
Renal Disorders Associated with Waldenström Macroglobulinaemia, IgM MGUS and IgM-Producing B-Cell Lymphoproliferative Disorders

Abstract
1. Introduction
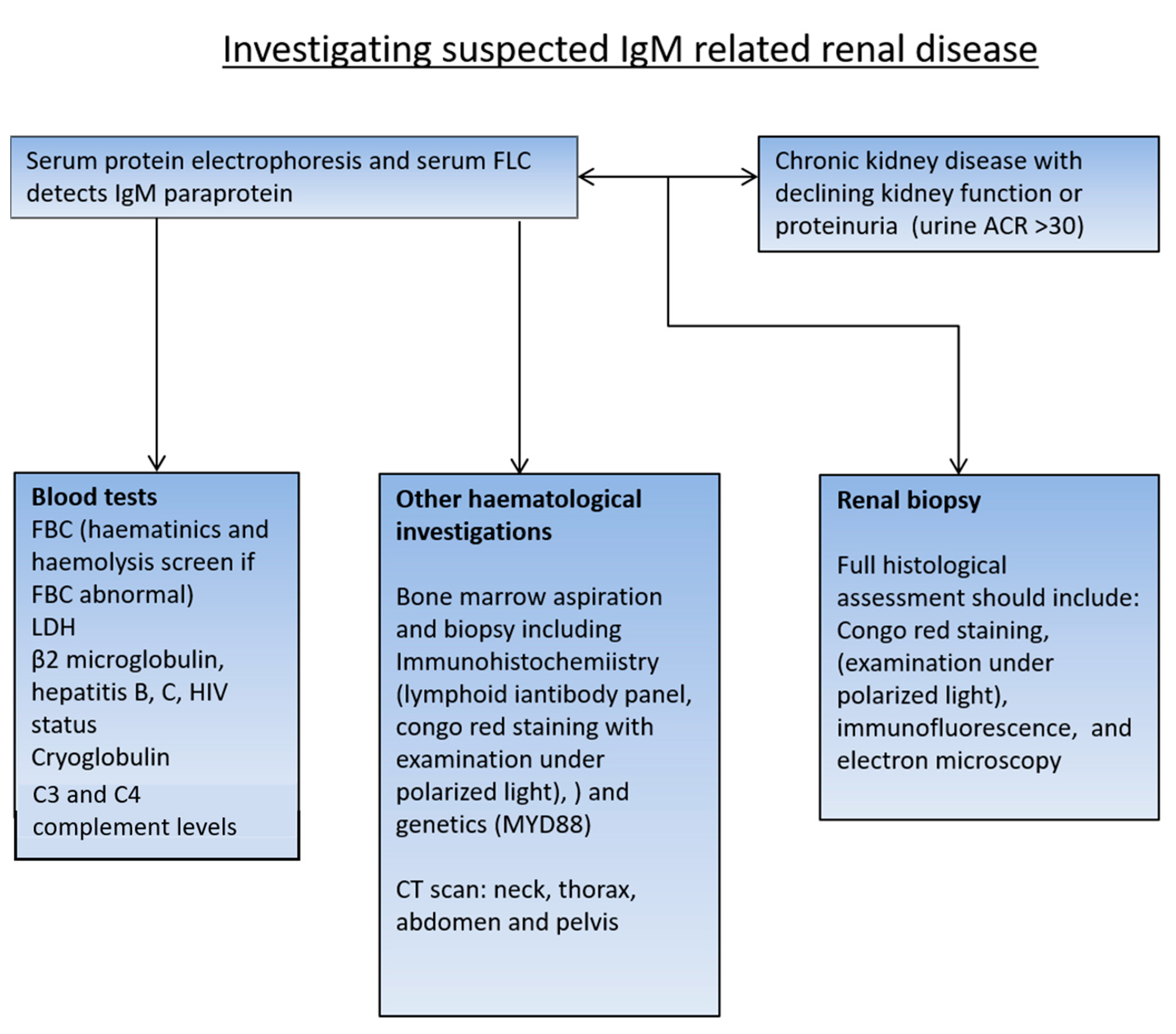
2. Investigating Renal Impairment in a Patient with WM or IgM MGUS
3. Patients with Symptomatic WM with a Significant Tumour Burden and Renal Impairment
4. Patients with Suspected Renal Amyloidosis
5. Patients with a Non-Amyloid form of MGRS and Serum Monoclonal IgM
6. Cryoglobulinaemic Glomerulopathy
7. Light-Chain Deposition Disease (LCDD)
8. Rarer MGRS
9. Other Renal Pathologies with Less Clear Relationships to WM and IgM MGUS
10. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Owen, R.G. Developing diagnostic criteria in Waldenstrom’s macroglobulinemia. Semin. Oncol. 2003, 30, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; World Health Organization; International Agency for Research on Cancer. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; World Health Organization Classification of Tumours; International Agency for Research on Cancer: Lyon, France, 2017. [Google Scholar]
- Higgins, L.; Nasr, S.H.; Said, S.M.; Kapoor, P.; Dingli, D.; King, R.L.; Rajkumar, S.V.; Kyle, R.A.; Kourelis, T.; Gertz, M.A.; et al. Kidney Involvement of Patients with Waldenström Macroglobulinemia and Other IgM-Producing B Cell Lymphoproliferative Disorders. Clin. J. Am. Soc. Nephrol. 2018, 13, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Vos, J.M.; Gustine, J.; Rennke, H.G.; Hunter, Z.; Manning, R.J.; Dubeau, T.E.; Meid, K.; Minnema, M.C.; Kersten, M.J.; Treon, S.P.; et al. Renal disease related to Waldenström macroglobulinaemia: Incidence, pathology and clinical outcomes. Br. J. Haematol. 2016, 175, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Chauvet, S.; Bridoux, F.; Ecotière, L.; Javaugue, V.; Sirac, C.; Arnulf, B.; Thierry, A.; Quellard, N.; Milin, S.; Bender, S.; et al. Kidney diseases associated with monoclonal immunoglobulin M-secreting B-cell lym-phoproliferative disorders: A case series of 35 patients. Am. J. Kidney Dis. 2015, 66, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Audard, V.; Georges, B.; Vanhille, P.; Toly, C.; Deroure, B.; Fakhouri, F.; Cuvelier, R.; Belenfant, X.; Surin, B.; Aucouturier, P.; et al. Renal lesions associated with IgM-secreting monoclonal proliferations: Revisiting the disease spectrum. Clin. J. Am. Soc. Nephrol. 2008, 3, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liang, D.; Liang, S.; Xu, F.; Huang, X.; Jiang, S.; Hou, J. Clinicopathologic characteristics and prognostic analysis of monoclonal gammopathy of renal significance (MGRS) in patients with IgM monoclonal gammopathy: A case series. Sci. Rep. 2022, 12, 16994. [Google Scholar] [CrossRef]
- Roberts, I.; Roufosse, C.; Sheaff, M. Tissue Pathway for Native Medical Renal Biopsies; Royal College of Pathologists: London, UK, 2019. [Google Scholar]
- Pratt, G.; El-Sharkawi, D.; Kothari, J.; D’Sa, S.; Auer, R.; McCarthy, H.; Krishna, R.; Miles, O.; Kyriakou, C.; Owen, R. Diagnosis and management of Waldenström macroglobulinaemia-A British Society for Haematology guideline. Br. J. Haematol. 2022, 197, 171–187. [Google Scholar] [CrossRef]
- Sidana, S.; Larson, D.P.; Greipp, P.T.; He, R.; McPhail, E.D.; Dispenzieri, A.; Murray, D.L.; Dasari, S.; Ansell, S.M.; Muchtar, E.; et al. IgM AL amyloidosis: Delineating disease biology and outcomes with clinical, genomic and bone marrow morphological features. Leukemia 2020, 34, 1373–1382. [Google Scholar] [CrossRef]
- Kumar, S.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Colby, C.; Laumann, K.; Zeldenrust, S.R.; Leung, N.; Dingli, D.; et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac bi-omarkers and serum free light chain measurements. J. Clin. Oncol. 2012, 30, 989–995. [Google Scholar] [CrossRef]
- Long, T.E.; Indridason, O.S.; Palsson, R.; Rognvaldsson, S.; Love, T.J.; Thorsteinsdottir, S.; Sverrisdottir, I.S.; Vidarsson, B.; Onundarson, P.T.; Agnarsson, B.A.; et al. Defining new reference intervals for serum free light chains in individuals with chronic kidney disease: Results of the iStopMM study. Blood Cancer J. 2022, 12, 133. [Google Scholar] [CrossRef]
- Hutchison, C.A.; Plant, T.; Drayson, M.; Cockwell, P.; Kountouri, M.; Basnayake, K.; Harding, S.; Bradwell, A.R.; Mead, G. Serum free light chain measurement aids the diagnosis of myeloma in patients with severe renal failure. BMC Nephrol. 2008, 9, 11–18. [Google Scholar] [CrossRef]
- Molina-Andújar, A.; Robles, P.; Cibeira, M.T.; Montagud-Marrahi, E.; Guillen, E.; Xipell, M.; Blasco, M.; Poch, E.; Rosiñol, L.; Bladé, J.; et al. The renal range of the κ/λ sFLC ratio: Best strategy to evaluate multiple myeloma in patients with chronic kidney disease. BMC Nephrol. 2020, 21, 111–117. [Google Scholar] [CrossRef]
- Kolopp-Sarda, M.N.; Nombel, A.; Miossec, P. Cryoglobulins Today: Detection and Immunologic Characteristics of 1675 Positive Samples From 13,439 Patients Obtained Over Six Years. Arthritis Rheumatol. 2019, 71, 1904–1912. [Google Scholar] [CrossRef]
- Javaugue, V.; Valeri, A.M.; Sathick, I.J.; Said, S.M.; Damgard, S.E.; Murray, D.L.; Klobucher, T.; Andeen, N.K.; Sethi, S.; Fervenza, F.C.; et al. The characteristics of seronegative and seropositive non-hepatitis-associated cryo-globulinemic glomerulonephritis. Kidney Int. 2022, 102, 382–394. [Google Scholar] [CrossRef]
- Zhang, L.L.; Cao, X.X.; Shen, K.N.; Han, H.X.; Zhang, C.L.; Qiu, Y.; Zhao, H.; Gao, X.M.; Feng, J.; Zhang, L.; et al. Clinical characteristics and treatment outcome of type I cryoglobulinemia in Chinese patients: A single-center study of 45 patients. Ann. Hematol. 2020, 99, 1735–1740. [Google Scholar] [CrossRef]
- Khwaja, J.; D’sa, S.; Minnema, M.C.; Kersten, M.J.; Wechalekar, A.; Vos, J.M. IgM monoclonal gammopathies of clinical significance: Diagnosis and management. Haematologica 2022, 107, 2037–2050. [Google Scholar] [CrossRef]
- Mills, J.R.; Barnidge, D.R.; Murray, D.L. Detecting monoclonal immunoglobulins in human serum using mass spectrometry. Methods 2015, 81, 56–65. [Google Scholar] [CrossRef]
- Wilhite, D.; Arfa, A.; Cotter, T.; Savage, N.M.; Bollag, R.J.; Singh, G. Multiple myeloma: Detection of free monoclonal light chains by modified immunofixation electrophoresis with antisera against free light chains. Pract. Lab. Med. 2021, 27, e00256. [Google Scholar] [CrossRef]
- Singh, G.; Bollag, R. Quantification by Ultrafiltration and Immunofixation Electrophoresis Testing for Monoclonal Serum Free Light Chains. Lab. Med. 2020, 51, 592–600. [Google Scholar] [CrossRef]
- Ravichandran, S.; Law, S.; Mahmood, S.; Wisniowski, B.; Foard, D.; Fontana, M.; Martinez-Naharro, A.; Whelan, C.; Hawkins, P.N.; Gillmore, J.D.; et al. Long-term outcomes in light chain deposition disease-analysis of a UK cohort. Am. J. Hematol. 2022, 97, E444–E446. [Google Scholar] [CrossRef]
- Da’as, N.; Polliack, A.; Cohen, Y.; Amir, G.; Darmon, D.; Kleinman, Y.; Goldfarb, A.W.; Ben-Yehuda, D. Kidney involvement and renal manifestations in non-Hodgkin’s lymphoma and lymphocytic leukemia: A retrospective study in 700 patients. Eur. J. Haematol. 2001, 67, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Kofman, T.; Zhang, S.Y.; Copie-Bergman, C.; Moktefi, A.; Raimbourg, Q.; Francois, H.; Karras, A.; Plaisier, E.; Painchart, B.; Favre, G.; et al. Minimal change nephrotic syndrome associated with non-Hodgkin lymphoid disorders: A retrospective study of 18 cases. Medicine 2014, 93, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Leeaphorn, N.; Kue-A-Pai, P.; Thamcharoen, N.; Ungprasert, P.; Stokes, M.B.; Knight, E.L. Prevalence of cancer in membranous nephropathy: A systematic review and me-ta-analysis of observational studies. Am. J. Nephrol. 2014, 40, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Lodhi, A.; Kumar, A.; Saqlain, M.U.; Suneja, M. Thrombotic microangiopathy associated with proteasome inhibitors. Clin. Kidney J. 2015, 8, 632–636. [Google Scholar] [CrossRef]
- Burstein, D.M.; Korbet, S.M.; Schwartz, M.M. Membranous Glomerulonephritis and Malignancy. Am. J. Kidney Dis. 1993, 22, 5–10. [Google Scholar] [CrossRef]
- Munyentwali, H.; Bouachi, K.; Audard, V.; Remy, P.; Lang, P.; Mojaat, R.; Deschênes, G.; Ronco, P.M.; Plaisier, E.M.; Dahan, K.Y. Rituximab is an efficient and safe treatment in adults with steroid-dependent minimal change disease. Kidney Int. 2013, 83, 511–516. [Google Scholar] [CrossRef]
- Sayed, R.H.; Gilbertson, J.A.; Hutt, D.F.; Lachmann, H.J.; Hawkins, P.N.; Bass, P.; Gillmore, J.D. Misdiagnosing renal amyloidosis as minimal change disease. Nephrol. Dial. Transplant. 2014, 29, 2120–2126. [Google Scholar] [CrossRef]
- Connor, T.M.; Aiello, V.; Griffith, M.; Cairns, T.; Roufosse, C.; Cook, H.T.; Pusey, C.D. The natural history of immunoglobulin M nephropathy in adults. Nephrol. Dial. Transplant. 2017, 32, 823–829. [Google Scholar] [CrossRef]


{kind=link}
| Disease/Renal Lesion | Clinical Symptoms | Renal Histology |
|---|---|---|
| Amyloidosis (AL, AH and AHL) | Renal: Proteinuria, low serum albumin, oedema and reduced eGFR Cardiac: Restrictive cardiomyopathy, shortness of breath, raised cardiac bio-markers Peripheral and/or autonomic neuropathy Easy bruising (peri orbital) and soft tissue infiltration (macroglossia, muscle stiffness and jaw claudication) | Organised fibrillar deposits LM and Congo red-positive deposits IF and IHC-positive deposits for kappa or lambda EM: randomly oriented fibrils (7–14 nm) |
| Cryoglobulinaemia/ Cryoglobulinaemic glomerulonephropathy | Renal: Isolated proteinuria or haematuria, and occasionally, nephrotic syndrome Cutaneous disease: Digital ischemia, Livedo reticularis and skin necrosis, erythematous macules and purpuric papules in lower limbs, and Reynaud’s phenomena Other manifestations: Peripheral neuropathy Hyperviscosity symptoms (blurred vision, headache, vertigo, nystagmus, confusion, stroke and coma) Arthralgia, fatigue and myalgia Pulmonary involvement: small airways disease, dyspnoea, cough and pleurisy Lymphadenopathy and splenomegaly Vasculitis of most internal organs possible | Organized microtubular or crystal deposits LM: membranoproliferative or endocapillary proliferative GN with monocyte infiltration and often immune thrombi IF: monotypic Ig granular deposits in glomeruli and vessels EM: microtubular extracellular electron-dense deposits and occasional intracellular crystals |
| Monoclonal immunoglobulin deposition disease (MIDD) LCDD, LHCDD and HCDD | Predominantly renal-limited (nephrotic syndrome and common hypertension) Cardiac involvement, cardiomyopathy and heart failure symptoms (rare) Liver involvement Peripheral nerve involvement possible | Non-organised deposits LM: nodular glomerulosclerosis (67% of cases) and thickened TBM IF: linear deposits along GBM, TBM and vessels EM: punctate electron-dense deposits along GBM, TBM and vessels |
| Immunotactoid glomerulonephritis | Renal isolated disease Proteinuria seen in 100% Haematuria is common Reduced eGFR in 50% of cases Low complements may be seen | Organised non-fibrillar deposits LM: atypical membranous, membranoproliferative, mesangial or endocapillary proliferative GN IF: monotypic IgG granular deposits in mesangium and GBM EM: glomerular microtubular deposits (14–60 nm) in parallel arrangement |
| Proliferative glomerulonephritis with monoclonal Ig deposits (PGNMID) | Renal isolated disease Nephrotic syndrome (50%) Haematuria (70%) Reduced eGFR (70%) | Non-organised deposits LM: membranoproliferative, endocapillary proliferative or membranous GN IF: monotypic Ig (mostly IgG3κ) granular deposits in mesangium and GBM EM: electron-dense deposits in glomeruli |
| Light-chain Proximal Tubulopathy | Renal isolated disease Fanconi syndrome, hypophosphataemia, renal glucosuria and/or aminoaciduria Muscle weakness, increased urine output and thirst | Organized crystals or inclusions LM: proximal tubular swelling IF: proximal tubular staining with κ (in crystalline variant) or λ (mostly in non-crystalline variant) EM: proximal tubular LC crystals or lysosomal inclusions |
| Fibrillary GN with monoclonal gammopathy | Renal isolated disease Proteinuria is common, and nephrotic syndrome is reported in 70% Haematuria is common Reduced eGFR in 50% of cases Low complements may be seen | Organised fibrillar deposits LM: MPGN, diffuse proliferative, mesangial proliferative or diffuse sclerosis IF: Smudged deposits; stain for IgG, C3, κ, or λ, and DNAJB9 EM: Randomly arranged fibrils (12–24 nm) |
| C3 glomerulopathy with monoclonal gammopathy | Renal isolated disease Hypertension Proteinuria Declining renal function Low complements in some patients | Non-organised deposits LM: membranoproliferative, mesangioproliferative or endocapillary proliferative GN IF: C3 granular deposits in mesangium and GBM (with paucity of Ig deposits) EM: ill-defined electron-dense deposits in C3GN and intramembranous and mesangial highly electron-dense deposits in dense-deposit disease |
| Thrombotic microangiopathy (TMA) | Unexplained anaemia and thrombocytopenia Hypertension, possibly severe | Light and electron microscopy: GBM duplication, mesangiolysis, subendothelial “fluff,” and thrombosis IF: no Ig deposits |
| Membranous nephropathy | Renal isolated disease Nephrotic syndrome is common Proteinuria is the predominant feature | LM: diffuse thickening of the GBM. “Spikes” of GBM extending among immune deposits may be seen on silver stain EM: subepithelial electron-dense deposits; effacement of foot processes and expansion of the GBM with deposition of extracellular matrix. IF: diffuse granular pattern of immunoglobulin (Ig) G and C3 staining along the GBM |
| Minimal change nephropathy | Renal isolated disease Nephrotic syndrome with rapid onset | LM: glomeruli appear normal EM: diffuse effacement of epithelial foot processes IF: no complement or immunoglobulin deposits |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pratt, G.; Giles, H.V.; Pinney, J.H. Renal Disorders Associated with Waldenström Macroglobulinaemia, IgM MGUS and IgM-Producing B-Cell Lymphoproliferative Disorders. Hemato 2023, 4, 184-195. https://doi.org/10.3390/hemato4020015
Pratt G, Giles HV, Pinney JH. Renal Disorders Associated with Waldenström Macroglobulinaemia, IgM MGUS and IgM-Producing B-Cell Lymphoproliferative Disorders. Hemato. 2023; 4(2):184-195. https://doi.org/10.3390/hemato4020015
Chicago/Turabian StylePratt, Guy, Hannah V. Giles, and Jennifer H. Pinney. 2023. "Renal Disorders Associated with Waldenström Macroglobulinaemia, IgM MGUS and IgM-Producing B-Cell Lymphoproliferative Disorders" Hemato 4, no. 2: 184-195. https://doi.org/10.3390/hemato4020015
APA StylePratt, G., Giles, H. V., & Pinney, J. H. (2023). Renal Disorders Associated with Waldenström Macroglobulinaemia, IgM MGUS and IgM-Producing B-Cell Lymphoproliferative Disorders. Hemato, 4(2), 184-195. https://doi.org/10.3390/hemato4020015