
MYD88 Wild Type in IgM Monoclonal Gammopathies: From Molecular Mechanisms to Clinical Challenges
 ,
,  and
and
Abstract
1. Introduction
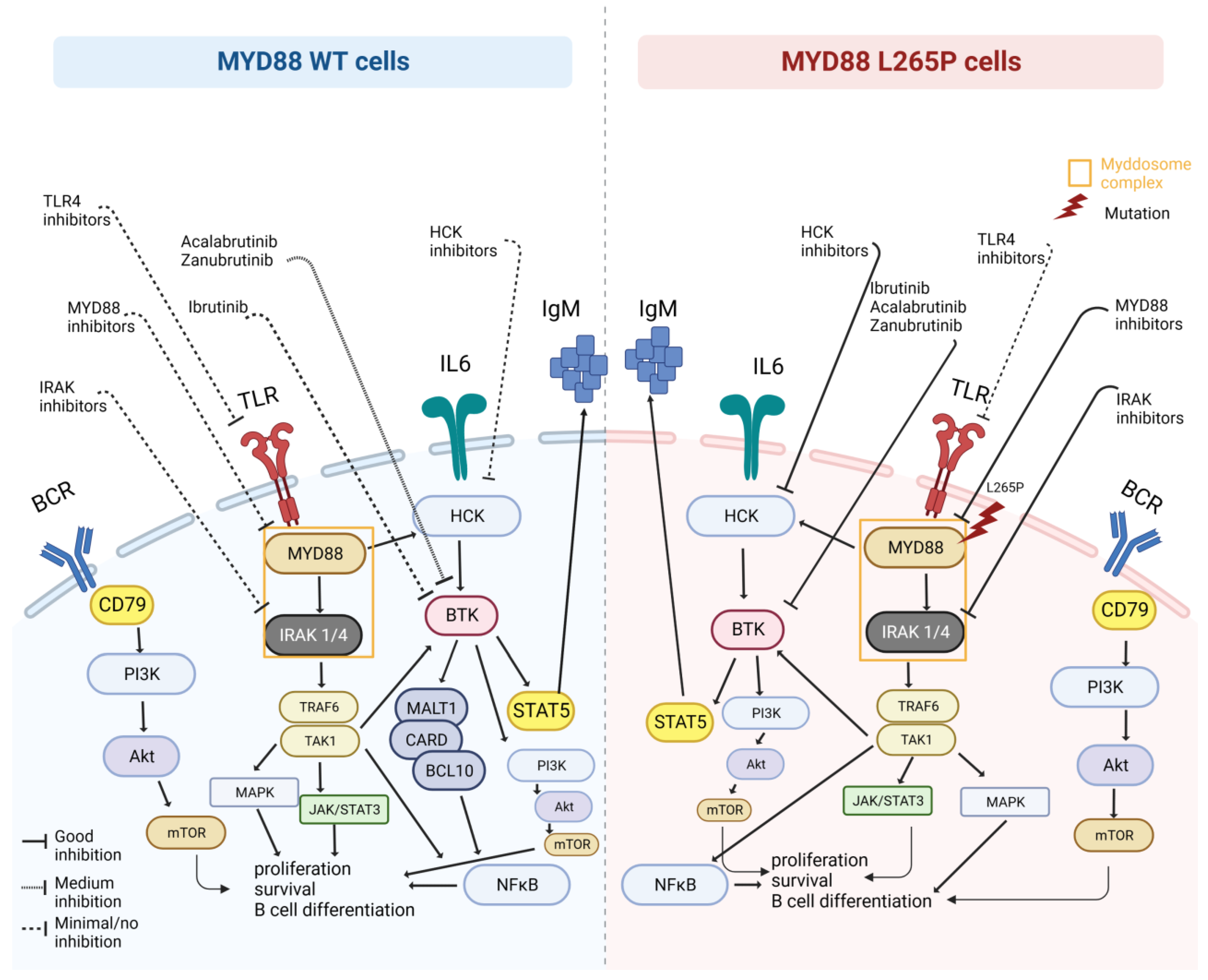
2. MYD88: Role, Pathway, Origin of Mutation
3. MYD88 Mutation Detection Assays
4. MYD88 Mutation Status in B-Cell Neoplasms
5. MYD88WT Genotype in IgM Monoclonal Gammopathies
6. MYD88WT and Related Genes
7. MYD88WT and Therapeutic Implications
8. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Guerrera, M.L.; Tsakmaklis, N.; Xu, L.; Yang, G.; Demos, M.; Kofides, A.; Chan, G.G.; Manning, R.J.; Liu, X.; Chen, J.G.; et al. MYD88 mutated and wild-type Waldenström’s Macroglobulinemia: Characterization of chromosome 6q gene losses and their mutual exclusivity with mutations in CXCR4. Haematologica 2018, 103, e408–e411. [Google Scholar] [CrossRef] [PubMed]
- García-Sanz, R.; Dogliotti, I.; Zaccaria, G.M.; Ocio, E.M.; Rubio, A.; Murillo, I.; Escalante, F.; Aguilera, C.; García-Mateo, A.; García de Coca, A.; et al. 6q deletion in Waldenström macroglobulinaemia negatively affects time to transformation and survival. Br. J. Haematol. 2021, 192, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Khac, F.; Lambert, J.; Chapiro, E.; Grelier, A.; Mould, S.; Barin, C.; Daudignon, A.; Gachard, N.; Struski, S.; Henry, C.; et al. Chromosomal aberrations and their prognostic value in a series of 174 untreated patients with Waldenström's macroglobulinemia. Haematologica 2013, 98, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Gentles, A.J.; Nair, R.V.; Irish, J.M.; Kihira, S.; Liu, C.L.; Kela, I.; Hopmans, E.S.; Myklebust, J.H.; Ji, H.; et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood 2013, 121, 1604–1611. [Google Scholar] [CrossRef]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [CrossRef]
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833. [Google Scholar] [CrossRef]
- Hamadeh, F.; MacNamara, S.P.; Aguilera, N.S.; Swerdlow, S.H.; Cook, J.R. MYD88 L265P mutation analysis helps define nodal lymphoplasmacytic lymphoma. Mod. Pathol. 2015, 28, 564–574. [Google Scholar] [CrossRef]
- Ondrejka, S.L.; Lin, J.J.; Warden, D.W.; Durkin, L.; Cook, J.R.; Hsi, E.D. MYD88 L265P somatic mutation: Its usefulness in the differential diagnosis of bone marrow involvement by B-cell lymphoproliferative disorders. Am. J. Clin. Pathol. 2013, 140, 387–394. [Google Scholar] [CrossRef]
- Patkar, N.; Subramanian, P.G.; Deshpande, P.; Ghodke, K.; Tembhare, P.; Mascarenhas, R.; Muranjan, A.; Chaudhary, S.; Bagal, B.; Gujral, S.; et al. MYD88 mutant lymphoplasmacytic lymphoma/Waldenström macroglobulinemia has distinct clinical and pathological features as compared to its mutation negative counterpart. Leuk. Lymphoma 2015, 56, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [PubMed]
- García-Abellás, P.; Ferrer Gómez, A.; Bueno Sacristán, D.; Piris Villaespesa, M.; Talavera Yagüe, M.; Reguero Callejas, M.E.; García-Cosío, M. Lymphoplasmacytic lymphoma and marginal zone lymphoma involving bone marrow: A diagnostic dilemma. Useful clinicopathological features to accurate the diagnosis. EJHaem 2022, 3, 1181–1187. [Google Scholar] [CrossRef]
- Treon, S.P.; Xu, L.; Hunter, Z. MYD88 Mutations and Response to Ibrutinib in Waldenström's Macroglobulinemia. N. Engl. J. Med. 2015, 373, 584–586. [Google Scholar] [CrossRef]
- Toshchakov, V.; Jones, B.W.; Perera, P.Y.; Thomas, K.; Cody, M.J.; Zhang, S.; Williams, B.R.; Major, J.; Hamilton, T.A.; Fenton, M.J.; et al. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat. Immunol. 2002, 3, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.; Janssens, S.; Brissoni, B.; Olivos, N.; Beyaert, R.; Tschopp, J. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J. Exp. Med. 2003, 197, 263–268. [Google Scholar] [CrossRef]
- Wesche, H.; Gao, X.; Li, X.; Kirschning, C.J.; Stark, G.R.; Cao, Z. IRAK-M is a novel member of the Pelle/interleukin-1 receptor-associated kinase (IRAK) family. J. Biol. Chem. 1999, 274, 19403–19410. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Kopp, E.; Stadlen, A.; Chen, C.; Ghosh, S.; Janeway, C.A., Jr. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell 1998, 2, 253–258. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Dubois, S.; Viailly, P.J.; Bohers, E.; Bertrand, P.; Ruminy, P.; Marchand, V.; Maingonnat, C.; Mareschal, S.; Picquenot, J.M.; Penther, D.; et al. Biological and Clinical Relevance of Associated Genomic Alterations in MYD88 L265P and non-L265P-Mutated Diffuse Large B-Cell Lymphoma: Analysis of 361 Cases. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2232–2244. [Google Scholar] [CrossRef]
- Zhan, C.; Qi, R.; Wei, G.; Guven-Maiorov, E.; Nussinov, R.; Ma, B. Conformational dynamics of cancer-associated MyD88-TIR domain mutant L252P (L265P) allosterically tilts the landscape toward homo-dimerization. Protein Eng. Des. Sel. PEDS 2016, 29, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Vyncke, L.; Bovijn, C.; Pauwels, E.; Van Acker, T.; Ruyssinck, E.; Burg, E.; Tavernier, J.; Peelman, F. Reconstructing the TIR Side of the Myddosome: A Paradigm for TIR-TIR Interactions. Structure 2016, 24, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, W.; Deng, Q.; Li, L.; Hsi, E.D.; Young, K.H.; Zhang, M.; Li, Y. MYD88 L265P Mutation in Lymphoid Malignancies. Cancer Res. 2018, 78, 2457–2462. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Hodge, L.S.; Secreto, F.J.; Manske, M.; Braggio, E.; Price-Troska, T.; Ziesmer, S.; Li, Y.; Johnson, S.H.; Hart, S.N.; et al. Activation of TAK1 by MYD88 L265P drives malignant B-cell Growth in non-Hodgkin lymphoma. Blood Cancer J. 2014, 4, e183. [Google Scholar] [CrossRef]
- Rousseau, S.; Martel, G. Gain-of-Function Mutations in the Toll-Like Receptor Pathway: TPL2-Mediated ERK1/ERK2 MAPK Activation, a Path to Tumorigenesis in Lymphoid Neoplasms? Front. Cell Dev. Biol. 2016, 4, 50. [Google Scholar] [CrossRef]
- Wang, J.Q.; Jeelall, Y.S.; Beutler, B.; Horikawa, K.; Goodnow, C.C. Consequences of the recurrent MYD88(L265P) somatic mutation for B cell tolerance. J. Exp. Med. 2014, 211, 413–426. [Google Scholar] [CrossRef]
- Hunter, Z.R.; Yang, G.; Xu, L.; Liu, X.; Castillo, J.J.; Treon, S.P. Genomics, Signaling, and Treatment of Waldenström Macroglobulinemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 994–1001. [Google Scholar] [CrossRef]
- Varettoni, M.; Zibellini, S.; Defrancesco, I.; Ferretti, V.V.; Rizzo, E.; Malcovati, L.; Gallì, A.; Porta, M.G.D.; Boveri, E.; Arcaini, L.; et al. Pattern of somatic mutations in patients with Waldenström macroglobulinemia or IgM monoclonal gammopathy of undetermined significance. Haematologica 2017, 102, 2077–2085. [Google Scholar] [CrossRef]
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.J.; Williams, P.M.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 2015, 21, 922–926. [Google Scholar] [CrossRef]
- de Groen, R.A.L.; Schrader, A.M.R.; Kersten, M.J.; Pals, S.T.; Vermaat, J.S.P. MYD88 in the driver’s seat of B-cell lymphomagenesis: From molecular mechanisms to clinical implications. Haematologica 2019, 104, 2337–2348. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, Y.; Liu, X.; Xu, L.; Cao, Y.; Manning, R.J.; Patterson, C.J.; Buhrlage, S.J.; Gray, N.; Tai, Y.T.; et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood 2013, 122, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Poulain, S.; Roumier, C.; Decambron, A.; Renneville, A.; Herbaux, C.; Bertrand, E.; Tricot, S.; Daudignon, A.; Galiègue-Zouitina, S.; Soenen, V.; et al. MYD88 L265P mutation in Waldenstrom macroglobulinemia. Blood 2013, 121, 4504–4511. [Google Scholar] [CrossRef]
- Varettoni, M.; Arcaini, L.; Zibellini, S.; Boveri, E.; Rattotti, S.; Riboni, R.; Corso, A.; Orlandi, E.; Bonfichi, M.; Gotti, M.; et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenstrom’s macroglobulinemia and related lymphoid neoplasms. Blood 2013, 121, 2522–2528. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hunter, Z.R.; Yang, G.; Zhou, Y.; Cao, Y.; Liu, X.; Morra, E.; Trojani, A.; Greco, A.; Arcaini, L.; et al. MYD88 L265P in Waldenström macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood 2013, 121, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, C.; Sebastián, E.; Chillón, M.C.; Giraldo, P.; Mariano Hernández, J.; Escalante, F.; González-López, T.J.; Aguilera, C.; de Coca, A.G.; Murillo, I.; et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenström’s macroglobulinemia. Leukemia 2013, 27, 1722–1728. [Google Scholar] [CrossRef]
- Drandi, D.; Genuardi, E.; Dogliotti, I.; Ferrante, M.; Jiménez, C.; Guerrini, F.; Schirico, M.L.; Mantoan, B.; Muccio, V.; Lia, G.; et al. Highly sensitive MYD88(L265P) mutation detection by droplet digital polymerase chain reaction in Waldenström macroglobulinemia. Haematologica 2018, 103, 1029–1037. [Google Scholar] [CrossRef]
- Bagratuni, T.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Mavrianou-Koutsoukou, N.; Liacos, C.; Patseas, D.; Kanellias, N.; Migkou, M.; Ziogas, D.C.; Eleutherakis-Papaiakovou, E.; et al. Detection of MYD88 and CXCR4 mutations in cell-free DNA of patients with IgM monoclonal gammopathies. Leukemia 2018, 32, 2617–2625. [Google Scholar] [CrossRef]
- Bagratuni, T.; Markou, A.; Patseas, D.; Mavrianou-Koutsoukou, N.; Aktypi, F.; Liacos, C.I.; Sklirou, A.D.; Theodorakakou, F.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; et al. Determination of MYD88L265P mutation fraction in IgM monoclonal gammopathies. Blood Adv. 2022, 6, 189–199. [Google Scholar] [CrossRef]
- Wang, C.Z.; Lin, J.; Qian, J.; Shao, R.; Xue, D.; Qian, W.; Xiao, G.F.; Deng, Z.Q.; Yang, J.; Li, Y.; et al. Development of high-resolution melting analysis for the detection of the MYD88 L265P mutation. Clin. Biochem. 2013, 46, 385–387. [Google Scholar] [CrossRef]
- Dogliotti, I.; Jiménez, C.; Varettoni, M.; Talaulikar, D.; Bagratuni, T.; Ferrante, M.; Pérez, J.; Drandi, D.; Puig, N.; Gilestro, M.; et al. Diagnostics in Waldenström’s macroglobulinemia: A consensus statement of the European Consortium for Waldenström’s Macroglobulinemia. Leukemia 2023, 37, 388–395. [Google Scholar] [CrossRef]
- Treon, S.P.; Xu, L.; Guerrera, M.L.; Jimenez, C.; Hunter, Z.R.; Liu, X.; Demos, M.; Gustine, J.; Chan, G.; Munshi, M.; et al. Genomic Landscape of Waldenström Macroglobulinemia and Its Impact on Treatment Strategies. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Hunter, Z.R.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Manning, R.J.; Tripsas, C.; Patterson, C.J.; Sheehy, P.; et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood 2014, 123, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Mithraprabhu, S.; Spencer, A. Circulating tumour DNA analysis in multiple myeloma. Oncotarget 2017, 8, 90610–90611. [Google Scholar] [CrossRef] [PubMed]
- Oberle, A.; Brandt, A.; Voigtlaender, M.; Thiele, B.; Radloff, J.; Schulenkorf, A.; Alawi, M.; Akyüz, N.; März, M.; Ford, C.T.; et al. Monitoring multiple myeloma by next-generation sequencing of V(D)J rearrangements from circulating myeloma cells and cell-free myeloma DNA. Haematologica 2017, 102, 1105–1111. [Google Scholar] [CrossRef]
- Rustad, E.H.; Coward, E.; Skytøen, E.R.; Misund, K.; Holien, T.; Standal, T.; Børset, M.; Beisvag, V.; Myklebost, O.; Meza-Zepeda, L.A.; et al. Monitoring multiple myeloma by quantification of recurrent mutations in serum. Haematologica 2017, 102, 1266–1272. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Cao, X.; Medeiros, L.J.; Xia, Y.; Wang, X.; Thomas, S.K.; Loghavi, S.; Li, X.; Shah, J.J.; Gustafson, S.A.; Weber, D.M.; et al. Clinicopathologic features and outcomes of lymphoplasmacytic lymphoma patients with monoclonal IgG or IgA paraprotein expression. Leuk. Lymphoma 2016, 57, 1104–1113. [Google Scholar] [CrossRef]
- Campo, E.; Swerdlow, S.H.; Harris, N.L.; Pileri, S.; Stein, H.; Jaffe, E.S. The 2008 WHO classification of lymphoid neoplasms and beyond: Evolving concepts and practical applications. Blood 2011, 117, 5019–5032. [Google Scholar] [CrossRef]
- Owen, R.G.; Treon, S.P.; Al-Katib, A.; Fonseca, R.; Greipp, P.R.; McMaster, M.L.; Morra, E.; Pangalis, G.A.; San Miguel, J.F.; Branagan, A.R.; et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: Consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin. Oncol. 2003, 30, 110–115. [Google Scholar] [CrossRef]
- Kyle, R.A.; Durie, B.G.; Rajkumar, S.V.; Landgren, O.; Blade, J.; Merlini, G.; Kröger, N.; Einsele, H.; Vesole, D.H.; Dimopoulos, M.; et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010, 24, 1121–1127. [Google Scholar] [CrossRef]
- Landgren, O.; Staudt, L. MYD88 L265P somatic mutation in IgM MGUS. N. Engl. J. Med. 2012, 367, 2255–2256, author reply 2256–2257. [Google Scholar] [CrossRef]
- Varettoni, M.; Zibellini, S.; Arcaini, L.; Boveri, E.; Rattotti, S.; Pascutto, C.; Mangiacavalli, S.; Gotti, M.; Pochintesta, L.; Paulli, M.; et al. MYD88 (L265P) mutation is an independent risk factor for progression in patients with IgM monoclonal gammopathy of undetermined significance. Blood 2013, 122, 2284–2285. [Google Scholar] [CrossRef] [PubMed]
- Varettoni, M.; Zibellini, S.; Boveri, E.; Klersy, C.; Candido, C.; Rattotti, S.; Ferretti, V.V.; Defrancesco, I.; Mangiacavalli, S.; Nizzoli, M.E.; et al. A risk-stratification model based on the initial concentration of the serum monoclonal protein and MYD88 mutation status identifies a subset of patients with IgM monoclonal gammopathy of undetermined significance at high risk of progression to Waldenström macroglobulinaemia or other lymphoproliferative disorders. Br. J. Haematol. 2019, 187, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Reid, R.; Friedberg, J.W. Management of marginal zone lymphoma. Oncology 2013, 27, 840,842,844. [Google Scholar] [PubMed]
- Insuasti-Beltran, G.; Gale, J.M.; Wilson, C.S.; Foucar, K.; Czuchlewski, D.R. Significance of MYD88 L265P Mutation Status in the Subclassification of Low-Grade B-Cell Lymphoma/Leukemia. Arch. Pathol. Lab. Med. 2015, 139, 1035–1041. [Google Scholar] [CrossRef]
- Gebauer, N.; Hardel, T.T.; Gebauer, J.; Bernard, V.; Merz, H.; Feller, A.C.; Rades, D.; Biersack, H.; Lehnert, H.; Thorns, C. Activating mutations affecting the NF-kappa B pathway and EZH2-mediated epigenetic regulation are rare events in primary mediastinal large B-cell lymphoma. Anticancer Res. 2014, 34, 5503–5507. [Google Scholar]
- Lee, J.H.; Jeong, H.; Choi, J.W.; Oh, H.; Kim, Y.S. Clinicopathologic significance of MYD88 L265P mutation in diffuse large B-cell lymphoma: A meta-analysis. Sci. Rep. 2017, 7, 1785. [Google Scholar] [CrossRef]
- Jallades, L.; Baseggio, L.; Sujobert, P.; Huet, S.; Chabane, K.; Callet-Bauchu, E.; Verney, A.; Hayette, S.; Desvignes, J.P.; Salgado, D.; et al. Exome sequencing identifies recurrent BCOR alterations and the absence of KLF2, TNFAIP3 and MYD88 mutations in splenic diffuse red pulp small B-cell lymphoma. Haematologica 2017, 102, 1758–1766. [Google Scholar] [CrossRef]
- Staiger, A.M.; Ott, M.M.; Parmentier, S.; Rosenwald, A.; Ott, G.; Horn, H.; Griese, E.U. Allele-specific PCR is a powerful tool for the detection of the MYD88 L265P mutation in diffuse large B cell lymphoma and decalcified bone marrow samples. Br. J. Haematol. 2015, 171, 145–148. [Google Scholar] [CrossRef]
- Onaindia, A.; Medeiros, L.J.; Patel, K.P. Clinical utility of recently identified diagnostic, prognostic, and predictive molecular biomarkers in mature B-cell neoplasms. Mod. Pathol. 2017, 30, 1338–1366. [Google Scholar] [CrossRef]
- Hung, S.S.; Meissner, B.; Chavez, E.A.; Ben-Neriah, S.; Ennishi, D.; Jones, M.R.; Shulha, H.P.; Chan, F.C.; Boyle, M.; Kridel, R.; et al. Assessment of Capture and Amplicon-Based Approaches for the Development of a Targeted Next-Generation Sequencing Pipeline to Personalize Lymphoma Management. J. Mol. Diagn. JMD 2018, 20, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Okosun, J.; Bödör, C.; Wang, J.; Araf, S.; Yang, C.Y.; Pan, C.; Boller, S.; Cittaro, D.; Bozek, M.; Iqbal, S.; et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet. 2014, 46, 176–181. [Google Scholar] [CrossRef]
- Baer, C.; Dicker, F.; Kern, W.; Haferlach, T.; Haferlach, C. Genetic characterization of MYD88-mutated lymphoplasmacytic lymphoma in comparison with MYD88-mutated chronic lymphocytic leukemia. Leukemia 2017, 31, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.J.; Lin, C.T.; Agathangelidis, A.; Lin, L.I.; Kuo, Y.Y.; Tien, H.F.; Ghia, P. Distinct molecular genetics of chronic lymphocytic leukemia in Taiwan: Clinical and pathogenetic implications. Haematologica 2017, 102, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.B.; Foad, R.M.; Abdel-Wahed, E. Lack of Associations between TLR9 and MYD88 Gene Polymorphisms and Risk of Chronic Lymphocytic Leukemia. Asian Pac. J. Cancer Prev. APJCP 2017, 18, 3245–3250. [Google Scholar] [CrossRef]
- Treon, S.P.; Cao, Y.; Xu, L.; Yang, G.; Liu, X.; Hunter, Z.R. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood 2014, 123, 2791–2796. [Google Scholar] [CrossRef]
- Chakraborty, R.; Novak, A.J.; Ansell, S.M.; Muchtar, E.; Kapoor, P.; Hayman, S.R.; Dispenzieri, A.; Buadi, F.K.; Lacy, M.Q.; King, R.L.; et al. First report of MYD88 L265P somatic mutation in IgM-associated light-chain amyloidosis. Blood 2016, 127, 2936–2938. [Google Scholar] [CrossRef]
- Abeykoon, J.P.; Paludo, J.; King, R.L.; Ansell, S.M.; Gertz, M.A.; LaPlant, B.R.; Halvorson, A.E.; Gonsalves, W.I.; Dingli, D.; Fang, H.; et al. MYD88 mutation status does not impact overall survival in Waldenström macroglobulinemia. Am. J. Hematol. 2018, 93, 187–194. [Google Scholar] [CrossRef]
- Treon, S.P.; Gustine, J.; Xu, L.; Manning, R.J.; Tsakmaklis, N.; Demos, M.; Meid, K.; Guerrera, M.L.; Munshi, M.; Chan, G.; et al. MYD88 wild-type Waldenstrom Macroglobulinaemia: Differential diagnosis, risk of histological transformation, and overall survival. Br. J. Haematol. 2018, 180, 374–380. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Garand, R.; Lodé, L.; Harousseau, J.L.; Bataille, R. Translocation t(11;14)(q13;q32) is the hallmark of IgM, IgE, and nonsecretory multiple myeloma variants. Blood 2003, 101, 1570–1571. [Google Scholar] [CrossRef]
- Qin, S.C.; Xia, Y.; Miao, Y.; Zhu, H.Y.; Wu, J.Z.; Fan, L.; Li, J.Y.; Xu, W.; Qiao, C. MYD88 mutations predict unfavorable prognosis in Chronic Lymphocytic Leukemia patients with mutated IGHV gene. Blood Cancer J. 2017, 7, 651. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Parry, M.; Rose-Zerilli, M.J.; Ljungström, V.; Gibson, J.; Wang, J.; Walewska, R.; Parker, H.; Parker, A.; Davis, Z.; Gardiner, A.; et al. Genetics and Prognostication in Splenic Marginal Zone Lymphoma: Revelations from Deep Sequencing. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 4174–4183. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A. Waldenström macroglobulinemia: 2019 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2019, 94, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Braggio, E. The MYDas touch of next-gen sequencing. Blood 2013, 121, 2373–2374. [Google Scholar] [CrossRef]
- Hunter, Z.R.; Xu, L.; Tsakmaklis, N.; Demos, M.G.; Kofides, A.; Jimenez, C.; Chan, G.G.; Chen, J.; Liu, X.; Munshi, M.; et al. Insights into the genomic landscape of MYD88 wild-type Waldenström macroglobulinemia. Blood Adv. 2018, 2, 2937–2946. [Google Scholar] [CrossRef]
- Hunter, Z.R.; Xu, L.; Yang, G.; Tsakmaklis, N.; Vos, J.M.; Liu, X.; Chen, J.; Manning, R.J.; Chen, J.G.; Brodsky, P.; et al. Transcriptome sequencing reveals a profile that corresponds to genomic variants in Waldenström macroglobulinemia. Blood 2016, 128, 827–838. [Google Scholar] [CrossRef]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beà, S.; González-Díaz, M.; et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef]
- Zanwar, S.; Abeykoon, J.P.; Durot, E.; King, R.; Perez Burbano, G.E.; Kumar, S.; Gertz, M.A.; Quinquenel, A.; Delmer, A.; Gonsalves, W.; et al. Impact of MYD88(L265P) mutation status on histological transformation of Waldenström Macroglobulinemia. Am. J. Hematol. 2020, 95, 274–281. [Google Scholar] [CrossRef]
- Wang, Y.; Gali, V.L.; Xu-Monette, Z.Y.; Sano, D.; Thomas, S.K.; Weber, D.M.; Zhu, F.; Fang, X.; Deng, M.; Zhang, M.; et al. Molecular and genetic biomarkers implemented from next-generation sequencing provide treatment insights in clinical practice for Waldenström macroglobulinemia. Neoplasia 2021, 23, 361–374. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Aguilar, A.; Idbaih, A.; Boisselier, B.; Habbita, N.; Rossetto, M.; Laurenge, A.; Bruno, A.; Jouvet, A.; Polivka, M.; Adam, C.; et al. Recurrent mutations of MYD88 and TBL1XR1 in primary central nervous system lymphomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 5203–5211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Chang, Q.; Zeng, L.; Gu, J.; Brown, S.; Basch, R.S. TBLR1 regulates the expression of nuclear hormone receptor co-repressors. BMC Cell Biol. 2006, 7, 31. [Google Scholar] [CrossRef]
- Parker, H.; An, Q.; Barber, K.; Case, M.; Davies, T.; Konn, Z.; Stewart, A.; Wright, S.; Griffiths, M.; Ross, F.M.; et al. The complex genomic profile of ETV6-RUNX1 positive acute lymphoblastic leukemia highlights a recurrent deletion of TBL1XR1. Genes Chromosomes Cancer 2008, 47, 1118–1125. [Google Scholar] [CrossRef]
- Zhang, J.; Mullighan, C.G.; Harvey, R.C.; Wu, G.; Chen, X.; Edmonson, M.; Buetow, K.H.; Carroll, W.L.; Chen, I.M.; Devidas, M.; et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Blood 2011, 118, 3080–3087. [Google Scholar] [CrossRef]
- Balaji, S.; Ahmed, M.; Lorence, E.; Yan, F.; Nomie, K.; Wang, M. NF-κB signaling and its relevance to the treatment of mantle cell lymphoma. J. Hematol. Oncol. 2018, 11, 83. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef]
- van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef]
- Carrassa, L.; Colombo, I.; Damia, G.; Bertoni, F. Targeting the DNA damage response for patients with lymphoma: Preclinical and clinical evidences. Cancer Treat. Rev. 2020, 90, 102090. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, E.; Agostinelli, C.; Imbrogno, E.; Iacobucci, I.; Casadei, B.; Brighenti, E.; Righi, S.; Fuligni, F.; Ghelli Luserna Di Rorà, A.; Ferrari, A.; et al. Constitutive activation of the DNA damage response pathway as a novel therapeutic target in diffuse large B-cell lymphoma. Oncotarget 2015, 6, 6553–6569. [Google Scholar] [CrossRef] [PubMed]
- de Miranda, N.F.; Peng, R.; Georgiou, K.; Wu, C.; Falk Sörqvist, E.; Berglund, M.; Chen, L.; Gao, Z.; Lagerstedt, K.; Lisboa, S.; et al. DNA repair genes are selectively mutated in diffuse large B cell lymphomas. J. Exp. Med. 2013, 210, 1729–1742. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [Google Scholar] [CrossRef]
- Enoch, T.; Norbury, C. Cellular responses to DNA damage: Cell-cycle checkpoints, apoptosis and the roles of p53 and ATM. Trends Biochem. Sci. 1995, 20, 426–430. [Google Scholar] [CrossRef]
- Fan, S.; el-Deiry, W.S.; Bae, I.; Freeman, J.; Jondle, D.; Bhatia, K.; Fornace, A.J., Jr.; Magrath, I.; Kohn, K.W.; O’Connor, P.M. p53 gene mutations are associated with decreased sensitivity of human lymphoma cells to DNA damaging agents. Cancer Res. 1994, 54, 5824–5830. [Google Scholar] [PubMed]
- Yarde, D.N.; Oliveira, V.; Mathews, L.; Wang, X.; Villagra, A.; Boulware, D.; Shain, K.H.; Hazlehurst, L.A.; Alsina, M.; Chen, D.T.; et al. Targeting the Fanconi anemia/BRCA pathway circumvents drug resistance in multiple myeloma. Cancer Res. 2009, 69, 9367–9375. [Google Scholar] [CrossRef]
- Li, N.; Lopez, M.A.; Linares, M.; Kumar, S.; Oliva, S.; Martinez-Lopez, J.; Xu, L.; Xu, Y.; Perini, T.; Senapedis, W.; et al. Dual PAK4-NAMPT Inhibition Impacts Growth and Survival, and Increases Sensitivity to DNA-Damaging Agents in Waldenström Macroglobulinemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 369–377. [Google Scholar] [CrossRef]
- Krzisch, D.; Guedes, N.; Boccon-Gibod, C.; Baron, M.; Bravetti, C.; Davi, F.; Armand, M.; Smagghe, L.; Caron, J.; Bernard, O.A.; et al. Cytogenetic and molecular abnormalities in Waldenström’s macroglobulinemia patients: Correlations and prognostic impact. Am. J. Hematol. 2021, 96, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Poulain, S.; Roumier, C.; Bertrand, E.; Renneville, A.; Caillault-Venet, A.; Doye, E.; Geffroy, S.; Sebda, S.; Nibourel, O.; Nudel, M.; et al. TP53 Mutation and Its Prognostic Significance in Waldenstrom’s Macroglobulinemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6325–6335. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Opat, S.; D’Sa, S.; Jurczak, W.; Lee, H.P.; Cull, G.; Owen, R.G.; Marlton, P.; Wahlin, B.E.; Sanz, R.G.; et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: The ASPEN study. Blood 2020, 136, 2038–2050. [Google Scholar] [CrossRef] [PubMed]
- Gustine, J.N.; Tsakmaklis, N.; Demos, M.G.; Kofides, A.; Chen, J.G.; Liu, X.; Munshi, M.; Guerrera, M.L.; Chan, G.G.; Patterson, C.J.; et al. TP53 mutations are associated with mutated MYD88 and CXCR4, and confer an adverse outcome in Waldenström macroglobulinaemia. Br. J. Haematol. 2019, 184, 242–245. [Google Scholar] [CrossRef]
- Xu, L.; Hunter, Z.R.; Tsakmaklis, N.; Cao, Y.; Yang, G.; Chen, J.; Liu, X.; Kanan, S.; Castillo, J.J.; Tai, Y.T.; et al. Clonal architecture of CXCR4 WHIM-like mutations in Waldenström Macroglobulinaemia. Br. J. Haematol. 2016, 172, 735–744. [Google Scholar] [CrossRef]
- Kaiser, L.M.; Hunter, Z.R.; Treon, S.P.; Buske, C. CXCR4 in Waldenström’s Macroglobulinema: Chances and challenges. Leukemia 2021, 35, 333–345. [Google Scholar] [CrossRef]
- Ansell, S.M.; Kyle, R.A.; Reeder, C.B.; Fonseca, R.; Mikhael, J.R.; Morice, W.G.; Bergsagel, P.L.; Buadi, F.K.; Colgan, J.P.; Dingli, D.; et al. Diagnosis and management of Waldenström macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines. Mayo Clin. Proc. 2010, 85, 824–833. [Google Scholar] [CrossRef]
- Treon, S.P.; Ioakimidis, L.; Soumerai, J.D.; Patterson, C.J.; Sheehy, P.; Nelson, M.; Willen, M.; Matous, J.; Mattern, J., 2nd; Diener, J.G.; et al. Primary therapy of Waldenström macroglobulinemia with bortezomib, dexamethasone, and rituximab: WMCTG clinical trial 05–180. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 3830–3835. [Google Scholar] [CrossRef]
- Paulus, A.; Ailawadhi, S.; Chanan-Khan, A. Novel therapeutic targets in Waldenstrom macroglobulinemia. Best Pract. Res. Clin. Haematol. 2016, 29, 216–228. [Google Scholar] [CrossRef]
- Grunenberg, A.; Buske, C. Monoclonal IgM Gammopathy and Waldenström’s Macroglobulinemia. Dtsch. Arztebl. Int. 2017, 114, 745–751. [Google Scholar] [CrossRef]
- Schuster, S.R.; Rajkumar, S.V.; Dispenzieri, A.; Morice, W.; Aspitia, A.M.; Ansell, S.; Kyle, R.; Mikhael, J. IgM multiple myeloma: Disease definition, prognosis, and differentiation from Waldenstrom’s macroglobulinemia. Am. J. Hematol. 2010, 85, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Kastritis, E.; Ghobrial, I.M. Waldenström’s macroglobulinemia: A clinical perspective in the era of novel therapeutics. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.J.; Treon, S.P. Toward personalized treatment in Waldenström macroglobulinemia. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Argyropoulos, K.V.; Vogel, R.; Ziegler, C.; Altan-Bonnet, G.; Velardi, E.; Calafiore, M.; Dogan, A.; Arcila, M.; Patel, M.; Knapp, K.; et al. Clonal B cells in Waldenström’s macroglobulinemia exhibit functional features of chronic active B-cell receptor signaling. Leukemia 2016, 30, 1116–1125. [Google Scholar] [CrossRef][Green Version]
- Treon, S.P.; Meid, K.; Gustine, J.; Yang, G.; Xu, L.; Liu, X.; Patterson, C.J.; Hunter, Z.R.; Branagan, A.R.; Laubach, J.P.; et al. Long-Term Follow-Up of Ibrutinib Monotherapy in Symptomatic, Previously Treated Patients With Waldenström Macroglobulinemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 565–575. [Google Scholar] [CrossRef]
- Ravi, G.; Kapoor, P. Current approach to Waldenström Macroglobulinemia. Cancer Treat. Res. Commun. 2022, 31, 100527. [Google Scholar] [CrossRef]
- Owen, R.G.; McCarthy, H.; Rule, S.; D’Sa, S.; Thomas, S.K.; Tournilhac, O.; Forconi, F.; Kersten, M.J.; Zinzani, P.L.; Iyengar, S.; et al. Acalabrutinib monotherapy in patients with Waldenström macroglobulinemia: A single-arm, multicentre, phase 2 study. Lancet Haematol. 2020, 7, e112–e121. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Sanz, R.G.; Lee, H.P.; Trneny, M.; Varettoni, M.; Opat, S.; D’Sa, S.; Owen, R.G.; Cull, G.; Mulligan, S.; et al. Zanubrutinib for the treatment of MYD88 wild-type Waldenström macroglobulinemia: A substudy of the phase 3 ASPEN trial. Blood Adv. 2020, 4, 6009–6018. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Salman, Z.; Buske, C. Ibrutinib and Rituximab in Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2018, 379, 1975–1976. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Trotman, J.; Tedeschi, A.; Matous, J.V.; Macdonald, D.; Tam, C.; Tournilhac, O.; Ma, S.; Oriol, A.; Heffner, L.T.; et al. Ibrutinib for patients with rituximab-refractory Waldenström’s macroglobulinaemia (iNNOVATE): An open-label substudy of an international, multicentre, phase 3 trial. Lancet Oncol. 2017, 18, 241–250. [Google Scholar] [CrossRef]
- Buske, C.; Jurczak, W.; Salem, J.E.; Dimopoulos, M.A. Managing Waldenström’s macroglobulinemia with BTK inhibitors. Leukemia 2023, 37, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Buske, C.; Tedeschi, A.; Trotman, J.; García-Sanz, R.; MacDonald, D.; Leblond, V.; Mahe, B.; Herbaux, C.; Matous, J.V.; Tam, C.S.; et al. Ibrutinib Plus Rituximab Versus Placebo Plus Rituximab for Waldenström’s Macroglobulinemia: Final Analysis From the Randomized Phase III iNNOVATE Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2022, 40, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Paludo, J.; Abeykoon, J.P.; Shreders, A.; Ansell, S.M.; Kumar, S.; Ailawadhi, S.; King, R.L.; Koehler, A.B.; Reeder, C.B.; Buadi, F.K.; et al. Bendamustine and rituximab (BR) versus dexamethasone, rituximab, and cyclophosphamide (DRC) in patients with Waldenström macroglobulinemia. Ann. Hematol. 2018, 97, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Laribi, K.; Poulain, S.; Willems, L.; Merabet, F.; Le Calloch, R.; Eveillard, J.R.; Herbaux, C.; Roos-Weil, D.; Chaoui, D.; Roussel, X.; et al. Bendamustine plus rituximab in newly-diagnosed Waldenström macroglobulinaemia patients. A study on behalf of the French Innovative Leukaemia Organization (FILO). Br. J. Haematol. 2019, 186, 146–149. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| MYD88WT (n) | TN (n) | ORR | MRR | ||
|---|---|---|---|---|---|
| Ibrutinib | 4 | 0 | 50% | 0 | Treon SP et al. J Clin Oncol 2020 [41] |
| Ibrutinib + Rituximab | 11 | 82% | 73% | Dimopoulos MA et al. N Engl J Med 2018 [119] | |
| Acalabrutinib | 14 | 1 | 79% | 64% | Owen R et al. Lancet Haematol 2020 [117] |
| Zanubrutinib | 26 | 5 | 81% | 50% | Dimopoulos MA et al. Blood Adv 2020 [118] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagratuni, T.; Papadimou, A.; Taouxi, K.; Dimopoulos, M.A.; Kastritis, E. MYD88 Wild Type in IgM Monoclonal Gammopathies: From Molecular Mechanisms to Clinical Challenges. Hemato 2023, 4, 259-272. https://doi.org/10.3390/hemato4030021
Bagratuni T, Papadimou A, Taouxi K, Dimopoulos MA, Kastritis E. MYD88 Wild Type in IgM Monoclonal Gammopathies: From Molecular Mechanisms to Clinical Challenges. Hemato. 2023; 4(3):259-272. https://doi.org/10.3390/hemato4030021
Chicago/Turabian StyleBagratuni, Tina, Alexandra Papadimou, Kostantina Taouxi, Meletios A. Dimopoulos, and Efstathios Kastritis. 2023. "MYD88 Wild Type in IgM Monoclonal Gammopathies: From Molecular Mechanisms to Clinical Challenges" Hemato 4, no. 3: 259-272. https://doi.org/10.3390/hemato4030021
APA StyleBagratuni, T., Papadimou, A., Taouxi, K., Dimopoulos, M. A., & Kastritis, E. (2023). MYD88 Wild Type in IgM Monoclonal Gammopathies: From Molecular Mechanisms to Clinical Challenges. Hemato, 4(3), 259-272. https://doi.org/10.3390/hemato4030021