
The Era of Genomic Research for Lymphoma: Looking Back and Forward


Abstract
:1. Introduction
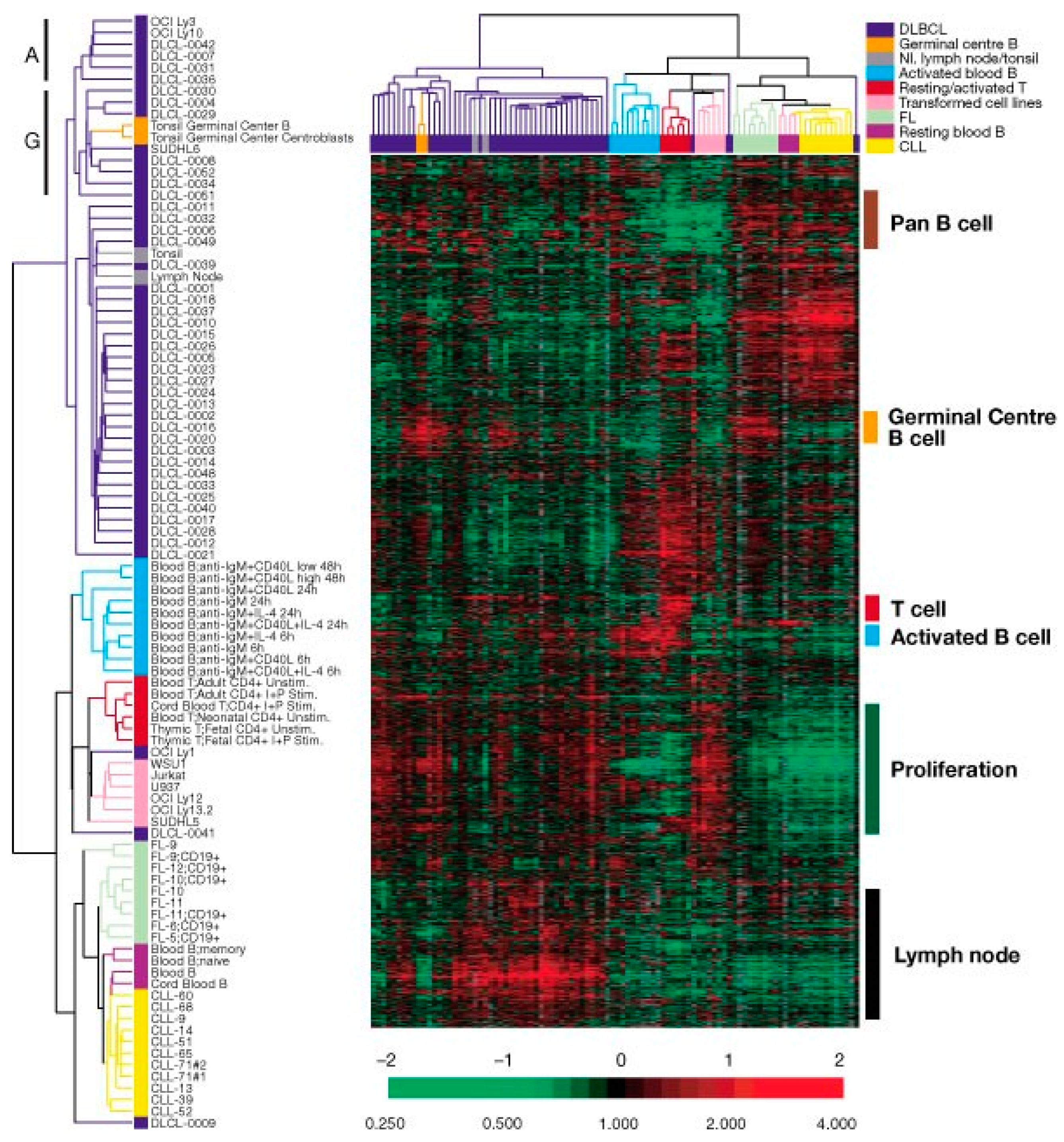
2. Gene Expression Profiling (GEP) Analysis
2.1. Diffuse Large B-Cell Lymphomas (DLBCLs)
2.2. Other B-Cell Lymphomas
2.3. Follicular Lymphoma (FL) and Transformed FL (t-FL)
2.4. Peripheral T-Cell Lymphoma (PTCL)
3. Global Genetic Analysis
3.1. The Study of Genomic Copy Number Abnormalities (gCNAs)
3.2. Mutation Analysis: Example on DLBCL
3.3. Mutation and gCNA Analyses: Peripheral T-Cell Lymphoma
3.4. Cooperativity of Genetic Alterations
4. The Integration of Multiomics Data
5. The Tumor Microenvironment
6. A New Diagnostic Platform
7. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saluz, H.P.; Iqbal, J.; Gino, V.L.; Andre, R.; Wu, Z. Fundamentals of DNA-chip/array technology for comparative gene-expression analysis. Curr. Sci. 2002, 83, 829–833. [Google Scholar]
- Freeman, W.M.; Robertson, D.J.; Vrana, K.E. Fundamentals of DNA hybridization arrays for gene expression analysis. Biotechniques 2000, 29, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, D.J.; Dong, H.; Byrne, M.C.; Follettie, M.T.; Gallo, M.V.; Chee, M.S.; Mittmann, M.; Wang, C.; Kobayashi, M.; Horton, H.; et al. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat. Biotechnol. 1996, 14, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Draghici, S.; Khatri, P.; Eklund, A.C.; Szallasi, Z. Reliability and reproducibility issues in DNA microarray measurements. Trends Genet. 2006, 22, 101–109. [Google Scholar] [CrossRef] [PubMed]
- van Hijum, S.A.; de Jong, A.; Baerends, R.J.; Karsens, H.A.; Kramer, N.E.; Larsen, R.; den Hengst, C.D.; Albers, C.J.; Kok, J.; Kuipers, O.P. A generally applicable validation scheme for the assessment of factors involved in reproducibility and quality of DNA-microarray data. BMC Genom. 2005, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- McShane, L.M.; Radmacher, M.D.; Freidlin, B.; Yu, R.; Li, M.C.; Simon, R. Methods for assessing reproducibility of clustering patterns observed in analyses of microarray data. Bioinformatics 2002, 18, 1462–1469. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Hsueh, H.M.; Delongchamp, R.R.; Lin, C.J.; Tsai, C.A. Reproducibility of microarray data: A further analysis of microarray quality control (MAQC) data. BMC Bioinform. 2007, 8, 412. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.V.; Chen, E.; Hasseman, J.P.; Liang, W.; Frank, B.C.; Wang, S.; Sharov, V.; Saeed, A.I.; White, J.; Li, J.; et al. Within the fold: Assessing differential expression measures and reproducibility in microarray assays. Genome Biol. 2002, 3, research0062. [Google Scholar] [PubMed]
- Iqbal, J.; d’Amore, F.; Hu, Q.; Chan, W.C.; Fu, K. Gene arrays in lymphoma: Where will they fit in? Curr. Hematol. Malig. Rep. 2006, 1, 129–136. [Google Scholar] [CrossRef]
- Iqbal, J.; Liu, Z.; Deffenbacher, K.; Chan, W.C. Gene expression profiling in lymphoma diagnosis and management. Best Pract. Res. Clin. Haematol. 2009, 22, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed]
- Schneider, V.A.; Graves-Lindsay, T.; Howe, K.; Bouk, N.; Chen, H.C.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-Nissen, F.; Albracht, D.; et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef]
- Carter, N.P. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat. Genet. 2007, 39, S16–S21. [Google Scholar] [CrossRef]
- Coughlin, C.R.; Scharer, G.H., 2nd; Shaikh, T.H. Clinical impact of copy number variation analysis using high-resolution microarray technologies: Advantages, limitations and concerns. Genome Med. 2012, 4, 80. [Google Scholar] [CrossRef]
- Zhang, F.; Gu, W.; Hurles, M.E.; Lupski, J.R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genom. Hum. Genet. 2009, 10, 451–481. [Google Scholar] [CrossRef]
- McCarroll, S.A.; Altshuler, D.M. Copy-number variation and association studies of human disease. Nat. Genet. 2007, 39, S37–S42. [Google Scholar] [CrossRef]
- Hinds, D.A.; Kloek, A.P.; Jen, M.; Chen, X.; Frazer, K.A. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat. Genet. 2006, 38, 82–85. [Google Scholar] [CrossRef]
- Chee, M.; Yang, R.; Hubbell, E.; Berno, A.; Huang, X.C.; Stern, D.; Winkler, J.; Lockhart, D.J.; Morris, M.S.; Fodor, S.P. Accessing genetic information with high-density DNA arrays. Science 1996, 274, 610–614. [Google Scholar] [CrossRef]
- Lashkari, D.A.; DeRisi, J.L.; McCusker, J.H.; Namath, A.F.; Gentile, C.; Hwang, S.Y.; Brown, P.O.; Davis, R.W. Yeast microarrays for genome wide parallel genetic and gene expression analysis. Proc. Natl. Acad. Sci. USA 1997, 94, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.; Schwager, C.; Hentze, S.; Ansorge, W.; Hentze, M.W.; Muckenthaler, M. Comparison of fluorescent tag DNA labeling methods used for expression analysis by DNA microarrays. Biotechniques 2002, 33, 620–628. [Google Scholar] [CrossRef]
- DeRisi, J.; Penland, L.; Bittner, M.; Meltzer, P.; Ray, M.; Chen, Y.; Su, Y.; Trent, J. Use of a cDNA microarray to analyse gene expression. Nat. Genet. 1996, 14, 457–460. [Google Scholar] [PubMed]
- Diehl, F.; Grahlmann, S.; Beier, M.; Hoheisel, J.D. Manufacturing DNA microarrays of high spot homogeneity and reduced background signal. Nucleic Acids Res. 2001, 29, E38. [Google Scholar] [CrossRef] [PubMed]
- Lipshutz, R.J.; Fodor, S.P.; Gingeras, T.R.; Lockhart, D.J. High density synthetic oligonucleotide arrays. Nat. Genet. 1999, 21, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947. [Google Scholar] [CrossRef]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef]
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 2001, 194, 1861–1874. [Google Scholar] [CrossRef]
- Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; Gascoyne, R.D.; Delabie, J.; Ott, G.; Muller-Hermelink, H.K.; Campo, E.; Braziel, R.M.; Jaffe, E.S.; et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004, 103, 275–282. [Google Scholar] [CrossRef]
- Choi, W.W.; Weisenburger, D.D.; Greiner, T.C.; Piris, M.A.; Banham, A.H.; Delabie, J.; Braziel, R.M.; Geng, H.; Iqbal, J.; Lenz, G.; et al. A new immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy. Clin. Cancer Res. 2009, 15, 5494–5502. [Google Scholar] [CrossRef]
- Meyer, P.N.; Fu, K.; Greiner, T.C.; Smith, L.M.; Delabie, J.; Gascoyne, R.D.; Ott, G.; Rosenwald, A.; Braziel, R.M.; Campo, E.; et al. Immunohistochemical methods for predicting cell of origin and survival in patients with diffuse large B-cell lymphoma treated with rituximab. J. Clin. Oncol. 2011, 29, 200–207. [Google Scholar] [CrossRef]
- Scott, D.W.; Wright, G.W.; Williams, P.M.; Lih, C.J.; Walsh, W.; Jaffe, E.S.; Rosenwald, A.; Campo, E.; Chan, W.C.; Connors, J.M.; et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 2014, 123, 1214–1217. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Shen, Y.; Huang, X.; Liu, Y.; Wake, L.; Liu, C.; Deffenbacher, K.; Lachel, C.M.; Wang, C.; Rohr, J.; et al. Global microRNA expression profiling uncovers molecular markers for classification and prognosis in aggressive B-cell lymphoma. Blood 2015, 125, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.L.; Trinh, D.L.; Scott, D.W.; Chu, A.; Krzywinski, M.; Zhao, Y.; Robertson, A.G.; Mungall, A.J.; Schein, J.; Boyle, M.; et al. Comprehensive miRNA sequence analysis reveals survival differences in diffuse large B-cell lymphoma patients. Genome Biol. 2015, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Xu, Y.; Zhang, J.; Zhang, P.; Yao, Z.; Yan, Z.; Wang, H.; Chu, J.; Yao, S.; Zhao, S.; et al. MiRNA-363-3p/DUSP10/JNK axis mediates chemoresistance by enhancing DNA damage repair in diffuse large B-cell lymphoma. Leukemia 2022, 36, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.; Savage, K.J.; Kutok, J.L.; Feuerhake, F.; Kurtin, P.; Mihm, M.; Wu, B.; Pasqualucci, L.; Neuberg, D.; Aguiar, R.C.; et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005, 105, 1851–1861. [Google Scholar] [CrossRef]
- Rosenwald, A.; Wright, G.; Leroy, K.; Yu, X.; Gaulard, P.; Gascoyne, R.D.; Chan, W.C.; Zhao, T.; Haioun, C.; Greiner, T.C.; et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med. 2003, 198, 851–862. [Google Scholar] [CrossRef]
- Savage, K.J.; Monti, S.; Kutok, J.L.; Cattoretti, G.; Neuberg, D.; De Leval, L.; Kurtin, P.; Dal Cin, P.; Ladd, C.; Feuerhake, F.; et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood 2003, 102, 3871–3879. [Google Scholar] [CrossRef] [PubMed]
- Vigano, E.; Gunawardana, J.; Mottok, A.; Van Tol, T.; Mak, K.; Chan, F.C.; Chong, L.; Chavez, E.; Woolcock, B.; Takata, K.; et al. Somatic IL4R mutations in primary mediastinal large B-cell lymphoma lead to constitutive JAK-STAT signaling activation. Blood 2018, 131, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Twa, D.D.; Chan, F.C.; Ben-Neriah, S.; Woolcock, B.W.; Mottok, A.; Tan, K.L.; Slack, G.W.; Gunawardana, J.; Lim, R.S.; McPherson, A.W.; et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood 2014, 123, 2062–2065. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Wright, G.; Rosenwald, A.; Steidl, C.; Gascoyne, R.D.; Connors, J.M.; Mottok, A.; Weisenburger, D.D.; Greiner, T.C.; Fu, K.; et al. Identification of Primary Mediastinal Large B-cell Lymphoma at Nonmediastinal Sites by Gene Expression Profiling. Am. J. Surg. Pathol. 2015, 39, 1322–1330. [Google Scholar] [CrossRef]
- Klein, U.; Gloghini, A.; Gaidano, G.; Chadburn, A.; Cesarman, E.; Dalla-Favera, R.; Carbone, A. Gene expression profile analysis of AIDS-related primary effusion lymphoma (PEL) suggests a plasmablastic derivation and identifies PEL-specific transcripts. Blood 2003, 101, 4115–4121. [Google Scholar] [CrossRef]
- Fan, W.; Bubman, D.; Chadburn, A.; Harrington, W.J., Jr.; Cesarman, E.; Knowles, D.M. Distinct subsets of primary effusion lymphoma can be identified based on their cellular gene expression profile and viral association. J. Virol. 2005, 79, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Yamaguchi, M.; Kim, S.; Morikawa, J.; Ogawa, S.; Ueno, S.; Suh, E.; Dougherty, E.; Shmulevich, I.; Shiku, H.; et al. Microarray reveals differences in both tumors and vascular specific gene expression in de novo CD5+ and CD5− diffuse large B-cell lymphomas. Cancer Res. 2003, 63, 60–66. [Google Scholar]
- Karnan, S.; Tagawa, H.; Suzuki, R.; Suguro, M.; Yamaguchi, M.; Okamoto, M.; Morishima, Y.; Nakamura, S.; Seto, M. Analysis of chromosomal imbalances in de novo CD5-positive diffuse large-B-cell lymphoma detected by comparative genomic hybridization. Genes Chromosomes Cancer 2004, 39, 77–81. [Google Scholar] [CrossRef]
- Jardin, F. Next generation sequencing and the management of diffuse large B-cell lymphoma: From whole exome analysis to targeted therapy. Discov. Med. 2014, 18, 51–65. [Google Scholar]
- Choi, J.W.; Kim, Y.; Lee, J.H.; Kim, Y.S. MYD88 expression and L265P mutation in diffuse large B-cell lymphoma. Hum. Pathol. 2013, 44, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Ramis-Zaldivar, J.E.; Gonzalez-Farre, B.; Nicolae, A.; Pack, S.; Clot, G.; Nadeu, F.; Mottok, A.; Horn, H.; Song, J.Y.; Fu, K.; et al. MAPK and JAK-STAT pathways dysregulation in plasmablastic lymphoma. Haematologica 2021, 106, 2682–2693. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.K.; Hoang, T.; Law, S.C.; Brosda, S.; O’Rourke, K.; Tobin, J.W.D.; Vari, F.; Murigneux, V.; Fink, L.; Gunawardana, J.; et al. EBV-associated primary CNS lymphoma occurring after immunosuppression is a distinct immunobiological entity. Blood 2021, 137, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Pham-Ledard, A.; Prochazkova-Carlotti, M.; Andrique, L.; Cappellen, D.; Vergier, B.; Martinez, F.; Grange, F.; Petrella, T.; Beylot-Barry, M.; Merlio, J.P. Multiple genetic alterations in primary cutaneous large B-cell lymphoma, leg type support a common lymphomagenesis with activated B-cell-like diffuse large B-cell lymphoma. Mod. Pathol. 2014, 27, 402–411. [Google Scholar] [CrossRef]
- Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; Chan, W.C.; Aoun, P.; Cochran, G.T.; Pan, Z.; Smith, L.M.; Lynch, J.C.; Bociek, R.G.; et al. Expression of PKC-beta or cyclin D2 predicts for inferior survival in diffuse large B-cell lymphoma. Mod. Pathol. 2005, 18, 1377–1384. [Google Scholar] [CrossRef]
- Perry, A.M.; Mitrovic, Z.; Chan, W.C. Biological prognostic markers in diffuse large B-cell lymphoma. Cancer Control 2012, 19, 214–226. [Google Scholar] [CrossRef]
- Young, K.H.; Leroy, K.; Moller, M.B.; Colleoni, G.W.; Sanchez-Beato, M.; Kerbauy, F.R.; Haioun, C.; Eickhoff, J.C.; Young, A.H.; Gaulard, P.; et al. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: An international collaborative study. Blood 2008, 112, 3088–3098. [Google Scholar] [CrossRef]
- Iqbal, J.; Meyer, P.N.; Smith, L.M.; Johnson, N.A.; Vose, J.M.; Greiner, T.C.; Connors, J.M.; Staudt, L.M.; Rimsza, L.; Jaffe, E.; et al. BCL2 predicts survival in germinal center B-cell-like diffuse large B-cell lymphoma treated with CHOP-like therapy and rituximab. Clin. Cancer Res. 2011, 17, 7785–7795. [Google Scholar] [CrossRef]
- Iqbal, J.; Neppalli, V.T.; Wright, G.; Dave, B.J.; Horsman, D.E.; Rosenwald, A.; Lynch, J.; Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; et al. BCL2 expression is a prognostic marker for the activated B-cell-like type of diffuse large B-cell lymphoma. J. Clin. Oncol. 2006, 24, 961–968. [Google Scholar] [CrossRef]
- Fu, K.; Weisenburger, D.D.; Choi, W.W.; Perry, K.D.; Smith, L.M.; Shi, X.; Hans, C.P.; Greiner, T.C.; Bierman, P.J.; Bociek, R.G.; et al. Addition of rituximab to standard chemotherapy improves the survival of both the germinal center B-cell-like and non-germinal center B-cell-like subtypes of diffuse large B-cell lymphoma. J. Clin. Oncol. 2008, 26, 4587–4594. [Google Scholar] [CrossRef]
- Rosenwald, A.; Wright, G.; Wiestner, A.; Chan, W.C.; Connors, J.M.; Campo, E.; Gascoyne, R.D.; Grogan, T.M.; Muller-Hermelink, H.K.; Smeland, E.B.; et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003, 3, 185–197. [Google Scholar] [CrossRef]
- Fu, K.; Weisenburger, D.D.; Greiner, T.C.; Dave, S.; Wright, G.; Rosenwald, A.; Chiorazzi, M.; Iqbal, J.; Gesk, S.; Siebert, R.; et al. Cyclin D1-negative mantle cell lymphoma: A clinicopathologic study based on gene expression profiling. Blood 2005, 106, 4315–4321. [Google Scholar] [CrossRef]
- Salaverria, I.; Royo, C.; Carvajal-Cuenca, A.; Clot, G.; Navarro, A.; Valera, A.; Song, J.Y.; Woroniecka, R.; Rymkiewicz, G.; Klapper, W.; et al. CCND2 rearrangements are the most frequent genetic events in cyclin D1(-) mantle cell lymphoma. Blood 2013, 121, 1394–1402. [Google Scholar] [CrossRef]
- Mozos, A.; Royo, C.; Hartmann, E.; De Jong, D.; Baro, C.; Valera, A.; Fu, K.; Weisenburger, D.D.; Delabie, J.; Chuang, S.S.; et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica 2009, 94, 1555–1562. [Google Scholar] [CrossRef]
- Scott, D.W.; Abrisqueta, P.; Wright, G.W.; Slack, G.W.; Mottok, A.; Villa, D.; Jares, P.; Rauert-Wunderlich, H.; Royo, C.; Clot, G.; et al. New Molecular Assay for the Proliferation Signature in Mantle Cell Lymphoma Applicable to Formalin-Fixed Paraffin-Embedded Biopsies. J. Clin. Oncol. 2017, 35, 1668–1677. [Google Scholar] [CrossRef]
- Clot, G.; Jares, P.; Gine, E.; Navarro, A.; Royo, C.; Pinyol, M.; Martin-Garcia, D.; Demajo, S.; Espinet, B.; Salar, A.; et al. A gene signature that distinguishes conventional and leukemic nonnodal mantle cell lymphoma helps predict outcome. Blood 2018, 132, 413–422. [Google Scholar] [CrossRef]
- Iqbal, J.; Shen, Y.; Liu, Y.; Fu, K.; Jaffe, E.S.; Liu, C.; Liu, Z.; Lachel, C.M.; Deffenbacher, K.; Greiner, T.C.; et al. Genome-wide miRNA profiling of mantle cell lymphoma reveals a distinct subgroup with poor prognosis. Blood 2012, 119, 4939–4948. [Google Scholar] [CrossRef]
- Sohani, A.R.; Hasserjian, R.P. Diagnosis of Burkitt Lymphoma and Related High-Grade B-Cell Neoplasms. Surg. Pathol. Clin. 2010, 3, 1035–1059. [Google Scholar] [CrossRef]
- Dave, S.S.; Fu, K.; Wright, G.W.; Lam, L.T.; Kluin, P.; Boerma, E.J.; Greiner, T.C.; Weisenburger, D.D.; Rosenwald, A.; Ott, G.; et al. Molecular diagnosis of Burkitt’s lymphoma. N. Engl. J. Med. 2006, 354, 2431–2442. [Google Scholar] [CrossRef]
- Hummel, M.; Bentink, S.; Berger, H.; Klapper, W.; Wessendorf, S.; Barth, T.F.; Bernd, H.W.; Cogliatti, S.B.; Dierlamm, J.; Feller, A.C.; et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N. Engl. J. Med. 2006, 354, 2419–2430. [Google Scholar] [CrossRef]
- Victora, G.D.; Dominguez-Sola, D.; Holmes, A.B.; Deroubaix, S.; Dalla-Favera, R.; Nussenzweig, M.C. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood 2012, 120, 2240–2248. [Google Scholar] [CrossRef]
- Bouska, A.; Bagvati, S.; Iqbal, J.; William, B.; Chan, W. Follicular Lymphoma: Recent Advances. In Cancer Growth and Progression; Springer: Berlin/Heidelberg, Germany, 2012; pp. 21–42. [Google Scholar]
- Glas, A.M.; Kersten, M.J.; Delahaye, L.J.; Witteveen, A.T.; Kibbelaar, R.E.; Velds, A.; Wessels, L.F.; Joosten, P.; Kerkhoven, R.M.; Bernards, R. Gene expression profiling in follicular lymphoma to assess clinical aggressiveness and to guide the choice of treatment. Blood 2005, 105, 301–307. [Google Scholar] [CrossRef]
- Nann, D.; Ramis-Zaldivar, J.E.; Muller, I.; Gonzalez-Farre, B.; Schmidt, J.; Egan, C.; Salmeron-Villalobos, J.; Clot, G.; Mattern, S.; Otto, F.; et al. Follicular lymphoma t(14; 18)-negative is genetically a heterogeneous disease. Blood Adv. 2020, 4, 5652–5665. [Google Scholar] [CrossRef]
- Leich, E.; Salaverria, I.; Bea, S.; Zettl, A.; Wright, G.; Moreno, V.; Gascoyne, R.D.; Chan, W.C.; Braziel, R.M.; Rimsza, L.M.; et al. Follicular lymphomas with and without translocation t(14;18) differ in gene expression profiles and genetic alterations. Blood 2009, 114, 826–834. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification: Pathology and Genetics of tumors of Haematopoietic and Lymphoid Tissues, 4th ed.; WHO, Ed.; IARC Press: Lyon, France, 2008. [Google Scholar]
- Dave, S.S.; Wright, G.; Tan, B.; Rosenwald, A.; Gascoyne, R.D.; Chan, W.C.; Fisher, R.I.; Braziel, R.M.; Rimsza, L.M.; Grogan, T.M.; et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N. Engl. J. Med. 2004, 351, 2159–2169. [Google Scholar] [CrossRef]
- Cerhan, J.R.; Wang, S.; Maurer, M.J.; Ansell, S.M.; Geyer, S.M.; Cozen, W.; Morton, L.M.; Davis, S.; Severson, R.K.; Rothman, N. Prognostic significance of host immune gene polymorphisms in follicular lymphoma survival. Blood 2007, 109, 5439–5446. [Google Scholar] [CrossRef]
- Huet, S.; Tesson, B.; Jais, J.P.; Feldman, A.L.; Magnano, L.; Thomas, E.; Traverse-Glehen, A.; Albaud, B.; Carrere, M.; Xerri, L.; et al. A gene-expression profiling score for prediction of outcome in patients with follicular lymphoma: A retrospective training and validation analysis in three international cohorts. Lancet Oncol. 2018, 19, 549–561. [Google Scholar] [CrossRef]
- Wang, W.; Corrigan-Cummins, M.; Hudson, J.; Maric, I.; Simakova, O.; Neelapu, S.S.; Kwak, L.W.; Janik, J.E.; Gause, B.; Jaffe, E.S.; et al. MicroRNA profiling of follicular lymphoma identifies microRNAs related to cell proliferation and tumor response. Haematologica 2012, 97, 586–594. [Google Scholar] [CrossRef]
- Leich, E.; Zamo, A.; Horn, H.; Haralambieva, E.; Puppe, B.; Gascoyne, R.D.; Chan, W.C.; Braziel, R.M.; Rimsza, L.M.; Weisenburger, D.D.; et al. MicroRNA profiles of t(14; 18)-negative follicular lymphoma support a late germinal center B-cell phenotype. Blood 2011, 118, 5550–5558. [Google Scholar] [CrossRef]
- Rudiger, T.; Weisenburger, D.D.; Anderson, J.R.; Armitage, J.O.; Diebold, J.; MacLennan, K.A.; Nathwani, B.N.; Ullrich, F.; Muller-Hermelink, H.K.; Non-Hodgkin’s Lymphoma Classification Project. Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): Results from the Non-Hodgkin’s Lymphoma Classification Project. Ann. Oncol. 2002, 13, 140–149. [Google Scholar] [CrossRef]
- Bellei, M.; Chiattone, C.S.; Luminari, S.; Pesce, E.A.; Cabrera, M.E.; de Souza, C.A.; Gabus, R.; Zoppegno, L.; Zoppegno, L.; Milone, J.; et al. T-cell lymphomas in South america and europe. Rev. Bras. Hematol. Hemoter. 2012, 34, 42–47. [Google Scholar] [CrossRef]
- Briski, R.; Feldman, A.L.; Bailey, N.G.; Lim, M.S.; Ristow, K.; Habermann, T.M.; Macon, W.R.; Inwards, D.J.; Colgan, J.P.; Nowakowski, G.S.; et al. The role of front-line anthracycline-containing chemotherapy regimens in peripheral T-cell lymphomas. Blood Cancer J. 2014, 4, e214. [Google Scholar] [CrossRef]
- Vose, J.; Armitage, J.; Weisenburger, D.; International TCLP. International peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes. J. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar] [PubMed]
- Sabattini, E.; Bacci, F.; Sagramoso, C.; Pileri, S.A. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: An overview. Pathologica 2010, 102, 83–87. [Google Scholar]
- Herek, T.A.; Iqbal, J. Molecular Classification of the Peripheral T-cell Lymphomas. In The Peripheral T-Cell Lymphomas; Wiley: New York, NY, USA, 2021; pp. 91–103. [Google Scholar]
- Cuadros, M.; Dave, S.S.; Jaffe, E.S.; Honrado, E.; Milne, R.; Alves, J.; Rodriguez, J.; Zajac, M.; Benitez, J.; Staudt, L.M.; et al. Identification of a proliferation signature related to survival in nodal peripheral T-cell lymphomas. J. Clin. Oncol. 2007, 25, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Yamaguchi, M.; Imai, H.; Kobayashi, T.; Tamaru, S.; Nishii, K.; Yuda, M.; Shiku, H.; Katayama, N. Gene expression profiling of peripheral T-cell lymphoma including gammadelta T-cell lymphoma. Blood 2009, 113, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Ballester, B.; Ramuz, O.; Gisselbrecht, C.; Doucet, G.; Loi, L.; Loriod, B.; Bertucci, F.; Bouabdallah, R.; Devilard, E.; Carbuccia, N.; et al. Gene expression profiling identifies molecular subgroups among nodal peripheral T-cell lymphomas. Oncogene 2006, 25, 1560–1570. [Google Scholar] [CrossRef]
- Huang, Y.; de Reynies, A.; de Leval, L.; Ghazi, B.; Martin-Garcia, N.; Travert, M.; Bosq, J.; Briere, J.; Petit, B.; Thomas, E.; et al. Gene expression profiling identifies emerging oncogenic pathways operating in extranodal NK/T-cell lymphoma, nasal-type. Blood 2009, 115, 1226–1237. [Google Scholar] [CrossRef]
- de Leval, L.; Rickman, D.S.; Thielen, C.; Reynies, A.; Huang, Y.L.; Delsol, G.; Lamant, L.; Leroy, K.; Briere, J.; Molina, T.; et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood 2007, 109, 4952–4963. [Google Scholar] [CrossRef] [PubMed]
- Piccaluga, P.P.; Agostinelli, C.; Califano, A.; Rossi, M.; Basso, K.; Zupo, S.; Went, P.; Klein, U.; Zinzani, P.L.; Baccarani, M.; et al. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. J. Clin. Investig. 2007, 117, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Piccaluga, P.P.; Agostinelli, C.; Califano, A.; Carbone, A.; Fantoni, L.; Ferrari, S.; Gazzola, A.; Gloghini, A.; Righi, S.; Rossi, M.; et al. Gene expression analysis of angioimmunoblastic lymphoma indicates derivation from T follicular helper cells and vascular endothelial growth factor deregulation. Cancer Res. 2007, 67, 10703–10710. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Weisenburger, D.D.; Chowdhury, A.; Tsai, M.Y.; Srivastava, G.; Greiner, T.C.; Kucuk, C.; Deffenbacher, K.; Vose, J.; Smith, L.; et al. Natural killer cell lymphoma shares strikingly similar molecular features with a group of non-hepatosplenic gammadelta T-cell lymphoma and is highly sensitive to a novel aurora kinase A inhibitor in vitro. Leukemia 2011, 25, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Weisenburger, D.D.; Greiner, T.C.; Vose, J.M.; McKeithan, T.; Kucuk, C.; Geng, H.; Deffenbacher, K.; Smith, L.; Dybkaer, K.; et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood 2010, 115, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Wright, G.; Wang, C.; Rosenwald, A.; Gascoyne, R.D.; Weisenburger, D.D.; Greiner, T.C.; Smith, L.; Guo, S.; Wilcox, R.A.; et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood 2014, 123, 2915–2923. [Google Scholar] [CrossRef]
- Tindemans, I.; Serafini, N.; Di Santo, J.P.; Hendriks, R.W. GATA-3 function in innate and adaptive immunity. Immunity 2014, 41, 191–206. [Google Scholar] [CrossRef]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef]
- Wang, T.; Feldman, A.L.; Wada, D.A.; Lu, Y.; Polk, A.; Briski, R.; Ristow, K.; Habermann, T.M.; Thomas, D.; Ziesmer, S.C.; et al. GATA-3 expression identifies a high-risk subset of PTCL, NOS with distinct molecular and clinical features. Blood 2014, 123, 3007–3015. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Paul, W.E. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 2010, 327, 1098–1102. [Google Scholar] [CrossRef]
- Wang, C.; McKeithan, T.W.; Gong, Q.; Zhang, W.; Bouska, A.; Rosenwald, A.; Gascoyne, R.D.; Wu, X.; Wang, J.; Muhammad, Z.; et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood 2015, 126, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Heavican, T.B.; Bouska, A.; Yu, J.; Lone, W.; Amador, C.; Gong, Q.; Zhang, W.; Li, Y.; Dave, B.J.; Nairismagi, M.L.; et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood 2019, 133, 1664–1676. [Google Scholar] [CrossRef] [PubMed]
- Lone, W.; Bouska, A.; Sharma, S.; Amador, C.; Saumyaranjan, M.; Herek, T.A.; Heavican, T.B.; Yu, J.; Lim, S.T.; Ong, C.K.; et al. Genome-Wide miRNA Expression Profiling of Molecular Subgroups of Peripheral T-cell Lymphoma. Clin. Cancer Res. 2021, 27, 6039–6053. [Google Scholar] [CrossRef]
- Amador, C.; Greiner, T.C.; Heavican, T.B.; Smith, L.M.; Galvis, K.T.; Lone, W.; Bouska, A.; D’Amore, F.; Pedersen, M.B.; Pileri, S.; et al. Reproducing the molecular subclassification of peripheral T-cell lymphoma-NOS by immunohistochemistry. Blood 2019, 134, 2159–2170. [Google Scholar] [CrossRef] [PubMed]
- Amador, C.; Bouska, A.; Wright, G.; Weisenburger, D.D.; Feldman, A.L.; Smith, L.; Greiner, T.C.; Pileri, S.T.; Abanelli, V.; Ott, G.; et al. Gene expression signatures for the accurate diagnosis of peripheral T-cell lymphoma entities in the routine clinical practice. J. Clin. Oncol. 2022; in print. [Google Scholar]
- Ng, S.B.; Selvarajan, V.; Huang, G.; Zhou, J.; Feldman, A.L.; Law, M.; Kwong, Y.L.; Shimizu, N.; Kagami, Y.; Aozasa, K.; et al. Activated oncogenic pathways and therapeutic targets in extranodal nasal-type NK/T cell lymphoma revealed by gene expression profiling. J. Pathol. 2011, 223, 496–510. [Google Scholar] [CrossRef]
- Iqbal, J.; Kucuk, C.; deLeeuw, R.J.; Srivastava, G.; Tam, W.; Geng, H.; Klinkebiel, D.; Christman, J.K.; Patel, K.; Cao, K.; et al. Genomic analyses reveal global functional alterations that promote tumor growth and novel tumor suppressor genes in natural killer-cell malignancies. Leukemia 2009, 23, 1139–1151. [Google Scholar] [CrossRef]
- Dufva, O.; Kankainen, M.; Kelkka, T.; Sekiguchi, N.; Awad, S.A.; Eldfors, S.; Yadav, B.; Kuusanmaki, H.; Malani, D.; Andersson, E.I.; et al. Aggressive natural killer-cell leukemia mutational landscape and drug profiling highlight JAK-STAT signaling as therapeutic target. Nat. Commun. 2018, 9, 1567. [Google Scholar] [CrossRef] [PubMed]
- Abate, F.; Todaro, M.; van der Krogt, J.A.; Boi, M.; Landra, I.; Machiorlatti, R.; Tabbo, F.; Messana, K.; Abele, C.; Barreca, A.; et al. A novel patient-derived tumorgraft model with TRAF1-ALK anaplastic large-cell lymphoma translocation. Leukemia 2015, 29, 1390–1401. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef]
- She, X.; Jiang, Z.; Clark, R.A.; Liu, G.; Cheng, Z.; Tuzun, E.; Church, D.M.; Sutton, G.; Halpern, A.L.; Eichler, E.E. Shotgun sequence assembly and recent segmental duplications within the human genome. Nature 2004, 431, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Wright, G.W.; Emre, N.C.; Kohlhammer, H.; Dave, S.S.; Davis, R.E.; Carty, S.; Lam, L.T.; Shaffer, A.L.; Xiao, W.; et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 13520–13525. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Hartert, K.; Tadros, S.; Fiskus, W.; Havranek, O.; Ma, M.C.J.; Bouska, A.; Heavican, T.; Kumar, D.; Deng, Q.; et al. Targetable genetic alterations of TCF4 (E2-2) drive immunoglobulin expression in diffuse large B cell lymphoma. Sci. Transl. Med. 2019, 11, eaav5599. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Sanger, W.G.; Horsman, D.E.; Rosenwald, A.; Pickering, D.L.; Dave, B.; Dave, S.; Xiao, L.; Cao, K.; Zhu, Q.; et al. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am. J. Pathol. 2004, 165, 159–166. [Google Scholar] [CrossRef]
- Iqbal, J.; Greiner, T.C.; Patel, K.; Dave, B.J.; Smith, L.; Ji, J.; Wright, G.; Sanger, W.G.; Pickering, D.L.; Jain, S.; et al. Distinctive patterns of BCL6 molecular alterations and their functional consequences in different subgroups of diffuse large B-cell lymphoma. Leukemia 2007, 21, 2332–2343. [Google Scholar] [CrossRef] [PubMed]
- Shaknovich, R.; Geng, H.; Johnson, N.A.; Tsikitas, L.; Cerchietti, L.; Greally, J.M.; Gascoyne, R.D.; Elemento, O.; Melnick, A. DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood 2010, 116, e81–e89. [Google Scholar] [CrossRef] [PubMed]
- Halldorsdottir, A.M.; Sander, B.; Goransson, H.; Isaksson, A.; Kimby, E.; Mansouri, M.; Rosenquist, R.; Ehrencrona, H. High-resolution genomic screening in mantle cell lymphoma—Specific changes correlate with genomic complexity, the proliferation signature and survival. Genes Chromosomes Cancer 2011, 50, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, E.M.; Campo, E.; Wright, G.; Lenz, G.; Salaverria, I.; Jares, P.; Xiao, W.; Braziel, R.M.; Rimsza, L.M.; Chan, W.C.; et al. Pathway discovery in mantle cell lymphoma by integrated analysis of high-resolution gene expression and copy number profiling. Blood 2010, 116, 953–961. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, R.J.; Davies, J.J.; Rosenwald, A.; Bebb, G.; Gascoyne, R.D.; Dyer, M.J.; Staudt, L.M.; Martinez-Climent, J.A.; Lam, W.L. Comprehensive whole genome array CGH profiling of mantle cell lymphoma model genomes. Hum. Mol. Genet. 2004, 13, 1827–1837. [Google Scholar] [CrossRef]
- Honma, K.; Tsuzuki, S.; Nakagawa, M.; Tagawa, H.; Nakamura, S.; Morishima, Y.; Seto, M. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood 2009, 114, 2467–2475. [Google Scholar] [CrossRef] [PubMed]
- Bea, S.; Valdes-Mas, R.; Navarro, A.; Salaverria, I.; Martin-Garcia, D.; Jares, P.; Gine, E.; Pinyol, M.; Royo, C.; Nadeu, F.; et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18250–18255. [Google Scholar] [CrossRef] [PubMed]
- Enjuanes, A.; Albero, R.; Clot, G.; Navarro, A.; Bea, S.; Pinyol, M.; Martin-Subero, J.I.; Klapper, W.; Staudt, L.M.; Jaffe, E.S.; et al. Genome-wide methylation analyses identify a subset of mantle cell lymphoma with a high number of methylated CpGs and aggressive clinicopathological features. Int. J. Cancer 2013, 133, 2852–2863. [Google Scholar]
- Leshchenko, V.V.; Kuo, P.Y.; Shaknovich, R.; Yang, D.T.; Gellen, T.; Petrich, A.; Yu, Y.; Remache, Y.; Weniger, M.A.; Rafiq, S.; et al. Genomewide DNA methylation analysis reveals novel targets for drug development in mantle cell lymphoma. Blood 2010, 116, 1025–1034. [Google Scholar] [CrossRef]
- Boerma, E.G.; Siebert, R.; Kluin, P.M.; Baudis, M. Translocations involving 8q24 in Burkitt lymphoma and other malignant lymphomas: A historical review of cytogenetics in the light of todays knowledge. Leukemia 2009, 23, 225–234. [Google Scholar] [CrossRef]
- Scholtysik, R.; Kreuz, M.; Klapper, W.; Burkhardt, B.; Feller, A.C.; Hummel, M.; Loeffler, M.; Rosolowski, M.; Schwaenen, C.; Spang, R.; et al. Detection of genomic aberrations in molecularly defined Burkitt’s lymphoma by array-based, high resolution, single nucleotide polymorphism analysis. Haematologica 2010, 95, 2047–2055. [Google Scholar] [CrossRef]
- Schiffman, J.D.; Lorimer, P.D.; Rodic, V.; Jahromi, M.S.; Downie, J.M.; Bayerl, M.G.; Sanmann, J.N.; Althof, P.A.; Sanger, W.G.; Barnette, P.; et al. Genome wide copy number analysis of paediatric Burkitt lymphoma using formalin-fixed tissues reveals a subset with gain of chromosome 13q and corresponding miRNA over expression. Br. J. Haematol. 2011, 155, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Salaverria, I.; Zettl, A.; Bea, S.; Hartmann, E.M.; Dave, S.S.; Wright, G.W.; Boerma, E.J.; Kluin, P.M.; Ott, G.; Chan, W.C.; et al. Chromosomal alterations detected by comparative genomic hybridization in subgroups of gene expression-defined Burkitt’s lymphoma. Haematologica 2008, 93, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Abate, F.; Ambrosio, M.R.; Mundo, L.; Laginestra, M.A.; Fuligni, F.; Rossi, M.; Zairis, S.; Gazaneo, S.; De Falco, G.; Lazzi, S.; et al. Distinct Viral and Mutational Spectrum of Endemic Burkitt Lymphoma. PLoS Pathog. 2015, 11, e1005158. [Google Scholar] [CrossRef] [PubMed]
- Navari, M.; Fuligni, F.; Laginestra, M.A.; Etebari, M.; Ambrosio, M.R.; Sapienza, M.R.; Rossi, M.; De Falco, G.; Gibellini, D.; Tripodo, C.; et al. Molecular signature of Epstein Barr virus-positive Burkitt lymphoma and post-transplant lymphoproliferative disorder suggest different roles for Epstein Barr virus. Front. Microbiol. 2014, 5, 728. [Google Scholar] [CrossRef]
- Grande, B.M.; Gerhard, D.S.; Jiang, A.; Griner, N.B.; Abramson, J.S.; Alexander, T.B.; Allen, H.; Ayers, L.W.; Bethony, J.M.; Bhatia, K.; et al. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood 2019, 133, 1313–1324. [Google Scholar] [CrossRef]
- Horsman, D.E.; Connors, J.M.; Pantzar, T.; Gascoyne, R.D. Analysis of secondary chromosomal alterations in 165 cases of follicular lymphoma with t(14;18). Genes Chromosomes Cancer 2001, 30, 375–382. [Google Scholar] [CrossRef]
- Cheung, K.J.; Delaney, A.; Ben-Neriah, S.; Schein, J.; Lee, T.; Shah, S.P.; Cheung, D.; Johnson, N.A.; Mungall, A.J.; Telenius, A.; et al. High resolution analysis of follicular lymphoma genomes reveals somatic recurrent sites of copy-neutral loss of heterozygosity and copy number alterations that target single genes. Genes Chromosomes Cancer 2010, 49, 669–681. [Google Scholar] [CrossRef]
- Ross, C.W.; Ouillette, P.D.; Saddler, C.M.; Shedden, K.A.; Malek, S.N. Comprehensive analysis of copy number and allele status identifies multiple chromosome defects underlying follicular lymphoma pathogenesis. Clin. Cancer Res. 2007, 13, 4777–4785. [Google Scholar] [CrossRef]
- Hoglund, M.; Sehn, L.; Connors, J.M.; Gascoyne, R.D.; Siebert, R.; Sall, T.; Mitelman, F.; Horsman, D.E. Identification of cytogenetic subgroups and karyotypic pathways of clonal evolution in follicular lymphomas. Genes Chromosomes Cancer 2004, 39, 195–204. [Google Scholar] [CrossRef]
- d’Amore, F.; Chan, E.; Iqbal, J.; Geng, H.; Young, K.; Xiao, L.; Hess, M.M.; Sanger, W.G.; Smith, L.; Wiuf, C.; et al. Clonal evolution in t(14;18)-positive follicular lymphoma, evidence for multiple common pathways, and frequent parallel clonal evolution. Clin Cancer Res. 2008, 14, 7180–7187. [Google Scholar] [CrossRef]
- Bouska, A.; McKeithan, T.W.; Deffenbacher, K.E.; Lachel, C.; Wright, G.W.; Iqbal, J.; Smith, L.M.; Zhang, W.; Kucuk, C.; Rinaldi, A.; et al. Genome-wide copy-number analyses reveal genomic abnormalities involved in transformation of follicular lymphoma. Blood 2014, 123, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Jaffe, E.S.; Longo, D.L.; Raffeld, M. MYC rearrangements in histologically progressed follicular lymphomas. Blood 1992, 80, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.A.; Yano, T.; Clark, H.M.; Harris, C.; Longo, D.L.; Jaffe, E.S.; Raffeld, M. p53 mutation is associated with progression in follicular lymphomas. Blood 1993, 82, 1994–2004. [Google Scholar] [CrossRef]
- Matolcsy, A.; Casali, P.; Warnke, R.A.; Knowles, D.M. Morphologic transformation of follicular lymphoma is associated with somatic mutation of the translocated Bcl-2 gene. Blood 1996, 88, 3937–3944. [Google Scholar] [CrossRef] [PubMed]
- Pinyol, M.; Cobo, F.; Bea, S.; Jares, P.; Nayach, I.; Fernandez, P.L.; Montserrat, E.; Cardesa, A.; Campo, E. p16(INK4a) gene inactivation by deletions, mutations, and hypermethylation is associated with transformed and aggressive variants of non-Hodgkin’s lymphomas. Blood 1998, 91, 2977–2984. [Google Scholar] [CrossRef]
- Martinez-Climent, J.A.; Alizadeh, A.A.; Segraves, R.; Blesa, D.; Rubio-Moscardo, F.; Albertson, D.G.; Garcia-Conde, J.; Dyer, M.J.; Levy, R.; Pinkel, D.; et al. Transformation of follicular lymphoma to diffuse large cell lymphoma is associated with a heterogeneous set of DNA copy number and gene expression alterations. Blood 2003, 101, 3109–3117. [Google Scholar] [CrossRef]
- Bouska, A.; Zhang, W.; Gong, Q.; Iqbal, J.; Scuto, A.; Vose, J.; Ludvigsen, M.; Fu, K.; Weisenburger, D.D.; Greiner, T.C.; et al. Combined copy number and mutation analysis identifies oncogenic pathways associated with transformation of follicular lymphoma. Leukemia 2017, 31, 83–91. [Google Scholar] [CrossRef]
- O’Riain, C.; O’Shea, D.M.; Yang, Y.; Le Dieu, R.; Gribben, J.G.; Summers, K.; Yeboah-Afari, J.; Bhaw-Rosun, L.; Fleischmann, C.; Mein, C.A.; et al. Array-based DNA methylation profiling in follicular lymphoma. Leukemia 2009, 23, 1858–1866. [Google Scholar] [CrossRef]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef]
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008, 319, 1676–1679. [Google Scholar] [CrossRef]
- Phelan, J.D.; Young, R.M.; Webster, D.E.; Roulland, S.; Wright, G.W.; Kasbekar, M.; Shaffer, A.L., 3rd; Ceribelli, M.; Wang, J.Q.; Schmitz, R.; et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 2018, 560, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Neumeister, P.; Goossens, T.; Nanjangud, G.; Chaganti, R.S.; Kuppers, R.; Dalla-Favera, R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 2001, 412, 341–346. [Google Scholar] [CrossRef]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494.e15. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jima, D.; Moffitt, A.B.; Liu, Q.; Czader, M.; Hsi, E.D.; Fedoriw, Y.; Dunphy, C.H.; Richards, K.L.; Gill, J.I.; et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood 2014, 123, 2988–2996. [Google Scholar] [CrossRef]
- Bodor, C.; Grossmann, V.; Popov, N.; Okosun, J.; O’Riain, C.; Tan, K.; Marzec, J.; Araf, S.; Wang, J.; Lee, A.M.; et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 2013, 122, 3165–3168. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Khiabanian, H.; Fangazio, M.; Vasishtha, M.; Messina, M.; Holmes, A.B.; Ouillette, P.; Trifonov, V.; Rossi, D.; Tabbo, F.; et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014, 6, 130–140. [Google Scholar] [CrossRef]
- Richter, J.; Schlesner, M.; Hoffmann, S.; Kreuz, M.; Leich, E.; Burkhardt, B.; Rosolowski, M.; Ammerpohl, O.; Wagener, R.; Bernhart, S.H.; et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [Google Scholar]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Arribas, A.J.; Campos-Martin, Y.; Gomez-Abad, C.; Algara, P.; Sanchez-Beato, M.; Rodriguez-Pinilla, M.S.; Montes-Moreno, S.; Martinez, N.; Alves-Ferreira, J.; Piris, M.A.; et al. Nodal marginal zone lymphoma: Gene expression and miRNA profiling identify diagnostic markers and potential therapeutic targets. Blood 2012, 119, e9–e21. [Google Scholar] [CrossRef]
- Spina, V.; Khiabanian, H.; Messina, M.; Monti, S.; Cascione, L.; Bruscaggin, A.; Spaccarotella, E.; Holmes, A.B.; Arcaini, L.; Lucioni, M.; et al. The genetics of nodal marginal zone lymphoma. Blood 2016, 128, 1362–1373. [Google Scholar] [CrossRef]
- Vela, V.; Juskevicius, D.; Dirnhofer, S.; Menter, T.; Tzankov, A. Mutational landscape of marginal zone B-cell lymphomas of various origin: Organotypic alterations and diagnostic potential for assignment of organ origin. Virchows Arch. 2022, 480, 403–413. [Google Scholar] [CrossRef]
- Tu, P.H.; Giannini, C.; Judkins, A.R.; Schwalb, J.M.; Burack, R.; O’Neill, B.P.; Yachnis, A.T.; Burger, P.C.; Scheithauer, B.W.; Perry, A. Clinicopathologic and genetic profile of intracranial marginal zone lymphoma: A primary low-grade CNS lymphoma that mimics meningioma. J. Clin. Oncol. 2005, 23, 5718–5727. [Google Scholar] [CrossRef] [PubMed]
- Moody, S.; Thompson, J.S.; Chuang, S.S.; Liu, H.; Raderer, M.; Vassiliou, G.; Wlodarska, I.; Wu, F.; Cogliatti, S.; Robson, A.; et al. Novel GPR34 and CCR6 mutation and distinct genetic profiles in MALT lymphomas of different sites. Haematologica 2018, 103, 1329–1336. [Google Scholar] [CrossRef]
- Wright, G.W.; Huang, D.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020, 37, 551–568.e14. [Google Scholar] [CrossRef]
- Song, J.Y.; Egan, C.; Bouska, A.C.; Zhang, W.; Gong, Q.; Venkataraman, G.; Herrera, A.F.; Chen, L.; Ottesen, R.; Niland, J.C.; et al. Genomic characterization of diffuse large B-cell lymphoma transformation of nodular lymphocyte-predominant Hodgkin lymphoma. Leukemia 2020, 34, 2238–2242. [Google Scholar] [CrossRef]
- Hartmann, S.; Schuhmacher, B.; Rausch, T.; Fuller, L.; Doring, C.; Weniger, M.; Lollies, A.; Weiser, C.; Thurner, L.; Rengstl, B.; et al. Highly recurrent mutations of SGK1, DUSP2 and JUNB in nodular lymphocyte predominant Hodgkin lymphoma. Leukemia 2016, 30, 844–853. [Google Scholar] [CrossRef]
- Iqbal, J.; Wilcox, R.; Naushad, H.; Rohr, J.; Heavican, T.B.; Wang, C.; Bouska, A.; Fu, K.; Chan, W.C.; Vose, J.M. Genomic signatures in T-cell lymphoma: How can these improve precision in diagnosis and inform prognosis? Blood Rev. 2016, 30, 89–100. [Google Scholar] [CrossRef]
- Iqbal, J.; Naushad, H.; Bi, C.; Yu, J.; Bouska, A.; Rohr, J.; Chao, W.; Fu, K.; Chan, W.C.; Vose, J.M. Genomic signatures in B-cell lymphoma: How can these improve precision in diagnosis and inform prognosis? Blood Rev. 2016, 30, 73–88. [Google Scholar] [CrossRef]
- Iqbal, J.; Amador, C.; McKeithan, T.W.; Chan, W.C. Molecular and Genomic Landscape of Peripheral T-Cell Lymphoma. Cancer Treat. Res. 2019, 176, 31–68. [Google Scholar]
- Cairns, R.A.; Iqbal, J.; Lemonnier, F.; Kucuk, C.; de Leval, L.; Jais, J.P.; Parrens, M.; Martin, A.; Xerri, L.; Brousset, P.; et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 2012, 119, 1901–1903. [Google Scholar] [CrossRef]
- Herek, T.A.; Bouska, A.; Lone, W.; Amador, C.; Heavican, T.B.; Sharma, S.; Greiner, T.C.; Smith, L.; Pileri, S.; Feldman, A.L.; et al. DNMT3A mutation defines a unique biological and prognostic subgroup in PTCL-NOS. Blood, 2022; in print. [Google Scholar]
- Cheng, S.; Zhang, W.; Inghirami, G.; Tam, W. Mutation analysis links angioimmunoblastic T-cell lymphoma to clonal hematopoiesis and smoking. eLife 2021, 10, e66395. [Google Scholar] [CrossRef]
- Palomero, T.; Couronne, L.; Khiabanian, H.; Kim, M.Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef]
- Manso, R.; Sanchez-Beato, M.; Monsalvo, S.; Gomez, S.; Cereceda, L.; Llamas, P.; Rojo, F.; Mollejo, M.; Menarguez, J.; Alves, J.; et al. The RHOA G17V gene mutation occurs frequently in peripheral T-cell lymphoma and is associated with a characteristic molecular signature. Blood 2014, 123, 2893–2894. [Google Scholar] [CrossRef]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, J.S.; Kim, J.; Kim, S.J.; Kim, W.S.; Lee, S.; Ko, Y.H.; Yoo, H.Y. A highly recurrent novel missense mutation in CD28 among angioimmunoblastic T-cell lymphoma patients. Haematologica 2015, 100, e505–e507. [Google Scholar] [CrossRef]
- Rohr, J.; Guo, S.; Huo, J.; Bouska, A.; Lachel, C.; Li, Y.; Simone, P.D.; Zhang, W.; Gong, Q.; Wang, C.; et al. Recurrent activating mutations of CD28 in peripheral T-cell lymphomas. Leukemia 2016, 30, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Vallois, D.; Dobay, M.P.; Morin, R.D.; Lemonnier, F.; Missiaglia, E.; Juilland, M.; Iwaszkiewicz, J.; Fataccioli, V.; Bisig, B.; Roberti, A.; et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood 2016, 128, 1490–1502. [Google Scholar] [CrossRef]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef]
- Wartewig, T.; Kurgyis, Z.; Keppler, S.; Pechloff, K.; Hameister, E.; Ollinger, R.; Maresch, R.; Buch, T.; Steiger, K.; Winter, C.; et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature 2017, 552, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Debackere, K.; Marcelis, L.; Demeyer, S.; Vanden Bempt, M.; Mentens, N.; Gielen, O.; Jacobs, K.; Broux, M.; Verhoef, G.; Michaux, L.; et al. Fusion transcripts FYN-TRAF3IP2 and KHDRBS1-LCK hijack T cell receptor signaling in peripheral T-cell lymphoma, not otherwise specified. Nat. Commun. 2021, 12, 3705. [Google Scholar] [CrossRef]
- Moon, C.S.; Reglero, C.; Cortes, J.R.; Quinn, S.A.; Alvarez, S.; Zhao, J.; Lin, W.W.; Cooke, A.J.; Abate, F.; Soderquist, C.R.; et al. FYN-TRAF3IP2 induces NF-kappaB signaling-driven peripheral T cell lymphoma. Nat. Cancer 2021, 2, 98–113. [Google Scholar] [CrossRef]
- Abate, F.; da Silva-Almeida, A.C.; Zairis, S.; Robles-Valero, J.; Couronne, L.; Khiabanian, H.; Quinn, S.A.; Kim, M.Y.; Laginestra, M.A.; Kim, C.; et al. Activating mutations and translocations in the guanine exchange factor VAV1 in peripheral T-cell lymphomas. Proc. Natl. Acad. Sci. USA 2017, 114, 764–769. [Google Scholar] [CrossRef]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef]
- Parrilla Castellar, E.R.; Jaffe, E.S.; Said, J.W.; Swerdlow, S.H.; Ketterling, R.P.; Knudson, R.A.; Sidhu, J.S.; Hsi, E.D.; Karikehalli, S.; Jiang, L.; et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood 2014, 124, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Rui, L.; Emre, N.C.; Kruhlak, M.J.; Chung, H.J.; Steidl, C.; Slack, G.; Wright, G.W.; Lenz, G.; Ngo, V.N.; Shaffer, A.L.; et al. Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell 2010, 18, 590–605. [Google Scholar] [CrossRef]
- Yan, J.; Li, B.; Lin, B.; Lee, P.T.; Chung, T.H.; Tan, J.; Bi, C.; Lee, X.T.; Selvarajan, V.; Ng, S.B.; et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood 2016, 128, 948–958. [Google Scholar] [CrossRef]
- Kucuk, C.; Jiang, B.; Hu, X.; Zhang, W.; Chan, J.K.; Xiao, W.; Lack, N.; Alkan, C.; Williams, J.C.; Avery, K.N.; et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat. Commun. 2015, 6, 6025. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Velusamy, T.; Rolland, D.; Sahasrabuddhe, A.A.; Chung, F.; Bailey, N.G.; Schrader, A.; Li, B.; Li, J.Z.; Ozel, A.B.; et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood 2014, 124, 1460–1472. [Google Scholar] [CrossRef]
- Dong, G.; Liu, X.; Wang, L.; Yin, W.; Bouska, A.; Gong, Q.; Shetty, K.; Chen, L.; Sharma, S.; Zhang, J.; et al. Genomic profiling identifies distinct genetic subtypes in extra-nodal natural killer/T-cell lymphoma. Leukemia 2022, 36, 2064–2075. [Google Scholar] [CrossRef]
- Watatani, Y.; Sato, Y.; Miyoshi, H.; Sakamoto, K.; Nishida, K.; Gion, Y.; Nagata, Y.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; et al. Molecular heterogeneity in peripheral T-cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia 2019, 33, 2867–2883. [Google Scholar] [CrossRef]
- Laurent, C.; Fazilleau, N.; Brousset, P. A novel subset of T-helper cells: Follicular T-helper cells and their markers. Haematologica 2010, 95, 356–358. [Google Scholar] [CrossRef]
- Dobay, M.P.; Lemonnier, F.; Missiaglia, E.; Bastard, C.; Vallois, D.; Jais, J.-P.; Scourzic, L.; Dupuy, A.; Fataccioli, V.; Pujals, A.; et al. Integrative clinicopathological and molecular analyses of angioimmunoblastic T-cell lymphoma and other nodal lymphomas of follicular helper T-cell origin. Haematologica 2017, 102, e148–e151. [Google Scholar] [CrossRef] [PubMed]
- Golloshi, R.; Sanders, J.T.; McCord, R.P. Iteratively improving Hi-C experiments one step at a time. Methods 2018, 142, 47–58. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar]
- Steen, C.B.; Luca, B.A.; Esfahani, M.S.; Azizi, A.; Sworder, B.J.; Nabet, B.Y.; Kurtz, D.M.; Liu, C.L.; Khameneh, F.; Advani, R.H.; et al. The landscape of tumor cell states and ecosystems in diffuse large B cell lymphoma. Cancer Cell 2021, 39, 1422–1437.e10. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, J.R. Phenotyping Multiple Subsets of Immune Cells In Situ in FFPE Tissue Sections: An Overview of Methodologies. Methods Mol. Biol. 2017, 1546, 75–99. [Google Scholar]
- Trapecar, M.; Khan, S.; Roan, N.R.; Chen, T.H.; Telwatte, S.; Deswal, M.; Pao, M.; Somsouk, M.; Deeks, S.G.; Hunt, P.W.; et al. An Optimized and Validated Method for Isolation and Characterization of Lymphocytes from HIV+ Human Gut Biopsies. AIDS Res. Hum. Retrovir. 2017, 33, S31–S39. [Google Scholar] [CrossRef]
- Scurrah, C.R.; Simmons, A.J.; Lau, K.S. Single-Cell Mass Cytometry of Archived Human Epithelial Tissue for Decoding Cancer Signaling Pathways. Methods Mol. Biol. 2019, 1884, 215–229. [Google Scholar]
- Yao, Y.; Liu, R.; Shin, M.S.; Trentalange, M.; Allore, H.; Nassar, A.; Kang, I.; Pober, J.S.; Montgomery, R.R. CyTOF supports efficient detection of immune cell subsets from small samples. J. Immunol. Methods 2014, 415, 1–5. [Google Scholar] [CrossRef]
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schuffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef]
- Chang, Q.; Ornatsky, O.I.; Siddiqui, I.; Loboda, A.; Baranov, V.I.; Hedley, D.W. Imaging Mass Cytometry. Cytom. A 2017, 91, 160–169. [Google Scholar] [CrossRef]
- Fincham, R.E.A.; Bashiri, H.; Lau, M.C.; Yeong, J. Editorial: Multiplex Immunohistochemistry/Immunofluorescence Technique: The Potential and Promise for Clinical Application. Front. Mol. Biosci. 2022, 9, 831383. [Google Scholar] [CrossRef]
- Spagnolo, D.M.; Gyanchandani, R.; Al-Kofahi, Y.; Stern, A.M.; Lezon, T.R.; Gough, A.; Meyer, D.E.; Ginty, F.; Sarachan, B.; Fine, J.; et al. Pointwise mutual information quantifies intratumor heterogeneity in tissue sections labeled with multiple fluorescent biomarkers. J. Pathol. Inform. 2016, 7, 47. [Google Scholar] [CrossRef]
- Norton, S.; Kemp, R. Computational Analysis of High-Dimensional Mass Cytometry Data from Clinical Tissue Samples. Methods Mol. Biol. 2019, 1989, 295–307. [Google Scholar]
- Hedlund, E.; Deng, Q. Single-cell RNA sequencing: Technical advancements and biological applications. Mol. Asp. Med. 2018, 59, 36–46. [Google Scholar] [CrossRef]
- Camara, P.G. Topological methods for genomics: Present and future directions. Curr. Opin. Syst. Biol. 2017, 1, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Hav, M.; Colombo, A.; Chavez, E.A.; Nissen, M.; Wang, X.; Miyata-Takata, T.; et al. Single-Cell Transcriptome Analysis Reveals Disease-Defining T-cell Subsets in the Tumor Microenvironment of Classic Hodgkin Lymphoma. Cancer Discov. 2020, 10, 406–421. [Google Scholar] [CrossRef]
- Rindler, K.; Jonak, C.; Alkon, N.; Thaler, F.M.; Kurz, H.; Shaw, L.E.; Stingl, G.; Weninger, W.; Halbritter, F.; Bauer, W.M.; et al. Single-cell RNA sequencing reveals markers of disease progression in primary cutaneous T-cell lymphoma. Mol. Cancer 2021, 20, 124. [Google Scholar] [CrossRef]
- Ysebaert, L.; Quillet-Mary, A.; Tosolini, M.; Pont, F.; Laurent, C.; Fournie, J.J. Lymphoma Heterogeneity Unraveled by Single-Cell Transcriptomics. Front. Immunol. 2021, 12, 597651. [Google Scholar] [CrossRef]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.J.; Pogrebniak, K.; Rueda, O.M.; Provenzano, E.; Grant, J.; Chin, S.F.; Tsui, D.W.; Marass, F.; Gale, D.; et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat. Commun. 2015, 6, 8760. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Bratman, S.V.; Newman, A.M.; Alizadeh, A.A.; Diehn, M. Potential clinical utility of ultrasensitive circulating tumor DNA detection with CAPP-Seq. Expert Rev. Mol. Diagn. 2015, 15, 715–719. [Google Scholar] [CrossRef]
- McDonald, B.R.; Contente-Cuomo, T.; Sammut, S.J.; Odenheimer-Bergman, A.; Ernst, B.; Perdigones, N.; Chin, S.F.; Farooq, M.; Mejia, R.; Cronin, P.A.; et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci. Transl. Med. 2019, 11, eaax7392. [Google Scholar] [CrossRef]
- Spina, V.; Bruscaggin, A.; Cuccaro, A.; Martini, M.; Di Trani, M.; Forestieri, G.; Manzoni, M.; Condoluci, A.; Arribas, A.; Terzi-Di-Bergamo, L.; et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood 2018, 131, 2413–2425. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Scherer, F.; Jin, M.C.; Soo, J.; Craig, A.F.M.; Esfahani, M.S.; Chabon, J.J.; Stehr, H.; Liu, C.L.; Tibshirani, R.; et al. Circulating Tumor DNA Measurements As Early Outcome Predictors in Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2018, 36, 2845–2853. [Google Scholar] [CrossRef]
- Henriksen, S.D.; Thorlacius-Ussing, O. Cell-Free DNA Methylation as Blood-Based Biomarkers for Pancreatic Adenocarcinoma-A Literature Update. Epigenomes 2021, 5, 8. [Google Scholar] [CrossRef]
- Lauer, E.M.; Mutter, J.; Scherer, F. Circulating tumor DNA in B-cell lymphoma: Technical advances, clinical applications, and perspectives for translational research. Leukemia 2022, 6, 1–14. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Esfahani, M.S.; Scherer, F.; Soo, J.; Jin, M.C.; Liu, C.L.; Newman, A.M.; Duhrsen, U.; Huttmann, A.; Casasnovas, O.; et al. Dynamic Risk Profiling Using Serial Tumor Biomarkers for Personalized Outcome Prediction. Cell 2019, 178, 699–713.e19. [Google Scholar] [CrossRef]
- Rossi, D.; Kurtz, D.M.; Roschewski, M.; Cavalli, F.; Zucca, E.; Wilson, W.H. The development of liquid biopsy for research and clinical practice in lymphomas: Report of the 15-ICML workshop on ctDNA. Hematol. Oncol. 2020, 38, 34–37. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Chapuy B. et al. Nat. Med. 2018 | Schmitz R. et al. NEJM. 2018 | COO Classification | Prognosis | Genetic Characteristics |
|---|---|---|---|---|
| Cluster 1 | BN2 | ABC or ABC + UC | F | BCL6 rearrangement; Notch pathway: Notch 2, SPEN, DTX1; NF-κB: A20, TNIP1, BCL10, PKCB; immune escape CD70, FAS, PDLI/L2 |
| Cluster 2 | N/C | Mixed | UF | TP53 biallelic abnormalities; CDKN2A/RB loss; miR17-92 gain; MCL1 gain |
| Cluster 3 | EZB | GCB | UF | BCL2 translocation, EZH2 mutation, cRel amplification, TNFRSF14 alteration, MEF2B, and common chromatin modifier mutation: MLL2, CREBBP, EP300; SIPR2 pathway; STAT6; mTOR; MiR17-92; PTEN |
| Cluster 4 | N/C | GCB | F | Histone core and linkers; immune evasion; GNA13, RHOA, SGK1; NF-κB; BRAF/STAT3 |
| Cluster 5 | MCD | ABC | UF | MYD88L265P, CD79B; 18p gain, PRDMI, CDKN2A, ETV6, BTG1/2, TBL1XR1; PIM1; immune editing, high cAID |
| N/C | N1 | ABC | UF | NOTCH1 mutation; IRF4, 1D3, BCOR, A20; plasmacytic phenotype |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, W.C.; Iqbal, J. The Era of Genomic Research for Lymphoma: Looking Back and Forward. Hemato 2022, 3, 485-507. https://doi.org/10.3390/hemato3030034
Chan WC, Iqbal J. The Era of Genomic Research for Lymphoma: Looking Back and Forward. Hemato. 2022; 3(3):485-507. https://doi.org/10.3390/hemato3030034
Chicago/Turabian StyleChan, Wing C., and Javeed Iqbal. 2022. "The Era of Genomic Research for Lymphoma: Looking Back and Forward" Hemato 3, no. 3: 485-507. https://doi.org/10.3390/hemato3030034
APA StyleChan, W. C., & Iqbal, J. (2022). The Era of Genomic Research for Lymphoma: Looking Back and Forward. Hemato, 3(3), 485-507. https://doi.org/10.3390/hemato3030034