Chalcogen–Nitrogen Bond: Insights into a Key Chemical Motif †

 ,
, 

 ,
, 
Abstract
:1. Introduction
2. Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Joshi, P.G.; More, M.S.; Jadhav, A.A.; Khanna, P.K. Materials and biological applications of 1,2,3-selenadiazoles: A review. Mater. Today Chem. 2020, 16, 100255. [Google Scholar] [CrossRef]
- Alberto, E.E.; Nascimento, V. Do; Braga, A.L. Catalytic application of selenium and tellurium compounds as glutathione peroxidase enzyme mimetics. J. Braz. Chem. Soc. 2010, 21, 2032–2041. [Google Scholar] [CrossRef]
- Barbosa, N.V.; Nogueira, C.W.; Nogara, P.A.; De Bem, A.F.; Aschner, M.; Rocha, J.B.T. Organoselenium compounds as mimics of selenoproteins and thiol modifier agents. Metallomics 2017, 9, 1703–1734. [Google Scholar] [CrossRef] [PubMed]
- Chivers, T.; Laitinen, R.S. Neutral binary chalcogen-nitrogen and ternary S,N,P molecules: New structures, bonding insights and potential applications. Dalt. Trans. 2020, 49, 6532–6547. [Google Scholar] [CrossRef] [PubMed]
- Engman, L.; Hallberg, A. Expedient Synthesis of Ebselen and Related Compounds. J. Org. Chem. 1989, 54, 2964–2966. [Google Scholar] [CrossRef]
- Sarma, B.K.; Manna, D.; Minoura, M.; Mugesh, G. Synthesis, structure, spirocyclization mechanism, and glutathione peroxidase-like antioxidant activity of stable spirodiazaselenurane and spirodiazatellurane. J. Am. Chem. Soc. 2010, 132, 5364–5374. [Google Scholar] [CrossRef]
- Zade, S.S.; Panda, S.; Tripathi, S.K.; Singh, H.B.; Wolmershäuser, G. Convenient synthesis, characterization and GPx-like catalytic activity of novel ebselen derivatives. Eur. J. Org. Chem. 2004, 2004, 3857–3864. [Google Scholar] [CrossRef]
- Kersting, B.; DeLion, M. Synthesis of benzisochalcogenol and -azole derivatives via ortho metalation of isophthalamides. Z. Naturforsch. B 1999, 54, 1042–1047. [Google Scholar] [CrossRef]
- Bhowmick, D.; Mugesh, G. Introduction of a catalytic triad increases the glutathione peroxidase-like activity of diaryl diselenides. Org. Biomol. Chem. 2015, 13, 9072–9082. [Google Scholar] [CrossRef]
- Sarma, B.K.; Mugesh, G. Antioxidant Activity of the Anti-Inflammatory Compound Ebselen: A Reversible Cyclization Pathway via Selenenic and Seleninic Acid Intermediates. Chem. Eur. J. 2008, 14, 10603–10614. [Google Scholar] [CrossRef]
- Singh, V.P.; Singh, H.B.; Butcher, R.J. Synthesis and glutathione peroxidase-like activities of isoselenazolines. Eur. J. Org. Chem. 2011, 3, 5485–5497. [Google Scholar] [CrossRef]
- Müller, A.; Cadenas, E.; Graf, P.; Sies, H. A novel biologically active seleno-organic compound-1. Glutathione peroxidase-like activity in vitro and antioxidant capacity of PZ 51 (Ebselen). Biochem. Pharmacol. 1984, 33, 3235–3239. [Google Scholar] [CrossRef]
- Sies, H. Ebselen, A selenoorganic compound as glutathione peroxidase mimic. Free Radic. Biol. Med. 1993, 14, 313–323. [Google Scholar] [CrossRef]
- Nogueira, C.W.; Rocha, J.B.T. Toxicology and pharmacology of selenium: Emphasis on synthetic organoselenium compounds. Arch. Toxicol. 2011, 85, 1313–1359. [Google Scholar] [CrossRef]
- Azad, G.K.; Tomar, R.S. Ebselen, a promising antioxidant drug: Mechanisms of action and targets of biological pathways. Mol. Biol. Rep. 2014, 41, 4865–4879. [Google Scholar] [CrossRef]
- Zhao, R.; Holmgren, A. A novel antioxidant mechanism of ebselen involving ebselen diselenide, a substrate of mammalian thioredoxin and thioredoxin reductase. J. Biol. Chem. 2002, 277, 39456–39462. [Google Scholar] [CrossRef]
- Antony, S.; Bayse, C.A. Modeling the mechanism of the glutathione peroxidase mimic ebselen. Inorg. Chem. 2011, 50, 12075–12084. [Google Scholar] [CrossRef]
- Dalla Tiezza, M.; Ribaudo, G.; Orian, L. Organodiselenides: Organic Catalysis and Drug Design Learning from Glutathione Peroxidase. Curr. Org. Chem. 2018, 23, 1381–1402. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Sies, H.; Parnham, M.J. Potential therapeutic use of ebselen for COVID-19 and other respiratory viral infections. Free Radic. Biol. Med. 2020, 156, 107–112. [Google Scholar] [CrossRef]
- Bjorgvinsson, M.; Roesky, H.W. The structures of compounds containing selenium-nitrogen and tellurium-nitrogen bonds. Polyhedron 1991, 10, 2353–2370. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; Da Guedes Silva, M.F.C.; Pombeiro, A.J.L. Chalcogen bonding in synthesis, catalysis and design of materials. Dalt. Trans. 2017, 46, 10121–10138. [Google Scholar] [CrossRef] [PubMed]
- Rendekova, J.; Vlasakova, D.; Arsenyan, P.; Vasiljeva, J.; Nasim, M.J.; Witek, K.; Dominguez-Alvarez, E.; Zeslawska, E.; Manikova, D.; Tejchman, W.; et al. The Selenium-Nitrogen Bond as Basis for Reactive Selenium Species with Pronounced Antimicrobial Activity. Curr. Org. Synth. 2017, 14, 1082–1090. [Google Scholar] [CrossRef]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- SCM. ADF2019.103, Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Johnson, B.G.; Gill, P.M.W.; Pople, J.A. The performance of a family of density functional methods. J. Chem. Phys. 1993, 98, 5612–5626. [Google Scholar] [CrossRef]
- Russo, T.V.; Martin, R.L.; Hay, P.J. Density functional calculations on first-row transition metals. J. Chem. Phys. 1994, 101, 7729–7737. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- van Lenthe, E.; Snijders, J.G.; Baerends, E.J. The zero-order regular approximation for relativistic effects: The effect of spin–orbit coupling in closed shell molecules. J. Chem. Phys. 1996, 105, 6505–6516. [Google Scholar] [CrossRef]
- Van Lenthe, E. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Baerends, E.J. Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistry. In Reviews in Computational Chemistry; Lipkovitz, K.B., Boyd, D.B., Eds.; Wiley-VCH: New York, NY, USA, 2000; Volume 15, pp. 1–86. ISBN 9780470125922. [Google Scholar]
- Bickelhaupt, F.M.; Houk, K.N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem. Int. Ed. 2017, 56, 10070–10086. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. How Well Can New-Generation Density Functionals Describe the Energetics of Bond-Dissociation Reactions Producing Radicals? J. Phys. Chem. A 2008, 112, 1095–1099. [Google Scholar] [CrossRef]
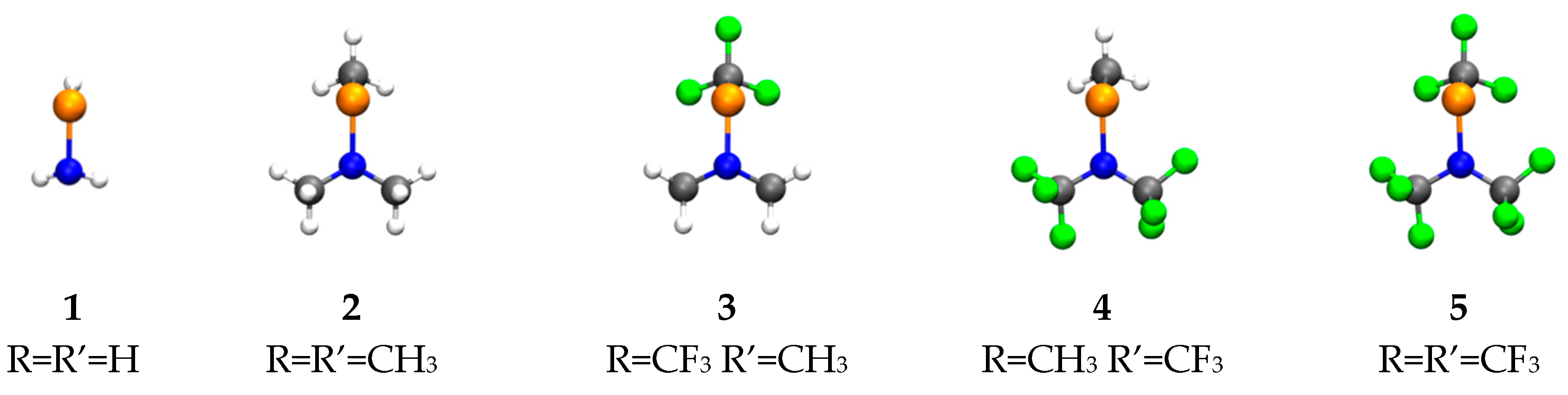

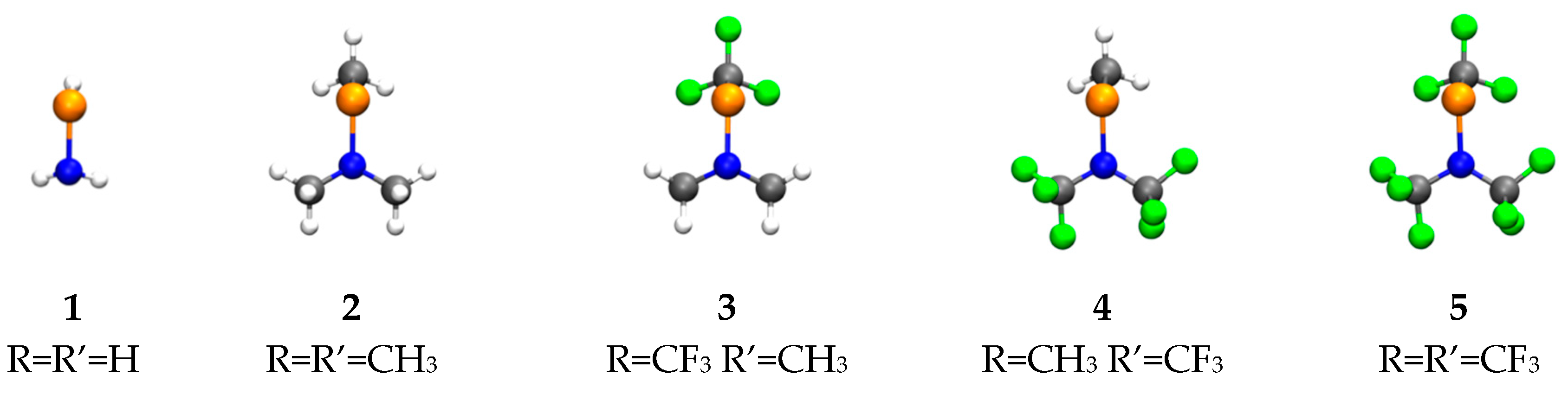{kind=link}
{kind=link}
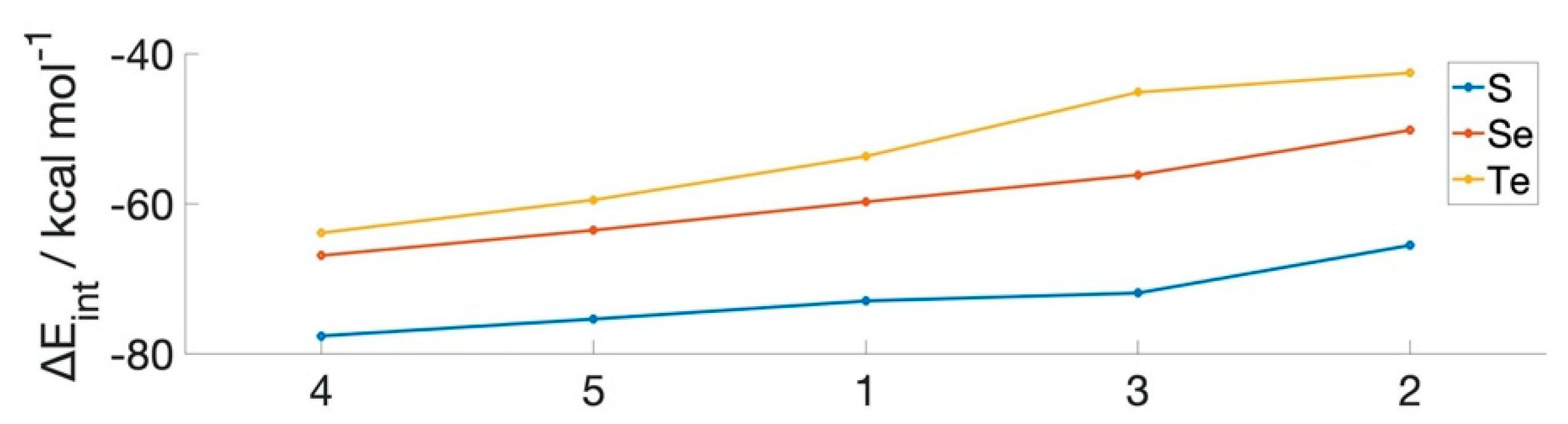
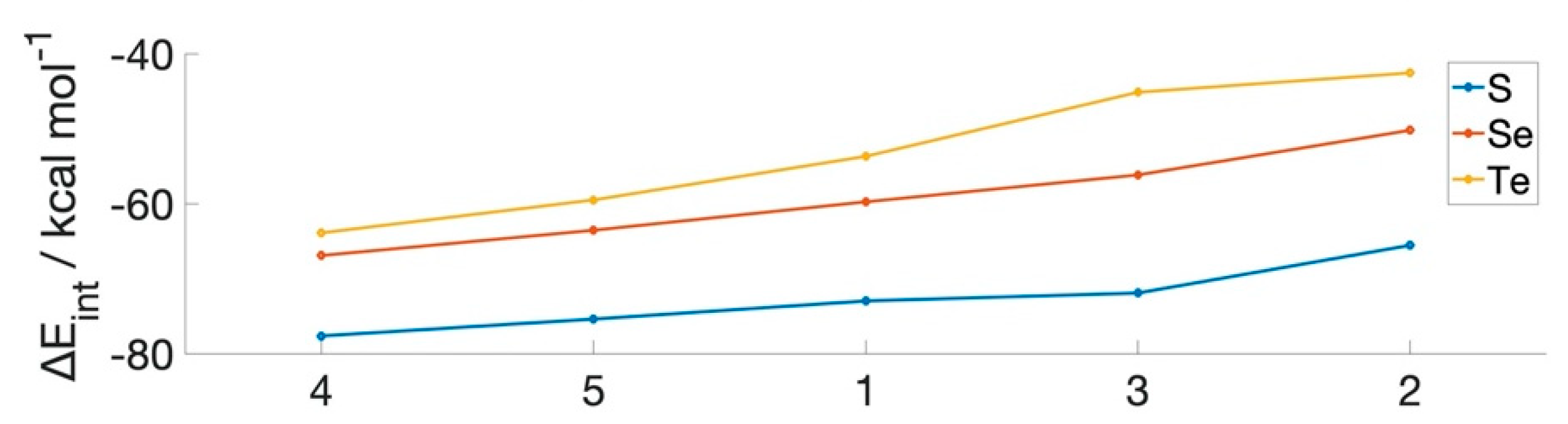
| RX–NR’2 | ∆Eint (kcal mol−1) | ||||
|---|---|---|---|---|---|
| Molecule | R | R’ | S | Se | Te |
| 1 | H | H | −73.0 | −59.8 | −53.7 |
| 2 | CH3 | CH3 | −65.5 | −50.2 | −42.6 |
| 3 | CF3 | CH3 | −71.9 | −56.2 | −45.1 |
| 4 | CH3 | CF3 | −77.6 | −66.9 | −63.9 |
| 5 | CF3 | CF3 | −75.4 | −63.5 | −59.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bortoli, M.; Madabeni, A.; Nogara, P.A.; Omage, F.B.; Ribaudo, G.; Zeppilli, D.; Rocha, J.B.T.; Orian, L. Chalcogen–Nitrogen Bond: Insights into a Key Chemical Motif. Chem. Proc. 2020, 2, 21. https://doi.org/10.3390/ECCS2020-07589
Bortoli M, Madabeni A, Nogara PA, Omage FB, Ribaudo G, Zeppilli D, Rocha JBT, Orian L. Chalcogen–Nitrogen Bond: Insights into a Key Chemical Motif. Chemistry Proceedings. 2020; 2(1):21. https://doi.org/10.3390/ECCS2020-07589
Chicago/Turabian StyleBortoli, Marco, Andrea Madabeni, Pablo Andrei Nogara, Folorunsho B. Omage, Giovanni Ribaudo, Davide Zeppilli, Joao Batista Teixeira Rocha, and Laura Orian. 2020. "Chalcogen–Nitrogen Bond: Insights into a Key Chemical Motif" Chemistry Proceedings 2, no. 1: 21. https://doi.org/10.3390/ECCS2020-07589
APA StyleBortoli, M., Madabeni, A., Nogara, P. A., Omage, F. B., Ribaudo, G., Zeppilli, D., Rocha, J. B. T., & Orian, L. (2020). Chalcogen–Nitrogen Bond: Insights into a Key Chemical Motif. Chemistry Proceedings, 2(1), 21. https://doi.org/10.3390/ECCS2020-07589