
Cold Agglutinin Disease: Improved Understanding of Pathogenesis Helps Define Targets for Therapy

 ,
,
Abstract
1. Introduction
2. Properties of Cold Agglutinins
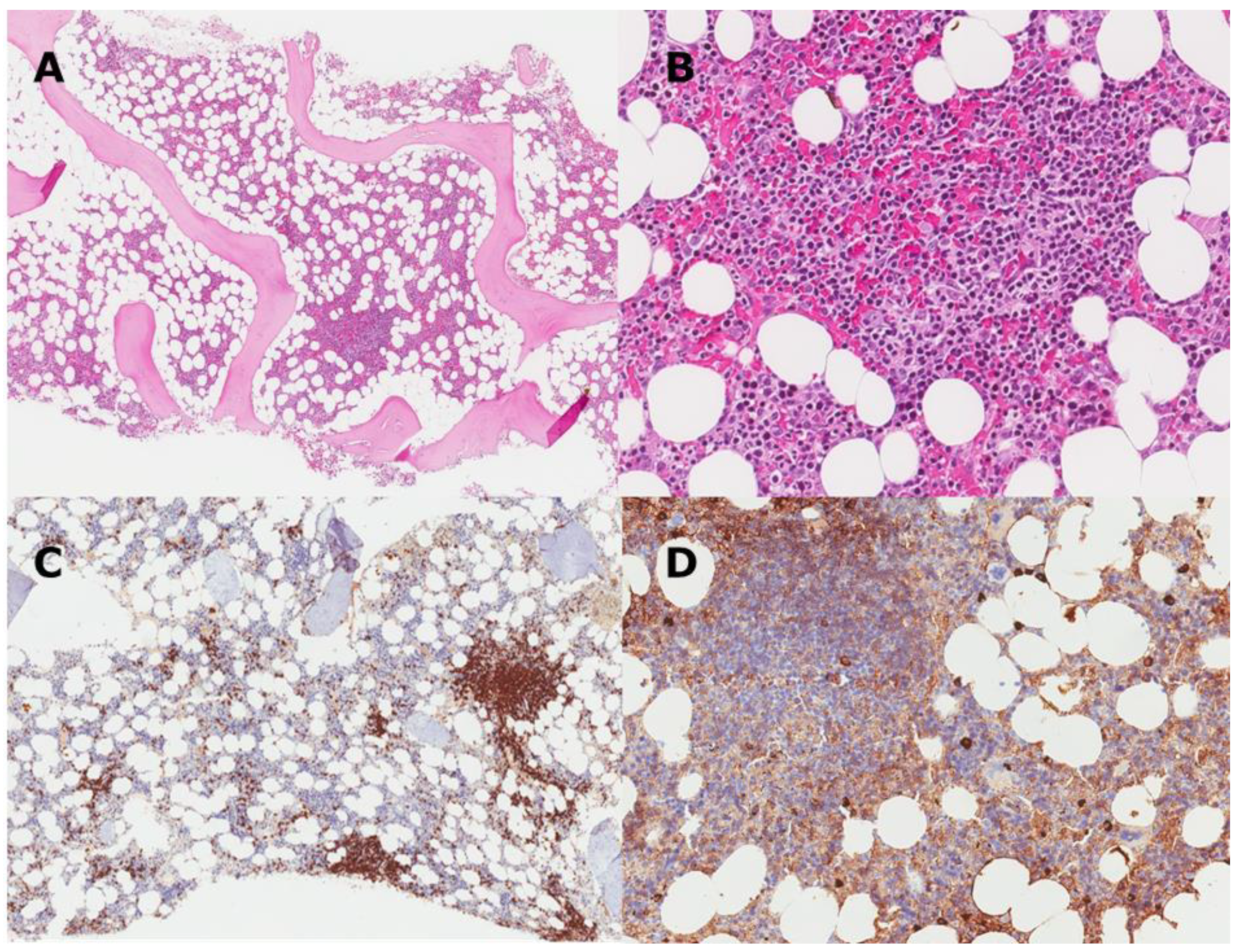
3. CAD as a Clonal Lymphoproliferative Disorder
4. CAD and Waldenström’s Macroglobulinemia: Distinction or Overlap?
5. CAD as a Complement-Mediated Hemolytic Anemia
6. Epidemiology and Clinical Features
7. Diagnosis
8. Management
9. B-Cell-Directed Therapies
10. Complement-Directed Therapies
11. Supportive Measures
11.1. Transfusion
11.2. Plasmapheresis
11.3. Folic Acid
11.4. Erythropoietin
11.5. Thrombosis Prophylaxis
12. Choice of Therapy
13. The Future
14. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berentsen, S. How I treat cold agglutinin disease. Blood 2021, 137, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Dacie, J. (Ed.) The auto-immune haemolytic anaemias: Introduction. In The Haemolytic Anaemias; Churchill Livingstone: London, UK, 1992; Volume 3, pp. 1–5. [Google Scholar]
- Sokol, R.J.; Hewitt, S.; Stamps, B.K. Autoimmune haemolysis: An 18-year study of 865 cases referred to a regional transfusion centre. Br. Med. J. 1981, 282, 2023–2027. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, W.; Fattizzo, B.; Zaninoni, A.; Radice, T.; Nichele, I.; Di Bona, E.; Lunghi, M.; Tassinari, C.; Alfinito, F.; Ferrari, A.; et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: A GIMEMA study of 308 patients. Blood 2014, 124, 2930–2936. [Google Scholar] [CrossRef] [PubMed]
- Jäger, U.; Barcellini, W.; Broome, C.M.; Gertz, M.A.; Hill, A.; Hill, Q.A.; Jilma, B.; Kuter, D.J.; Michel, M.; Montillo, M.; et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: Recommendations from the First International Consensus Meeting. Blood Rev. 2020, 41, 100648. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, S.; Barcellini, W. Autoimmune Hemolytic Anemias. N. Engl. J. Med. 2021, 385, 1407–1419. [Google Scholar] [CrossRef]
- Landsteiner, K. Über Beziehungen zwischen dem Blutserum und den Körperzellen. Münch Med. Wschr. 1903, 50, 1812–1814. [Google Scholar]
- Rosenthal, F.; Corten, M. Über das Phänomen der Autohämagglutination und über die Eigenscaften der Kältehämagglutinine. Folia Haematol. 1937, 58, 64–90. [Google Scholar]
- Rørvik, K. The syndrome of high-titre cold haemagglutination; a survey and a case report. Acta Med. Scand. 1954, 148, 299–308. [Google Scholar] [CrossRef]
- Schubothe, H. The cold hemagglutinin disease. Semin. Hematol. 1966, 3, 27–47. [Google Scholar]
- Harboe, M.; van Furth, R.; Schubothe, H.; Lind, K.; Evans, R.S. Exclusive occurrence of K chains in isolated cold haemagglutinins. Scand. J. Haematol. 1965, 2, 259–266. [Google Scholar] [CrossRef]
- Patriquin, C.J.; Pavenski, K. O, wind, if winter comes… Will symptoms be far behind?: Exploring the seasonality (or lack thereof) and management of cold agglutinin disease. Transfusion 2022, 62, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, S. Cold agglutinin disease. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Hill, Q.A.; Hill, A.; Berentsen, S. Defining autoimmune hemolytic anemia: A systematic review of the terminology used for diagnosis and treatment. Blood Adv. 2019, 3, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, S.; Tjønnfjord, G.E. Diagnosis and treatment of cold agglutinin mediated autoimmune hemolytic anemia. Blood Rev. 2012, 26, 107–115. [Google Scholar] [CrossRef]
- Hill, Q.A.; Stamps, R.; Massey, E.; Grainger, J.D.; Provan, D.; Hill, A.; British Society for Haematology, G. Guidelines on the management of drug-induced immune and secondary autoimmune, haemolytic anaemia. Br. J. Haematol. 2017, 177, 208–220. [Google Scholar] [CrossRef]
- Quinn, R.; Murakhovskaya, I. SARS-CoV-2 and Autoimmune Cytopenia. Hemato 2021, 2, 463–476. [Google Scholar] [CrossRef]
- Randen, U.; Trøen, G.; Tierens, A.; Steen, C.; Warsame, A.; Beiske, K.; Tjønnfjord, G.E.; Berentsen, S.; Delabie, J. Primary cold agglutinin-associated lymphoproliferative disease: A B-cell lymphoma of the bone marrow distinct from lymphoplasmacytic lymphoma. Haematologica 2014, 99, 497–504. [Google Scholar] [CrossRef]
- Ulvestad, E.; Berentsen, S.; Bø, K.; Shammas, F.V. Clinical immunology of chronic cold agglutinin disease. Eur. J. Haematol. 1999, 63, 259–266. [Google Scholar] [CrossRef]
- Berentsen, S.; Hill, A.; Hill, Q.A.; Tvedt, T.H.A.; Michel, M. Novel insights into the treatment of complement-mediated hemolytic anemias. Ther. Adv. Hematol. 2019, 10. [Google Scholar] [CrossRef]
- Berentsen, S. New insights in the pathogenesis and therapy of cold agglutinin-mediated autoimmune hemolytic anemia. Front. Immunol. 2020, 11, 590. [Google Scholar] [CrossRef]
- Winter, O.; Dame, C.; Jundt, F.; Hiepe, F. Pathogenic long-lived plasma cells and their survival niches in autoimmunity, malignancy, and allergy. J. Immunol. 2012, 189, 5105–5111. [Google Scholar] [CrossRef] [PubMed]
- El-Ayoubi, A.; Wang, J.Q.; Hein, N.; Talaulikar, D. Role of plasma cells in Waldenstrom macroglobulinaemia. Pathology 2017, 49, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Tomkins, O.; Berentsen, S.; Arulogun, S.; Sekhar, M.; D’Sa, S. Daratumumab for disabling cold agglutinin disease refractory to B-cell directed therapy. Am. J. Hematol. 2020, 95, E293–E295. [Google Scholar] [CrossRef] [PubMed]
- Harboe, M.; Deverill, J. Immunochemical Properties of Cold Haemagglutinins. Scand. J. Haematol. 1964, 1, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, S.; Barcellini, W.; D’Sa, S.; Randen, U.; Tvedt, T.H.A.; Fattizzo, B.; Haukas, E.; Kell, M.; Brudevold, R.; Dahm, A.E.A.; et al. Cold agglutinin disease revisited: A multinational, observational study of 232 patients. Blood 2020, 136, 480–488. [Google Scholar] [CrossRef]
- Silberstein, L.E.; Berkman, E.M.; Schreiber, A.D. Cold hemagglutinin disease associated with IgG cold-reactive antibody. Ann. Intern. Med. 1987, 106, 238–242. [Google Scholar] [CrossRef]
- Christenson, W.N.; Dacie, J.V.; Croucher, B.E.; Charlwood, P.A. Electrophoretic studies on sera containing high-titre cold haemagglutinins: Identification of the antibody as the cause of an abnormal gamma 1 peak. Br. J. Haematol. 1957, 3, 262–275. [Google Scholar] [CrossRef]
- Chadebech, P.; Michel, M.; Janvier, D.; Yamada, K.; Copie-Bergman, C.; Bodivit, G.; Bensussan, A.; Fournie, J.J.; Godeau, B.; Bierling, P.; et al. IgA-mediated human autoimmune hemolytic anemia as a result of hemagglutination in the spleen, but independent of complement activation and FcalphaRI. Blood 2010, 116, 4141–4147. [Google Scholar] [CrossRef]
- Sefland, Ø.; Randen, U.; Berentsen, S. Development of Multiple Myeloma of the IgA Type in a Patient with Cold Agglutinin Disease: Transformation or Coincidence? Case Rep. Hematol. 2019, 2019, 1610632. [Google Scholar] [CrossRef]
- Issitt, P.D. I blood group system and its relationship to disease. J. Med. Lab. Technol. 1968, 25, 1–6. [Google Scholar]
- Pruzanski, W.; Faird, N.; Keystone, E.; Armstrong, M. The influence of homogeneous cold agglutinins on polymorphonuclear and mononuclear phagocytes. Clin. Immunol. Immunopathol. 1975, 4, 277–285. [Google Scholar] [CrossRef]
- Pruzanski, W.; Farid, N.; Keystone, E.; Armstrong, M.; Greaves, M.F. The influence of homogeneous cold agglutinins on human B and T lymphocytes. Clin. Immunol. Immunopathol. 1975, 4, 248–257. [Google Scholar] [CrossRef]
- Dunstan, R.A.; Simpson, M.B.; Rosse, W.F. The presence of the Ii blood group system on human platelets. Am. J. Clin. Pathol. 1984, 82, 74–77. [Google Scholar] [CrossRef]
- Berentsen, S. Neutrophil aggregation on the peripheral blood smear in a patient with cold agglutinin disease. Ann. Hematol. 2017, 96, 1767–1768. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, L.E.; Robertson, G.A.; Harris, A.C.; Moreau, L.; Besa, E.; Nowell, P.C. Etiologic aspects of cold agglutinin disease: Evidence for cytogenetically defined clones of lymphoid cells and the demonstration that an anti-Pr cold autoantibody is derived from a chromosomally aberrant B cell clone. Blood 1986, 67, 1705–1709. [Google Scholar] [CrossRef] [PubMed]
- Mohanan, N.; Henry, N.; Rafi, A.M.; Innah, S.J. Anti IH: An antibody worth mention. Asian J. Transfus. Sci. 2016, 10, 152–154. [Google Scholar] [CrossRef]
- Malecka, A.; Trøen, G.; Tierens, A.; Østlie, I.; Malecki, J.; Randen, U.; Berentsen, S.; Tjønnfjord, G.E.; Delabie, J.M. Immunoglobulin heavy and light chain gene features are correlated with primary cold agglutinin disease onset and activity. Haematologica 2016, 101, e361–e364. [Google Scholar] [CrossRef]
- Pascual, V.; Victor, K.; Spellerberg, M.; Hamblin, T.J.; Stevenson, F.K.; Capra, J.D. VH restriction among human cold agglutinins. The VH4-21 gene segment is required to encode anti-I and anti-i specificities. J. Immunol. 1992, 149, 2337–2344. [Google Scholar]
- Thorpe, S.J.; Boult, C.E.; Stevenson, F.K.; Scott, M.L.; Sutherland, J.; Spellerberg, M.B.; Natvig, J.B.; Thompson, K.M. Cold agglutinin activity is common among human monoclonal IgM Rh system antibodies using the V4-34 heavy chain variable gene segment. Transfusion 1997, 37, 1111–1116. [Google Scholar] [CrossRef]
- Pascual, V.; Capra, J.D. VH4-21, a human VH gene segment overrepresented in the autoimmune repertoire. Arthritis Rheum. 1992, 35, 11–18. [Google Scholar] [CrossRef]
- Li, Y.; Spellerberg, M.B.; Stevenson, F.K.; Capra, J.D.; Potter, K.N. The I binding specificity of human VH 4-34 (VH 4-21) encoded antibodies is determined by both VH framework region 1 and complementarity determining region 3. J. Mol. Biol. 1996, 256, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Potter, K.N.; Hobby, P.; Klijn, S.; Stevenson, F.K.; Sutton, B.J. Evidence for involvement of a hydrophobic patch in framework region 1 of human V4-34-encoded Igs in recognition of the red blood cell I antigen. J. Immunol. 2002, 169, 3777–3782. [Google Scholar] [CrossRef] [PubMed]
- Sabouri, Z.; Schofield, P.; Horikawa, K.; Spierings, E.; Kipling, D.; Randall, K.L.; Langley, D.; Roome, B.; Vazquez-Lombardi, R.; Rouet, R.; et al. Redemption of autoantibodies on anergic B cells by variable-region glycosylation and mutation away from self-reactivity. Proc. Natl. Acad. Sci. USA 2014, 111, E2567–E2575. [Google Scholar] [CrossRef]
- Capra, J.D.; Kehoe, J.M.; Williams, R.C., Jr.; Feizi, T.; Kunkel, H.G. Light chain sequences of human IgM cold agglutinins (variable-region subgroups amino-acid sequence-kappa light chain-N-terminal). Proc. Natl. Acad. Sci. USA 1972, 69, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Gergely, J.; Wang, A.C.; Fudenberg, H.H. Chemical analyses of variable regions of heavy and light chains of cold agglutinins. Vox Sang. 1973, 24, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Ruzickova, S.; Pruss, A.; Odendahl, M.; Wolbart, K.; Burmester, G.R.; Scholze, J.; Dorner, T.; Hansen, A. Chronic lymphocytic leukemia preceded by cold agglutinin disease: Intraclonal immunoglobulin light-chain diversity in V(H)4-34 expressing single leukemic B cells. Blood 2002, 100, 3419–3422. [Google Scholar] [CrossRef]
- Silberstein, L.E.; Jefferies, L.C.; Goldman, J.; Friedman, D.; Moore, J.S.; Nowell, P.C.; Roelcke, D.; Pruzanski, W.; Roudier, J.; Silverman, G.J. Variable region gene analysis of pathologic human autoantibodies to the related i and I red blood cell antigens. Blood 1991, 78, 2372–2386. [Google Scholar] [CrossRef]
- Röth, A.; Fryzek, J.; Jiang, X.; Reichert, H.; Patel, P.; Su, J.; Morales Arias, J.; Broome, C.M. Complement-mediated hemolysis persists year round in patients with cold agglutinin disease. Transfusion 2022, 62, 51–59. [Google Scholar] [CrossRef]
- Rosse, W.F.; Adams, J.P. The variability of hemolysis in the cold agglutinin syndrome. Blood 1980, 56, 409–416. [Google Scholar] [CrossRef]
- Stone, M.J.; McElroy, Y.G.; Pestronk, A.; Reynolds, J.L.; Newman, J.T.; Tong, A.W. Human monoclonal macroglobulins with antibody activity. Semin. Oncol. 2003, 30, 318–324. [Google Scholar] [CrossRef]
- Randall, T.D.; King, L.B.; Corley, R.B. The biological effects of IgM hexamer formation. Eur. J. Immunol. 1990, 20, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Petrusic, V.; Zivkovic, I.; Stojanovic, M.; Stojicevic, I.; Marinkovic, E.; Inic-Kanada, A.; Dimitijevic, L. Antigenic specificity and expression of a natural idiotope on human pentameric and hexameric IgM polymers. Immunol. Res. 2011, 51, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Petrusic, V.; Zivkovic, I.; Stojanovic, M.; Stojicevic, I.; Marinkovic, E.; Dimitrijevic, L. Hexameric immunoglobulin M in humans: Desired or unwanted? Med. Hypotheses 2011, 77, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Dacie, J. (Ed.) Auto-immune haemolytic anaemia (AIHA): Cold-antibody syndromes II: Immunochemistry and specificity of the antibodies serum complement in autoimmune haemolytic anaemia. In The Haemolytic Anaemias; Churchill Livingstone: London, UK, 1992; Volume 3, pp. 240–295. [Google Scholar]
- Jain, M.D.; Cabrerizo-Sanchez, R.; Karkouti, K.; Yau, T.; Pendergrast, J.M.; Cserti-Gazdewich, C.M. Seek and you shall find—But then what do you do? Cold agglutinins in cardiopulmonary bypass and a single-center experience with cold agglutinin screening before cardiac surgery. Transfus. Med. Rev. 2013, 27, 65–73. [Google Scholar] [CrossRef]
- Berentsen, S.; Bø, K.; Shammas, F.V.; Myking, A.O.; Ulvestad, E. Chronic cold agglutinin disease of the “idiopathic” type is a premalignant or low-grade malignant lymphoproliferative disease. APMIS 1997, 105, 354–362. [Google Scholar] [CrossRef]
- Berentsen, S.; Ulvestad, E.; Langholm, R.; Beiske, K.; Hjorth-Hansen, H.; Ghanima, W.; Sørbø, J.H.; Tjønnfjord, G.E. Primary chronic cold agglutinin disease: A population based clinical study of 86 patients. Haematologica 2006, 91, 460–466. [Google Scholar]
- Swiecicki, P.L.; Hegerova, L.T.; Gertz, M.A. Cold agglutinin disease. Blood 2013, 122, 1114–1121. [Google Scholar] [CrossRef]
- Naresh, K.N.; Rossi, D.; Chen, X.; Berentsen, S.; Randen, U. Cold agglutinin disease. In WHO Classification of Haematolymphoid Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2022; p. 349. Available online: https://tumourclassification.iarc.who.int/chaptercontent/363/349 (accessed on 15 September 2022).
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, S.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A Report from the Clinical Advisory Committee. Blood 2022. online ahead of print. [Google Scholar] [CrossRef]
- Malecka, A.; Delabie, J.; Østlie, I.; Tierens, A.; Randen, U.; Berentsen, S.; Tjønnfjord, G.E.; Trøen, G. Cold agglutinin–associated B-cell lymphoproliferative disease shows highly recurrent gains of chromosome 3 and 12 or 18. Blood Adv. 2020, 4, 993–996. [Google Scholar] [CrossRef]
- Michaux, L.; Dierlamm, J.; Wlodarska, I.; Stul, M.; Bosly, A.; Delannoy, A.; Louwagie, A.; Mecucci, C.; Cassiman, J.J.; van den Berghe, H.; et al. Trisomy 3 is a consistent chromosome change in malignant lymphoproliferative disorders preceded by cold agglutinin disease. Br. J. Haematol. 1995, 91, 421–424. [Google Scholar] [CrossRef]
- Gordon, J.; Silberstein, L.; Moreau, L.; Nowell, P.C. Trisomy 3 in cold agglutinin disease. Cancer Genet. Cytogenet. 1990, 46, 89–92. [Google Scholar] [CrossRef]
- Malecka, A.; Trøen, G.; Delabie, J.; Malecki, J.; Østlie, I.; Tierens, A.; Randen, U.; Berentsen, S.; Tjønnfjord, G.E. The mutational landscape of cold agglutinin disease: CARD11 and CXCR4 mutations are correlated with lower hemoglobin levels. Am. J. Hematol. 2021, 96, E279–E283. [Google Scholar] [CrossRef]
- Malecka, A.; Trøen, G.; Tierens, A.; Østlie, I.; Malecki, J.; Randen, U.; Wang, J.; Berentsen, S.; Tjønnfjord, G.E.; Delabie, J.M.A. Frequent somatic mutations of KMT2D (MLL2) and CARD11 genes in primary cold agglutinin disease. Br. J. Haematol. 2018, 183, 838–842. [Google Scholar] [CrossRef]
- De Tute, R.; Rawstron, A.; Evans, P.; Owen, R. Cold agglutinin disease is a phenotypically distinct clonal B-cell disorder. Clin. Lymphoma Myeloma Leuk. 2015, 15, e184. [Google Scholar] [CrossRef]
- Arthold, C.; Skrabs, C.; Mitterbauer-Hohendanner, G.; Thalhammer, R.; Simonitsch-Klupp, I.; Panzer, S.; Valent, P.; Lechner, K.; Jäger, U.; Sillaber, C. Cold antibody autoimmune hemolytic anemia and lymphoproliferative disorders: A retrospective study of 20 patients including clinical, hematological, and molecular findings. Wien. Klin. Wochenschr. 2014, 126, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.G.; Treon, S.P.; Al-Katib, A.; Fonseca, R.; Greipp, P.R.; McMaster, M.L.; Morra, E.; Pangalis, G.A.; San Miguel, J.F.; Branagan, A.R.; et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: Consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin. Oncol. 2003, 30, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.J.; Merlini, G.; Pascual, V. Autoantibody activity in Waldenstrom’s macroglobulinemia. Clin. Lymphoma 2005, 5, 225–229. [Google Scholar] [CrossRef]
- Stone, M.J.; Berentsen, S. Hyperviscosity syndrome, cold agglutinin hemolytic anemia, and cryoglobulinemia. In Waldenström’s Macroglobulinemia; Leblond, V., Treon, S.P., Dimopoulos, M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 171–183. [Google Scholar]
- Khwaja, J.; D’Sa, S.; Minnema, M.C.; Kersten, M.J.; Wechalekar, A.; Vos, J.M. IgM monoclonal gammopathies of clinical significance: Diagnosis and management. Haematologica, 2022; online ahead of print. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Hamilos, G.; Zervas, K.; Symeonidis, A.; Kouvatseas, G.; Roussou, P.; Gika, D.; Karmiris, T.; Bourantas, K.; Zomas, A.; et al. Survival and prognostic factors after initiation of treatment in Waldenstrom’s macroglobulinemia. Ann. Oncol. 2003, 14, 1299–1305. [Google Scholar] [CrossRef]
- Berentsen, S. Cold agglutinin-mediated autoimmune hemolytic anemia in Waldenstrom’s macroglobulinemia. Clin. Lymphoma Myeloma 2009, 9, 110–112. [Google Scholar] [CrossRef]
- Gertz, M.A. Updates on the Diagnosis and Management of Cold Autoimmune Hemolytic Anemia. Hematol. Oncol. Clin. N. Am. 2022, 36, 341–352. [Google Scholar] [CrossRef]
- Owen, R.G.; Rawstron, A.; de Tute, R.M. Waldenström Macroglobulinaemia: Pathological Features and Diagnostic Assessment. In Waldenström’s Macroglobulinemia; Leblond, V., Treon, S., Dimopoulos, M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 3–19. [Google Scholar]
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hunter, Z.R.; Yang, G.; Zhou, Y.; Cao, Y.; Liu, X.; Morra, E.; Trojani, A.; Greco, A.; Arcaini, L.; et al. MYD88 L265P in Waldenstrom macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood 2013, 121, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Rose, E.L.; Singh, A.; Hussain, S.; Stagliano, N.E.; Parry, G.C.; Panicker, S. TNT003, an inhibitor of the serine protease C1s, prevents complement activation induced by cold agglutinins. Blood 2014, 123, 4015–4022. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, S. Complement Activation and Inhibition in Autoimmune Hemolytic Anemia: Focus on Cold Agglutinin Disease. Semin. Hematol. 2018, 55, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Jalink, M.; de Boer, E.C.W.; Evers, D.; Havinga, M.Q.; Vos, J.M.I.; Zeerleder, S.; de Haas, M.; Jongerius, I. Halting targeted and collateral damage to red blood cells by the complement system. Semin. Immunopathol. 2021, 43, 799–816. [Google Scholar] [CrossRef]
- Jaffe, C.J.; Atkinson, J.P.; Frank, M.M. The role of complement in the clearance of cold agglutinin-sensitized erythrocytes in man. J. Clin. Investig. 1976, 58, 942–949. [Google Scholar] [CrossRef]
- Barcellini, W. Immune Hemolysis: Diagnosis and Treatment Recommendations. Semin. Hematol. 2015, 52, 304–312. [Google Scholar] [CrossRef]
- Hill, Q.A.; Stamps, R.; Massey, E.; Grainger, J.D.; Provan, D.; Hill, A.; Haematology, B.S.F. The diagnosis and management of primary autoimmune haemolytic anaemia. Br. J. Haematol. 2017, 176, 395–411. [Google Scholar] [CrossRef]
- Ulvestad, E.; Berentsen, S.; Mollnes, T.E. Acute phase haemolysis in chronic cold agglutinin disease. Scand. J. Immunol. 2001, 54, 239–242. [Google Scholar] [CrossRef]
- Li, Z.; Shao, Z.; Xu, Y.; Shen, L.; Chen, G.; Zhang, Y.; Chu, Y. Subclasses of warm autoantibody IgG in patients with autoimmune hemolytic anemia and their clinical implications. Chin. Med. J. 1999, 112, 805–808. [Google Scholar] [PubMed]
- Berentsen, S.; Sundic, T. Red blood cell destruction in autoimmune hemolytic anemia: Role of complement and potential new targets for therapy. Biomed. Res. Int. 2015, 2015, 363278. [Google Scholar] [CrossRef] [PubMed]
- Volanakis, J.E. Transcriptional regulation of complement genes. Annu. Rev. Immunol. 1995, 13, 277–305. [Google Scholar] [CrossRef]
- Hansen, D.L.; Berentsen, S.; Fattizzo, B.; Hansen, P.L.; Barcellini, W.; Frederiksen, H. Seasonal variation in the incidence of cold agglutinin disease in Norway, Denmark, and Italy. Am. J. Hematol. 2021, 96, E262–E265. [Google Scholar] [CrossRef]
- Hansen, D.L.; Møller, S.; Berentsen, S.; Frederiksen, H. Mortality in cold agglutinin disease shows seasonal pattern. Transfusion 2022, 62, 1460–1461. [Google Scholar] [CrossRef]
- Lyckholm, L.J.; Edmond, M.B. Images in clinical medicine. Seasonal hemolysis due to cold-agglutinin syndrome. N. Engl. J. Med. 1996, 334, 437. [Google Scholar] [CrossRef]
- Tjønnfjord, E.; Vengen, O.A.; Berentsen, S.; Tjønnfjord, G.E. Prophylactic use of eculizumab during surgery in chronic cold agglutinin disease. BMJ Case Rep. 2017. [Google Scholar] [CrossRef]
- Tvedt, T.H.A.; Steien, E.; Øvrebø, B.; Haaverstad, R.; Hobbs, W.; Wardecki, M.; Tjønnfjord, G.E.; Berentsen, S.A. Sutimlimab, an investigational C1s inhibitor, effectively prevents exacerbation of hemolytic anemia in a patient with cold agglutinin disease undergoing major surgery. Am. J. Hematol. 2022, 97, E51–E54. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, S. Cold agglutinins: Fending off the attack. Blood 2019, 133, 885–886. [Google Scholar] [CrossRef]
- Berentsen, S.; Barcellini, W.; D’Sa, S.; Jilma, B. Sutimlimab for treatment of cold agglutinin disease: Why, how and for whom? Immunotherapy 2022. online ahead of print. [Google Scholar] [CrossRef]
- Röth, A.; Barcellini, W.; D’Sa, S.; Miyakawa, Y.; Broome, C.M.; Michel, M.; Kuter, D.J.; Jilma, B.; Tvedt, T.H.A.; Fruebis, J.; et al. Sutimlimab in Cold Agglutinin Disease. N. Engl. J. Med. 2021, 384, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.S.; Beaumont, J.L.; Ogale, S.; Brunetta, P.; Cella, D. Validation of the functional assessment of chronic illness therapy-fatigue scale in patients with moderately to severely active systemic lupus erythematosus, participating in a clinical trial. J. Rheumatol. 2011, 38, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, R.A.; Young, N.S.; Antonioli, E.; Risitano, A.M.; Schrezenmeier, H.; Schubert, J.; Gaya, A.; Coyle, L.; de Castro, C.; Fu, C.L.; et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood 2008, 111, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Röth, A.; Berentsen, S.; Barcellini, W.; D’Sa, S.; Jilma, B.; Michel, M.; Weitz, I.C.; Yamaguchi, M.; Nishimura, J.I.; Vos, J.M.I.; et al. Sutimlimab in patients with cold agglutinin disease: Results of the randomized placebo-controlled phase 3 CADENZA trial. Blood 2022, 140, 980–991. [Google Scholar] [CrossRef]
- Broome, C.M.; Cunningham, J.M.; Mullins, M.; Jiang, X.; Bylsma, L.C.; Fryzek, J.P.; Rosenthal, A. Increased risk of thrombotic events in cold agglutinin disease: A 10-year retrospective analysis. Res. Pract. Thromb. Haemost. 2020, 4, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Bylsma, L.C.; Gulbech Ording, A.; Rosenthal, A.; Ozturk, B.; Fryzek, J.P.; Arias, J.M.; Roth, A.; Berentsen, S. Occurrence, thromboembolic risk, and mortality in Danish patients with cold agglutinin disease. Blood Adv. 2019, 3, 2980–2985. [Google Scholar] [CrossRef] [PubMed]
- Kamesaki, T.; Nishimura, J.I.; Wada, H.; Yu, E.; Tsao, E.; Morales, J.; Kanakura, Y. Demographic characteristics, thromboembolism risk, and treatment patterns for patients with cold agglutinin disease in Japan. Int. J. Hematol. 2020, 112, 307–315. [Google Scholar] [CrossRef]
- Röth, A.; Bommer, M.; Huttmann, A.; Herich-Terhurne, D.; Kuklik, N.; Rekowski, J.; Lenz, V.; Schrezenmeier, H.; Duhrsen, U. Eculizumab in cold agglutinin disease (DECADE): An open-label, prospective, bicentric, nonrandomized phase 2 trial. Blood Adv. 2018, 2, 2543–2549. [Google Scholar] [CrossRef]
- Chandesris, M.O.; Schleinitz, N.; Ferrera, V.; Bernit, E.; Mazodier, K.; Gayet, S.; Chiaroni, J.M.; Veit, V.; Kaplanski, G.; Harle, J.R. Cold agglutinins, clinical presentation and significance; retrospective analysis of 58 patients. Rev. Med. Interne 2004, 25, 856–865. (In French) [Google Scholar] [CrossRef]
- Berentsen, S.; Röth, A.; Randen, U.; Jilma, B.; Tjønnfjord, G.E. Cold agglutinin disease: Current challenges and further prospects. J. Blood Med. 2019, 10, 93–103. [Google Scholar] [CrossRef]
- Dacie, J. (Ed.) Treatment and prognosis of cold-antibody AIHA. In The Haemolytic Anaemias; Churchill Livingstone: London, UK, 1992; Volume 3, pp. 502–520. [Google Scholar]
- Overman, R.A.; Gourlay, M.L.; Deal, C.L.; Farley, J.F.; Brookhart, M.A.; Layton, J.B. Fracture rate associated with quality metric-based anti-osteoporosis treatment in glucocorticoid-induced osteoporosis. Osteoporos. Int. 2015, 26, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, S.; Ulvestad, E.; Gjertsen, B.T.; Hjorth-Hansen, H.; Langholm, R.; Knutsen, H.; Ghanima, W.; Shammas, F.V.; Tjonnfjord, G.E. Rituximab for primary chronic cold agglutinin disease: A prospective study of 37 courses of therapy in 27 patients. Blood 2004, 103, 2925–2928. [Google Scholar] [CrossRef] [PubMed]
- Schöllkopf, C.; Kjeldsen, L.; Bjerrum, O.W.; Mourits-Andersen, H.T.; Nielsen, J.L.; Christensen, B.E.; Jensen, B.A.; Pedersen, B.B.; Taaning, E.B.; Klausen, T.W.; et al. Rituximab in chronic cold agglutinin disease: A prospective study of 20 patients. Leuk. Lymphoma 2006, 47, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, W.; Fattizzo, B.; Zaninoni, A. Current and emerging treatment options for autoimmune hemolytic anemia. Expert Rev. Clin. Immunol. 2018, 14, 857–872. [Google Scholar] [CrossRef]
- Jaeger, U. Hot therapy for cold agglutinin disease. Blood 2017, 130, 392–393. [Google Scholar] [CrossRef][Green Version]
- Berentsen, S.; Randen, U.; Vågan, A.M.; Hjorth-Hansen, H.; Vik, A.; Dalgaard, J.; Jacobsen, E.M.; Thoresen, A.S.; Beiske, K.; Tjønnfjord, G.E. High response rate and durable remissions following fludarabine and rituximab combination therapy for chronic cold agglutinin disease. Blood 2010, 116, 3180–3184. [Google Scholar] [CrossRef]
- Berentsen, S.; Randen, U.; Oksman, M.; Birgens, H.; Tvedt, T.H.A.; Dalgaard, J.; Galteland, E.; Haukas, E.; Brudevold, R.; Sørbø, J.H.; et al. Bendamustine plus rituximab for chronic cold agglutinin disease: Results of a Nordic prospective multicenter trial. Blood 2017, 130, 537–541. [Google Scholar] [CrossRef]
- Del Giudice, I.; Matutes, E.; Osuji, N.; Parry-Jones, N.; Swansbury, J.; Catovsky, D. Delayed response to fludarabine in lymphoplasmacytic lymphoma/Waldenström’s macroglobulinemia. Haematologica 2005, 90, 268–270. [Google Scholar]
- Rossi, G.; Gramegna, D.; Paoloni, F.; Fattizzo, B.; Binda, F.; D’Adda, M.; Farina, M.; Lucchini, E.; Mauro, F.R.; Salvi, F.; et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: A phase 2 prospective GIMEMA study. Blood 2018, 132, 547–550. [Google Scholar] [CrossRef]
- Jalink, M.; Berentsen, S.; Castillo, J.J.; Treon, S.P.; Cruijsen, M.; Fattizzo, B.; Cassin, R.; Fotiou, D.; Kastritis, E.; De Haas, M.; et al. Effect of ibrutinib treatment on hemolytic anemia and acrocyanosis in cold agglutinin disease/cold agglutinin syndrome. Blood 2021, 138, 2002–2005. [Google Scholar] [CrossRef]
- Röth, A.; Hüttmann, A.; Rother, R.P.; Dührsen, U.; Philipp, T. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood 2009, 113, 3885–3886. [Google Scholar] [CrossRef] [PubMed]
- Wouters, D.; Stephan, F.; Strengers, P.; de Haas, M.; Brouwer, C.; Hagenbeek, A.; van Oers, M.H.; Zeerleder, S. C1-esterase inhibitor concentrate rescues erythrocytes from complement-mediated destruction in autoimmune hemolytic anemia. Blood 2013, 121, 1242–1244. [Google Scholar] [CrossRef] [PubMed]
- Tesfaye, A.; Broome, C. A Novel Approach for Treatment of Cold Agglutinin Syndrome-Related Severe Hemolysis. J. Hematol. 2016, 5, 30–33. [Google Scholar] [CrossRef]
- De Boer, E.C.V.; Jalink, M.; Delvasto-Nuñes, L.; Meulenbroek, E.M.; Baas, I.; Janssen, S.R.; Dijkman, E.E.M.; Gelderman, K.A.; Wouters, D.; Engel, M.D.; et al. Peritransfusional C1-inhibitor in patients with severe complement-mediated autoimmune hemolytic anemia: An open-label phase 2 trial. Presented at the European Hematology Association 2022 Congress. HemaSphere 2022, 6, 2690. [Google Scholar]
- Varela, J.C.; Tomlinson, S. Complement: An overview for the clinician. Hematol. Oncol. Clin. N. Am. 2015, 29, 409–427. [Google Scholar] [CrossRef]
- Jäger, U.; D’Sa, S.; Schörgenhofer, C.; Bartko, J.; Derhaschnig, U.; Sillaber, C.; Jilma-Stohlawetz, P.; Fillitz, M.; Schenk, T.; Patou, G.; et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: A first-in-human trial. Blood 2019, 133, 893–901. [Google Scholar] [CrossRef]
- Röth, A.; Barcellini, W.; D’Sa, S.; Miyakawa, Y.; Broome, C.M.; Michel, M.; Kuter, D.J.; Jilma, B.; Tvedt, T.H.A.; Weitz, I.C.; et al. Complement C1s inhibition with sutimlimab results in durable response in cold agglutinin disease: CARDINAL study 1-year interim follow-up results. Haematologica 2022, 107, 1698. [Google Scholar] [CrossRef]
- Gelbenegger, G.; Schoergenhofer, C.; Derhaschnig, U.; Buchtele, N.; Sillaber, C.; Fillitz, M.; Schenk, T.M.; D’Sa, S.; Cartwright, R.; Gilbert, J.C.; et al. Inhibition of complement C1s in patients with cold agglutinin disease: Lessons learned from a named patient program. Blood Adv. 2020, 4, 997–1005. [Google Scholar] [CrossRef]
- Gelbenegger, G.; Jaeger, U.; Fillitz, M.; D’Sa, S.; Cartwright, R.; Shafer, F.; Wardecki, M.; Wang, J.; Schoergenhofer, C.; Jilma, B. Sustained sutimlimab response for 3 years in patients with cold agglutinin disease: A phase I, open-label, extension trial. Br. J. Haematol. 2022, 198, e59–e62. [Google Scholar] [CrossRef]
- Hillmen, P.; Szer, J.; Weitz, I.; Roth, A.; Hochsmann, B.; Panse, J.; Usuki, K.; Griffin, M.; Kiladjian, J.J.; de Castro, C.; et al. Pegcetacoplan versus Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. N. Engl. J. Med. 2021, 384, 1028–1037. [Google Scholar] [CrossRef]
- Grossi, F.; Shum, M.K.; Gertz, M.A.; Roman, E.; Deschatelets, P.; Hamdani, M.; Stout, F.; Francois, C.G. Inhibition of C3 with APL-2 results in normalisation of markers of intravascular and extravascular hemolysis in patients with autoimmune hemolytic anemia (AIHA). Blood 2018, 132, 3623. [Google Scholar] [CrossRef]
- Grossi, F.V.; Bedwell, P.; Deschatelets, P.; Edis, L.; Francois, C.G.; Johnson, P.J.; Richardson, H.J.; Tan, L.; Vega, C.A.; Lickliter, J. APL-2, a complement C3 inhibitor for the potential treatment of paroxysmal nocturnal hemoglobinuria (PNH): Phase I data from two completed studies in healthy volunteers. Blood 2016, 128, 1251. [Google Scholar] [CrossRef]
- Aggarwal, R.; Sestak, A.L.; D’Souza, A.; Dillon, S.P.; Namjou, B.; Scofield, R.H. Complete complement deficiency in a large cohort of familial systemic lupus erythematosus. Lupus 2010, 19, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.; Padmanabhan, A.; Aqui, N.; Balogun, R.A.; Connelly-Smith, L.; Delaney, M.; Dunbar, N.M.; Witt, V.; Wu, Y.; Shaz, B.H. Guidelines on the Use of Therapeutic Apheresis in Clinical Practice-Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Seventh Special Issue. J. Clin. Apher. 2016, 31, 149–162. [Google Scholar] [CrossRef]
- Von Baeyer, H. Plasmapheresis in immune hematology: Review of clinical outcome data with respect to evidence-based medicine and clinical experience. Ther. Apher. Dial. 2003, 7, 127–140. [Google Scholar] [CrossRef]
- Berentsen, S. How I manage patients with cold agglutinin disease. Br. J. Haematol. 2018, 181, 320–330. [Google Scholar] [CrossRef]
- Liu, Y.K. Folic acid deficiency in sickle cell anaemia. Scand. J. Haematol. 1975, 14, 71–79. [Google Scholar] [CrossRef]
- Fattizzo, B.; Michel, M.; Zaninoni, A.; Giannotta, J.; Guillet, S.; Frederiksen, H.; Vos, J.M.I.; Mauro, F.R.; Jilma, B.; Patriarca, A.; et al. Efficacy of recombinant erythropoietin in autoimmune hemolytic anemia: A multicenter international study. Haematologica 2021, 106, 622–625. [Google Scholar] [CrossRef]
- Salama, A.; Hartnack, D.; Lindemann, H.W.; Lange, H.J.; Rummel, M.; Loew, A. The effect of erythropoiesis-stimulating agents in patients with therapy-refractory autoimmune hemolytic anemia. Transfus. Med. Hemother. 2014, 41, 462–468. [Google Scholar] [CrossRef]
- Zaninoni, A.; Giannotta, J.; Galli, A.; Artuso, R.; Bianchi, P.; Malcovati, L.; Barcellini, W.; Fattizzo, B. The Immunomodulatory Effect and Clinical Efficacy of Daratumumab in a Patient with Cold Agglutinin Disease. Front. Immunol. 2021, 12, 649441. [Google Scholar] [CrossRef]
- Lansita, J.A.; Mease, K.M.; Qiu, H.; Yednock, T.; Sankaranarayanan, S.; Kramer, S. Nonclinical Development of ANX005: A Humanized Anti-C1q Antibody for Treatment of Autoimmune and Neurodegenerative Diseases. Int. J. Toxicol. 2017, 36, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Qiu, H.; Kendall, L.; Saltarelli, M.; Yednock, T.; Sankaranarayanan, S. ANX005, an Inhibitory Antibody Against C1q, Blocks Complement Activation Triggered by Cold Agglutinins in Human Disease. Blood 2016, 128, 1265. [Google Scholar] [CrossRef]
- Van de Walle, I.; Silence, K.; Budding, K.; Van de Ven, L.; Dijkxhoorn, K.; de Zeeuw, E.; Yildiz, C.; Gabriels, S.; Percier, J.M.; Wildemann, J.; et al. ARGX-117, a therapeutic complement inhibiting antibody targeting C2. J. Allergy Clin. Immunol. 2021, 147, 1420–1429.e7. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | CAD | LPL |
|---|---|---|
| Growth pattern | Small nodules, no diffuse infiltrates or intrasinusoidal growth | Nodular and/or diffuse |
| Location | Intertrabecular infiltrates | Paratrabecular/intertrabecular infiltrates |
| Clonal cells | Small B-cells and usually plasma cells | Small B-cells, lymphoplasmacytoid cells, plasma cells |
| Plasma cells | Widely spread in the parenchyma | Admixed in the infiltrates |
| Mast cells | No mast cell infiltration | Mast cell infiltration present |
| Level | Criteria | Procedures and Comments |
|---|---|---|
| Required for diagnosis | Chronic hemolysis | As assessed by high bilirubin, low haptoglobin, high LDH, and, often, high absolute reticulocyte count |
| Monospecific DAT strongly positive for C3d | DAT is usually negative for IgG, but occasionally weakly positive | |
| Cold agglutinin titer ≥ 64 at 4 °C | Specimen must be kept at 37–38 °C from sampling until plasma/serum has been removed from the cells/clot | |
| No overt malignant disease or relevant infection | Clinical assessment for malignancy. Radiology as required. Exclude recent infection with Mycoplasma or Epstein–Barr virus | |
| Confirmative criteria not required for diagnosis | Monoclonal IgMκ in plasma/serum (or, rarely, IgG or λ phenotype) | Specimen must be kept at 37–38 °C from sampling until plasma/serum has been removed from the cells/clot |
| Ratio between κ- and λ-positive B-cells > 3.5 (or, rarely, <0.9) | Flow cytometry in bone marrow aspirate | |
| Cold agglutinin-associated lymphoproliferative disorder by histopathology | Bone marrow biopsy | |
| MYD88 L265P mutation not found | As assessed in bone marrow |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berentsen, S.; D’Sa, S.; Randen, U.; Małecka, A.; Vos, J.M.I. Cold Agglutinin Disease: Improved Understanding of Pathogenesis Helps Define Targets for Therapy. Hemato 2022, 3, 574-594. https://doi.org/10.3390/hemato3040040
Berentsen S, D’Sa S, Randen U, Małecka A, Vos JMI. Cold Agglutinin Disease: Improved Understanding of Pathogenesis Helps Define Targets for Therapy. Hemato. 2022; 3(4):574-594. https://doi.org/10.3390/hemato3040040
Chicago/Turabian StyleBerentsen, Sigbjørn, Shirley D’Sa, Ulla Randen, Agnieszka Małecka, and Josephine M. I. Vos. 2022. "Cold Agglutinin Disease: Improved Understanding of Pathogenesis Helps Define Targets for Therapy" Hemato 3, no. 4: 574-594. https://doi.org/10.3390/hemato3040040
APA StyleBerentsen, S., D’Sa, S., Randen, U., Małecka, A., & Vos, J. M. I. (2022). Cold Agglutinin Disease: Improved Understanding of Pathogenesis Helps Define Targets for Therapy. Hemato, 3(4), 574-594. https://doi.org/10.3390/hemato3040040