–13126. [Google Scholar] [CrossRef]
- Wanka, V.; Fottner, M.; Cigler, M.; Lang, K. Genetic Code Expansion Approaches to Decipher the Ubiquitin Code. Chem. Rev. 2024, 124, 11544–11584. [Google Scholar] [CrossRef]
- de Veer, S.J.; Craik, D.J.; Rehm, F.B.H. Highly Efficient Transpeptidase-Catalyzed Isopeptide Ligation. J. Am. Chem. Soc. 2025, 147, 557–565. [Google Scholar] [CrossRef]
- Gundemir, S.; Colak, G.; Tucholski, J.; Johnson, G.V.W. Transglutaminase 2: A Molecular Swiss Army Knife. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2012, 1823, 406–419. [Google Scholar] [CrossRef]
- Keillor, J.W.; Clouthier, C.M.; Apperley, K.Y.P.; Akbar, A.; Mulani, A. Acyl Transfer Mechanisms of Tissue Transglutaminase. Bioorg. Chem. 2014, 57, 186–197. [Google Scholar] [CrossRef]
- Sarkar, N.K.; Clarke, D.D.; Waelsch, H. An Enzymically Catalyzed Incorporation of Amines into Proteins. Biochim. Biophys. Acta 1957, 25, 451–452. [Google Scholar] [CrossRef]
- Pincus, J.H.; Waelsch, H. The Specificity of Transglutaminase: II. Structural Requirements of the Amine Substrate. Arch. Biochem. Biophys. 1968, 126, 44–52. [Google Scholar] [CrossRef]
- Griffin, M.; Casadio, R.; Bergamini, C.M. Transglutaminases: Nature’s Biological Glues. Biochem. J. 2002, 368, 377–396. [Google Scholar] [CrossRef] [PubMed]
- Aaron, L.; Torsten, M. Microbial Transglutaminase: A New Potential Player in Celiac Disease. Clin. Immunol. 2019, 199, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Wang, X.; Ye, J.; Rao, S.; Zhou, J.; Du, G.; Liu, S. Enhanced Thermostability and Catalytic Activity of Streptomyces Mobaraenesis Transglutaminase by Rationally Engineering Its Flexible Regions. J. Agric. Food Chem. 2023, 71, 6366–6375. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Yang, P.; Zhou, J.; Du, G.; Liu, S. Efficient Production of a Thermostable Mutant of Transglutaminase by Streptomyces Mobaraensis. J. Agric. Food Chem. 2024, 72, 4207–4216. [Google Scholar] [CrossRef]
- Marx, C.K.; Hertel, T.C.; Pietzsch, M. Random Mutagenesis of a Recombinant Microbial Transglutaminase for the Generation of Thermostable and Heat-Sensitive Variants. J. Biotechnol. 2008, 136, 156–162. [Google Scholar] [CrossRef]
- Buettner, K.; Hertel, T.C.; Pietzsch, M. Increased Thermostability of Microbial Transglutaminase by Combination of Several Hot Spots Evolved by Random and Saturation Mutagenesis. Amino Acids 2012, 42, 987–996. [Google Scholar] [CrossRef]
- Yokoyama, K.; Utsumi, H.; Nakamura, T.; Ogaya, D.; Shimba, N.; Suzuki, E.; Taguchi, S. Screening for Improved Activity of a Transglutaminase from Streptomyces Mobaraensis Created by a Novel Rational Mutagenesis and Random Mutagenesis. Appl. Microbiol. Biotechnol. 2010, 87, 2087–2096. [Google Scholar] [CrossRef]
- Wang, X.; Du, J.; Zhao, B.; Wang, H.; Rao, S.; Du, G.; Zhou, J.; Chen, J.; Liu, S. Significantly Improving the Thermostability and Catalytic Efficiency of Streptomyces Mobaraenesis Transglutaminase through Combined Rational Design. J. Agric. Food Chem. 2021, 69, 15268–15278. [Google Scholar] [CrossRef]
- Malešević, M.; Migge, A.; Hertel, T.C.; Pietzsch, M. A Fluorescence-Based Array Screen for Transglutaminase Substrates. ChemBioChem 2015, 16, 1169–1174. [Google Scholar] [CrossRef]
- Wang, X.; Xu, K.; Fu, H.; Chen, Q.; Zhao, B.; Zhao, X.; Zhou, J. Enhancing Substrate Specificity of Microbial Transglutaminase for Precise Nanobody Labeling. Synth. Syst. Biotechnol. 2025, 10, 185–193. [Google Scholar] [CrossRef]
- Siegel, M.; Khosla, C. Transglutaminase 2 Inhibitors and Their Therapeutic Role in Disease States. Pharmacol. Ther. 2007, 115, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Sadiki, A.; Liu, S.; Vaidya, S.R.; Kercher, E.M.; Lang, R.T.; McIsaac, J.; Spring, B.Q.; Auclair, J.R.; Zhou, Z.S. Site-Specific Conjugation of Native Antibody: Transglutaminase-Mediated Modification of a Conserved Glutamine While Maintaining the Primary Sequence and Core Fc Glycan via Trimming with an Endoglycosidase. Bioconjug. Chem. 2024, 35, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Dickgiesser, S.; Rieker, M.; Mueller-Pompalla, D.; Schröter, C.; Tonillo, J.; Warszawski, S.; Raab-Westphal, S.; Kühn, S.; Knehans, T.; Könning, D.; et al. Site-Specific Conjugation of Native Antibodies Using Engineered Microbial Transglutaminases. Bioconjug. Chem. 2020, 31, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Spidel, J.L.; Vaessen, B.; Albone, E.F.; Cheng, X.; Verdi, A.; Kline, J.B. Site-Specific Conjugation to Native and Engineered Lysines in Human Immunoglobulins by Microbial Transglutaminase. Bioconjug. Chem. 2017, 28, 2471–2484. [Google Scholar] [CrossRef]
- Lambert, J.M.; Morris, C.Q. Antibody–Drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Adv. Ther. 2017, 34, 1015–1035. [Google Scholar] [CrossRef]
- Bolzati, C.; Spolaore, B. Enzymatic Methods for the Site-Specific Radiolabeling of Targeting Proteins. Molecules 2021, 26, 3492. [Google Scholar] [CrossRef]
- Jeger, S.; Zimmermann, K.; Blanc, A.; Grünberg, J.; Honer, M.; Hunziker, P.; Struthers, H.; Schibli, R. Site-Specific and Stoichiometric Modification of Antibodies by Bacterial Transglutaminase. Angew. Chem. Int. Ed. 2010, 49, 9995–9997. [Google Scholar] [CrossRef]
- Dick, L.W., Jr.; Qiu, D.; Mahon, D.; Adamo, M.; Cheng, K.-C. C-Terminal Lysine Variants in Fully Human Monoclonal Antibodies: Investigation of Test Methods and Possible Causes. Biotechnol. Bioeng. 2008, 100, 1132–1143. [Google Scholar] [CrossRef]
- Kuraishi, C.; Sakamoto, J.; Yamazaki, K.; Susa, Y.; Kuhara, C.; Soeda, T. Production of Restructured Meat Using Microbial Transglutaminase without Salt or Cooking. J. Food Sci. 1997, 62, 488–490. [Google Scholar] [CrossRef]
- Feng, Y.; Liang, X.; Zhang, J.; Shi, P.; Cao, C.; Zhang, H.; Liu, Q.; Kong, B. Underlying Mechanisms and Combined Effects of Transglutaminase and κ-Carrageenan on the Quality Profiles and In Vitro Digestibility of Frankfurters. Food Hydrocoll. 2024, 147, 109344. [Google Scholar] [CrossRef]
- Vasić, K.; Knez, Ž.; Leitgeb, M. Transglutaminase in Foods and Biotechnology. Int. J. Mol. Sci. 2023, 24, 12402. [Google Scholar] [CrossRef] [PubMed]
- Zinina, O.; Merenkova, S.; Galimov, D.; Okuskhanova, E.; Rebezov, M.; Khayrullin, M.; Anichkina, O. Effects of Microbial Transglutaminase on Technological, Rheological, and Microstructural Indicators of Minced Meat with the Addition of Plant Raw Materials. Int. J. Food Sci. 2020, 2020, 8869401. [Google Scholar] [CrossRef] [PubMed]
- Baugreet, S.; Kerry, J.P.; Brodkorb, A.; Gomez, C.; Auty, M.; Allen, P.; Hamill, R.M. Optimisation of Plant Protein and Transglutaminase Content in Novel Beef Restructured Steaks for Older Adults by Central Composite Design. Meat Sci. 2018, 142, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Chau, V.; Tobias, J.W.; Bachmair, A.; Marriott, D.; Ecker, D.J.; Gonda, D.K.; Varshavsky, A. A Multiubiquitin Chain Is Confined to Specific Lysine in a Targeted Short-Lived Protein. Science 1989, 243, 1576–1583. [Google Scholar] [CrossRef]
- Berndsen, C.E.; Wolberger, C. New Insights into Ubiquitin E3 Ligase Mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef]
- Ramachandran, S.; Ciulli, A. Building Ubiquitination Machineries: E3 Ligase Multi-Subunit Assembly and Substrate Targeting by PROTACs and Molecular Glues. Curr. Opin. Struct. Biol. 2021, 67, 110–119. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Toma-Fukai, S.; Shimizu, T. Structural Diversity of Ubiquitin E3 Ligase. Molecules 2021, 26, 6682. [Google Scholar] [CrossRef]
- Jeong, Y.; Oh, A.-R.; Jung, Y.H.; Gi, H.; Kim, Y.U.; Kim, K. Targeting E3 Ubiquitin Ligases and Their Adaptors as a Therapeutic Strategy for Metabolic Diseases. Exp. Mol. Med. 2023, 55, 2097–2104. [Google Scholar] [CrossRef]
- Wang, X.S.; Cotton, T.R.; Trevelyan, S.J.; Richardson, L.W.; Lee, W.T.; Silke, J.; Lechtenberg, B.C. The Unifying Catalytic Mechanism of the RING-between-RING E3 Ubiquitin Ligase Family. Nat. Commun. 2023, 14, 168. [Google Scholar] [CrossRef]
- Lechtenberg, B.C.; Rajput, A.; Sanishvili, R.; Dobaczewska, M.K.; Ware, C.F.; Mace, P.D.; Riedl, S.J. Structure of a HOIP/E2~ubiquitin Complex Reveals RBR E3 Ligase Mechanism and Regulation. Nature 2016, 529, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Timms, R.T.; Mena, E.L.; Leng, Y.; Li, M.Z.; Tchasovnikarova, I.A.; Koren, I.; Elledge, S.J. Author Correction: Defining E3 Ligase–Substrate Relationships through Multiplex CRISPR Screening. Nat. Cell Biol. 2024, 26, 305. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Ciulli, A. E3 Ligase Ligands for PROTACs: How They Were Found and How to Discover New Ones. SLAS Discov. 2021, 26, 484–502. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, Present and Future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef]
- Moreau, K.; Coen, M.; Zhang, A.X.; Pachl, F.; Castaldi, M.P.; Dahl, G.; Boyd, H.; Scott, C.; Newham, P. Proteolysis-Targeting Chimeras in Drug Development: A Safety Perspective. Br. J. Pharmacol. 2020, 177, 1709–1718. [Google Scholar] [CrossRef]
- Ye, P.; Chi, X.; Cha, J.-H.; Luo, S.; Yang, G.; Yan, X.; Yang, W.-H. Potential of E3 Ubiquitin Ligases in Cancer Immunity: Opportunities and Challenges. Cells 2021, 10, 3309. [Google Scholar] [CrossRef]
- Zou, Y.; Ma, D.; Wang, Y. The PROTAC Technology in Drug Development. Cell Biochem. Funct. 2019, 37, 21–30. [Google Scholar] [CrossRef]
- Li, R.; Liu, M.; Yang, Z.; Li, J.; Gao, Y.; Tan, R. Proteolysis-Targeting Chimeras (PROTACs) in Cancer Therapy: Present and Future. Molecules 2022, 27, 8828. [Google Scholar] [CrossRef]
- Pasieka, A.; Diamanti, E.; Uliassi, E.; Laura Bolognesi, M. Click Chemistry and Targeted Degradation: A Winning Combination for Medicinal Chemists? ChemMedChem 2023, 18, e202300422. [Google Scholar] [CrossRef]
- Yang, C.; Tripathi, R.; Wang, B. Click Chemistry in the Development of PROTACs. RSC Chem. Biol. 2024, 5, 189–197. [Google Scholar] [CrossRef]
- Chang, M.; Gao, F.; Pontigon, D.; Gnawali, G.; Xu, H.; Wang, W. Bioorthogonal PROTAC Prodrugs Enabled by On-Target Activation. J. Am. Chem. Soc. 2023, 145, 14155–14163. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Hou, B.; Zhu, Q.; Yang, L.; Jiang, X.; Zou, Z.; Li, X.; Xu, T.; Zheng, M.; Chen, Y.-H.; et al. Author Correction: Engineered Bioorthogonal POLY-PROTAC Nanoparticles for Tumour-Specific Protein Degradation and Precise Cancer Therapy. Nat. Commun. 2022, 13, 4978. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.; Zhao, X.; Dai, Y.; Zhang, X.; Zhang, Q.; Wu, Y.; Hu, D.; Li, J. ClickRNA-PROTAC for Tumor-Selective Protein Degradation and Targeted Cancer Therapy. J. Am. Chem. Soc. 2024, 146, 27382–27391. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, P.S. Degrader-Antibody Conjugates. Chem. Soc. Rev. 2022, 51, 3886–3897. [Google Scholar] [CrossRef]
- Guo, Y.; Li, X.; Xie, Y.; Wang, Y. What Influences the Activity of Degrader−Antibody Conjugates (DACs). Eur. J. Med. Chem. 2024, 268, 116216. [Google Scholar] [CrossRef]
- Maneiro, M.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody–PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312. [Google Scholar] [CrossRef]
- Pillow, T.H.; Adhikari, P.; Blake, R.A.; Chen, J.; Del Rosario, G.; Deshmukh, G.; Figueroa, I.; Gascoigne, K.E.; Kamath, A.V.; Kaufman, S.; et al. Antibody Conjugation of a Chimeric BET Degrader Enables in Vivo Activity. ChemMedChem 2020, 15, 17–25. [Google Scholar] [CrossRef]
- Cotton, A.D.; Nguyen, D.P.; Gramespacher, J.A.; Seiple, I.B.; Wells, J.A. Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc. 2021, 143, 593–598. [Google Scholar] [CrossRef]
- Gramespacher, J.A.; Cotton, A.D.; Burroughs, P.W.W.; Seiple, I.B.; Wells, J.A. Roadmap for Optimizing and Broadening Antibody-Based PROTACs for Degradation of Cell Surface Proteins. ACS Chem. Biol. 2022, 17, 1259–1268. [Google Scholar] [CrossRef]
- Kane, P.M.; Yamashiro, C.T.; Wolczyk, D.F.; Neff, N.; Goebl, M.; Stevens, T.H. Protein Splicing Converts the Yeast TFP1 Gene Product to the 69-kdDSubunit of the Vacuolar H+-Adenosine Triphosphatase. Science 1990, 250, 651–657. [Google Scholar] [CrossRef]
- Hirata, R.; Ohsumk, Y.; Nakano, A.; Kawasaki, H.; Suzuki, K.; Anraku, Y. Molecular Structure of a Gene, VMA1, Encoding the Catalytic Subunit of H(+)-Translocating Adenosine Triphosphatase from Vacuolar Membranes of Saccharomyces Cerevisiae. J. Biol. Chem. 1990, 265, 6726–6733. [Google Scholar] [CrossRef] [PubMed]
- Perler, F.B.; Davis, E.O.; Dean, G.E.; Gimble, F.S.; Jack, W.E.; Neff, N.; Noren, C.J.; Thorner, J.; Belfort, M. Protein Splicing Elements: Inteins and Exteins--a Definition of Terms and Recommended Nomenclature. Nucleic Acids Res. 1994, 22, 1125. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Zhong, B.; Dai, Z. Protein Splicing of Inteins: A Powerful Tool in Synthetic Biology. Front. Bioeng. Biotechnol. 2022, 10, 810180. [Google Scholar] [CrossRef] [PubMed]
- Nanda, A.; Nasker, S.S.; Mehra, A.; Panda, S.; Nayak, S. Inteins in Science: Evolution to Application. Microorganisms 2020, 8, 2004. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.W.; Belfort, M.; Lennon, C.W. Inteins—Mechanism of Protein Splicing, Emerging Regulatory Roles, and Applications in Protein Engineering. Front. Microbiol. 2023, 14, 1305848. [Google Scholar] [CrossRef]
- Friedel, K.; Popp, M.A.; Matern, J.C.J.; Gazdag, E.M.; Thiel, I.V.; Volkmann, G.; Blankenfeldt, W.; Mootz, H.D. A Functional Interplay between Intein and Extein Sequences in Protein Splicing Compensates for the Essential Block B Histidine. Chem. Sci. 2019, 10, 239–251. [Google Scholar] [CrossRef]
- Volkmann, G.; Mootz, H.D. Recent Progress in Intein Research: From Mechanism to Directed Evolution and Applications. Cell Mol. Life Sci. 2012, 70, 1185–1206. [Google Scholar] [CrossRef]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B.H. Synthesis of Proteins by Native Chemical Ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef]
- Pinto, F.; Thornton, E.L.; Wang, B. An Expanded Library of Orthogonal Split Inteins Enables Modular Multi-Peptide Assemblies. Nat. Commun. 2020, 11, 1529. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritsema, Y.; Li, H.; Zheng, Q. Protein Ligases: Nature’s Gift for Protein/Peptide Synthesis. BioChem 2025, 5, 11. https://doi.org/10.3390/biochem5020011
Ritsema Y, Li H, Zheng Q. Protein Ligases: Nature’s Gift for Protein/Peptide Synthesis. BioChem. 2025; 5(2):11. https://doi.org/10.3390/biochem5020011
Chicago/Turabian StyleRitsema, Yvonne, Huapeng Li, and Qingfei Zheng. 2025. "Protein Ligases: Nature’s Gift for Protein/Peptide Synthesis" BioChem 5, no. 2: 11. https://doi.org/10.3390/biochem5020011
APA StyleRitsema, Y., Li, H., & Zheng, Q. (2025). Protein Ligases: Nature’s Gift for Protein/Peptide Synthesis. BioChem, 5(2), 11. https://doi.org/10.3390/biochem5020011