1. Introduction
Type 2 Diabetes Mellitus (T2DM) remains one of the most significant public health challenges globally, driven by rising obesity rates and sedentary lifestyles. Characterized by chronic hyperglycemia due to insulin resistance and progressive beta-cell dysfunction, which involves endoplasmic reticulum (ER) stress, mitochondrial dysfunction, oxidative stress, and chronic low-grade inflammation. These factors contribute to impaired insulin synthesis, vesicular trafficking, and increased beta-cell apoptosis, T2DM affects hundreds of millions worldwide and is associated with severe complications, including cardiovascular disease, renal failure, and blindness. Despite advances in diabetes management, the progressive decline in beta-cell function remains a critical barrier to long-term glycemic control, highlighting the need for novel therapeutic strategies [
1,
2,
3].
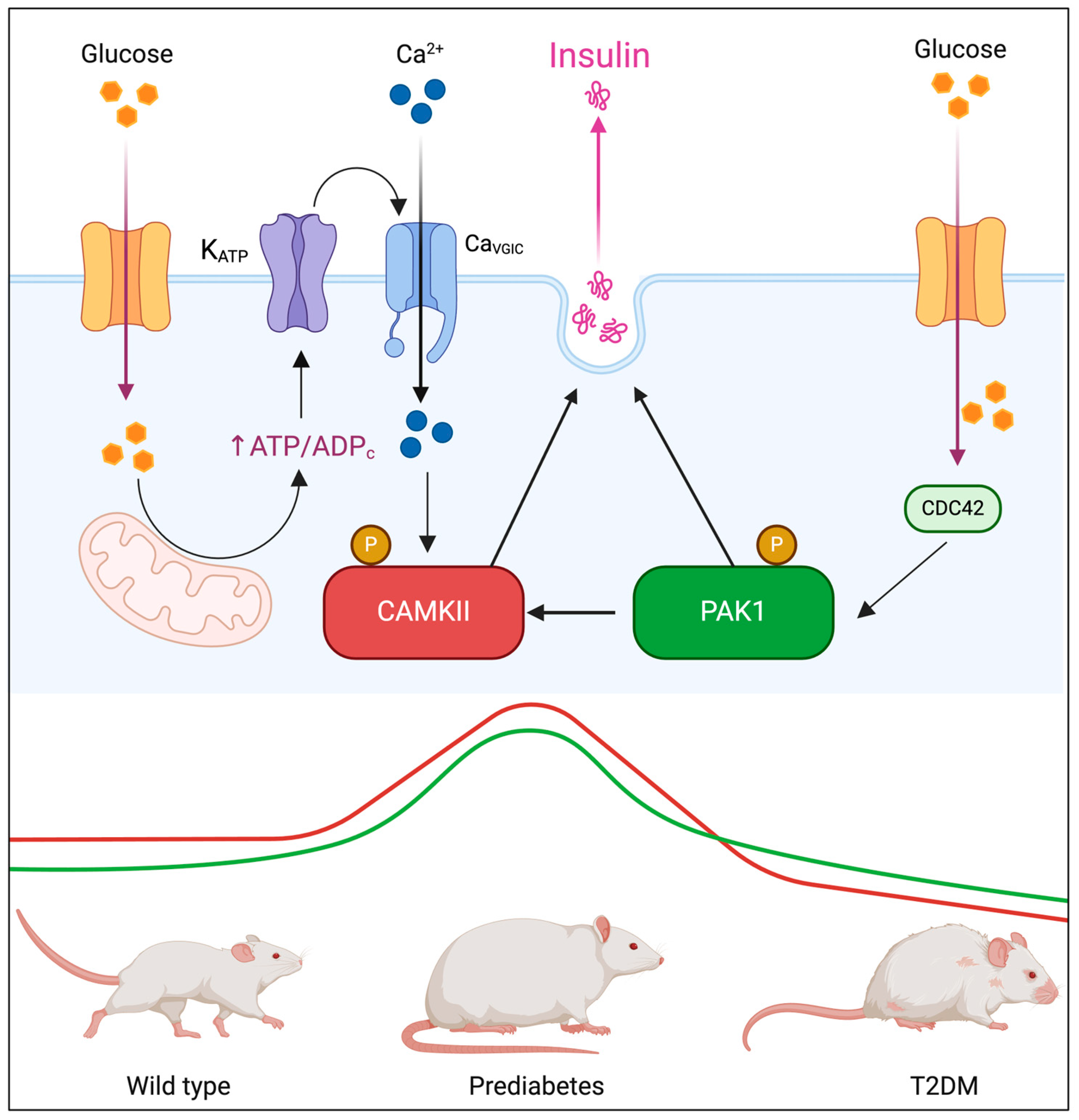
Insulin secretion from pancreatic beta-cells is tightly regulated through a biphasic mechanism. The first phase involves a rapid release of pre-stored insulin granules in response to glucose intake, while the second phase consists of sustained insulin secretion, necessary for prolonged glucose homeostasis. Several well-characterized proteins orchestrate these processes, including ATP-sensitive potassium channels (K_ATP), SNARE complex proteins (Syntaxin-1, SNAP-25, VAMP2), and L-type and P/Q-type voltage-gated calcium channels, which regulate vesicle docking, priming, and fusion. However, recent evidence suggests that kinases involved in cytoskeletal remodeling and intracellular signaling, such as p21-activated kinase 1 (PAK1) and calcium/calmodulin-dependent protein kinase II (CAMKII), may also play fundamental roles in insulin granule mobilization and secretion. Given their potential involvement in different phases of biphasic secretion, understanding how PAK1 and CAMKII interact may provide new insights into β-cell function and dysfunction in T2DM [
4,
5,
6,
7].
PAK1 is a serine/threonine kinase activated by the small GTPases Cdc42 and Rac1, which regulates various physiological processes, including cytoskeletal remodeling, cell motility, and gene transcription [
8,
9,
10]. Dysregulation of PAK1 has been implicated in oncogenesis, cardiovascular diseases, neurological disorders and, more recently, its role has begun to be explored in the exocrine pancreas, where it remains significantly under-studied, but its role in pancreatic beta-cells has only recently been explored [
11,
12,
13,
14]. Emerging evidence suggests that Pak1 is particularly important for the second phase of insulin secretion, where it regulates cytoskeletal remodeling and actin-dependent vesicle trafficking, facilitating insulin granule exocytosis in response to prolonged glucose stimulation. This has been demonstrated in PAK1 knockout mice, which exhibit selective impairments in the second phase of insulin release [
7,
15,
16,
17].
PAK1 knockout (
PAK1−/−) mice exhibit significant impairments in glucose-stimulated insulin secretion, primarily affecting the sustained second phase. These mice develop fasting hyperglycemia, glucose intolerance, and a reduction in beta-cell mass, underscoring the essential role of PAK1 in beta-cell function. At the molecular level, PAK1 promotes insulin biogenesis by enhancing the transcription of
PDX1,
NEUROD1, and
INS, contributing to increased insulin content and beta-cell survival under metabolic stress [
18,
19].
Calcium/calmodulin-dependent protein kinase II (CAMKII) is a multifunctional serine/threonine kinase that plays a central role in various cellular processes, including synaptic plasticity, vascular homeostasis, and metabolic regulation. In pancreatic beta-cells, CAMKII serves as a calcium sensor, amplifying Ca
2+ influx triggered by glucose and other secretagogues, thereby enhancing insulin granule exocytosis [
20,
21,
22]. CAMKII has been extensively studied in first-phase insulin secretion, where it facilitates the rapid fusion of insulin-containing vesicles with the plasma membrane. Pharmacological inhibition or genetic deletion of CAMKII in beta-cells significantly reduces insulin release and impairs glucose tolerance, highlighting its role in glucose-stimulated insulin secretion [
23,
24].
In the context of T2DM, chronic hyperglycemia is associated with persistent CAMKII activation in pancreatic islets and metabolic tissues. This prolonged activation contributes to beta-cell dysfunction, oxidative stress, and impaired insulin secretion, exacerbating diabetes progression. Moreover, CAMKII hyperactivation in the vascular system leads to vascular remodeling, endothelial dysfunction, and inflammation, further aggravating diabetes-related complications [
25]. Our research group has demonstrated that PAK1 directly phosphorylates and activates CAMKII, suggesting a functional link between these kinases in insulin secretion [
26]. Given that PAK1 is primarily implicated in the second phase of insulin secretion, while CAMKII is a key regulator of the first phase, their interaction may represent a previously unrecognized mechanism coordinating insulin granule mobilization across both phases. Understanding the PAK1-CAMKII axis could provide novel insights into beta-cell adaptation under metabolic stress and uncover potential therapeutic targets for preserving insulin secretion in T2DM. Based on this rationale, we hypothesized that PAK1 and CAMKII cooperatively regulate insulin secretion through coordinated cytoskeletal and calcium-dependent mechanisms, and that their dysregulation contributes to β-cell failure during diabetes progression. To test this, we investigated their colocalization, interaction, and activation in murine β-cells under glucose stimulation, assessed their expression dynamics in diabetic mouse models and human pancreatic islets, and examined the functional impact of their inhibition on insulin secretion. Our integrated approach aims to uncover the role of the PAK1–CAMKII axis in β-cell physiology and its relevance in the pathogenesis of T2DM.
2. Materials and Methods
2.1. Cell Culture and Glucose-Stimulated Insulin Secretion Assay (GSIS)
Beta-TC-6 murine insulinoma cells (ATCC, Manassas, VA, USA) were cultured in Dulbecco’s Modified Eagle Medium High Glucose (DMEM, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 15% fetal bovine serum (FBS) (FBS, Gibco, Thermo Fisher Scientific, Waltham, MA, USA), 50 U/mL penicillin, and 50 μg/mL streptomycin (Gibco, Thermo Fisher Scientific, Waltham, MA, USA). Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO
2. For glucose-stimulated insulin secretion (GSIS) assays cells were preincubated for 4 h in high-glucose DMEM (15% FBS) containing the kinase inhibitors FRAX-1036 (PAK1 inhibitor, 5 μM, MedChemExpress, Monmouth Junction, NJ, USA) and KN93 (CAMKII inhibitor, 20 μM, Sigma-Aldrich, St. Louis, MO, USA). Cell viability was assessed using the MTT assay after 24 h of inhibitor treatment, confirming no significant reduction in viability compared to vehicle-treated controls (DMSO 0.1%, Sigma-Aldrich, St. Louis, MO, USA) (
p > 0.05, Student’s
t-test). After preincubation, cells were washed with Krebs-Ringer buffer prepared in-house according to standard protocols, containing 0 mM glucose, and a supernatant sample was collected (baseline secretion). Subsequently, cells were incubated for 10 min in Krebs-Ringer buffer with 4 mM glucose, and another supernatant sample was collected (basal secretion). Finally, cells were exposed to either 4 mM or 20 mM glucose for 60 min, and a final supernatant sample was collected to measure glucose-stimulated insulin secretion. Insulin secretion was normalized to basal insulin levels. Experiments were performed using 50,000 cells per well to ensure appropriate protein content for ELISA normalization. To ensure physiologically relevant glucose stimulation, 4 mM and 20 mM glucose were used to represent normoglycemic and hyperglycemic conditions, respectively. These concentrations are commonly used in β-cell models and reflect the functional insulin secretion range of Beta-TC-6 cells, which respond to glucose from 2–3 mM and reach maximal secretion between 10–20 mM [
27,
28].
2.2. Immunofluorescence and Confocal Microscopy
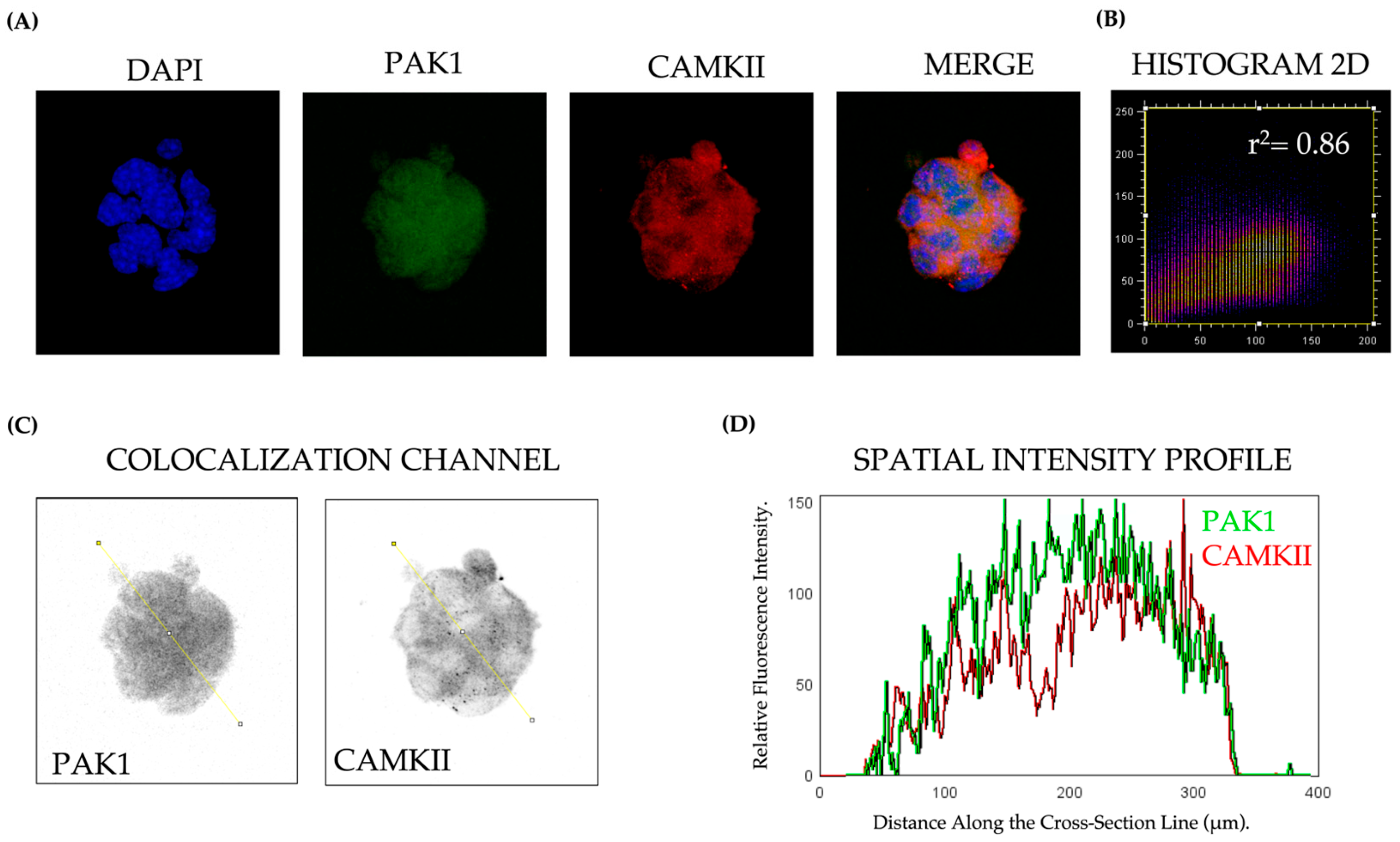
Beta-TC-6 cells (5000 per well) were plated onto sterile glass coverslips previously treated with poly-L-lysine (Sigma-Aldrich) to promote cell adhesion and maintained overnight in high-glucose DMEM supplemented with 15% FBS at 37 °C in a humidified 5% CO2 incubator. Cells were then fixed with 4% paraformaldehyde (PFA) for 15 min and permeabilized using 0.2% Triton X-100 for 10 min. After blocking with 5% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) for 1 h, cells were incubated with primary antibodies at room temperature for 2 h. The following primary antibodies were used: PAK1 (1:500, Cat# 2602, Cell Signaling Technology, Boston, MA, USA), Phospho-PAK1 (Ser199/204, 1:500, Cat# 2601, Cell Signaling Technology), CAMKII (1:500, Cat# AB134041, Abcam, Cambridge, UK), Phospho-CAMKII (Thr287, 1:500, Cat# 3361, Cell Signaling Technology). After PBS washes, cells were incubated with Alexa Fluor 488 or 594-conjugated secondary antibodies (1:500, Thermo Fisher Scientific, Waltham, MA, USA) for 1 h in the dark. Nuclei were counterstained with DAPI (Sigma-Aldrich), and images were captured using a Leica TCS SP8x confocal microscope (Leica Microsystems, Wetzlar, Germany). Colocalization analysis was performed using ImageJ software version 1.53t (NIH, Bethesda, MD, USA) with the Coloc2 plugin
2.3. Proximity Ligation Assay (PLA)
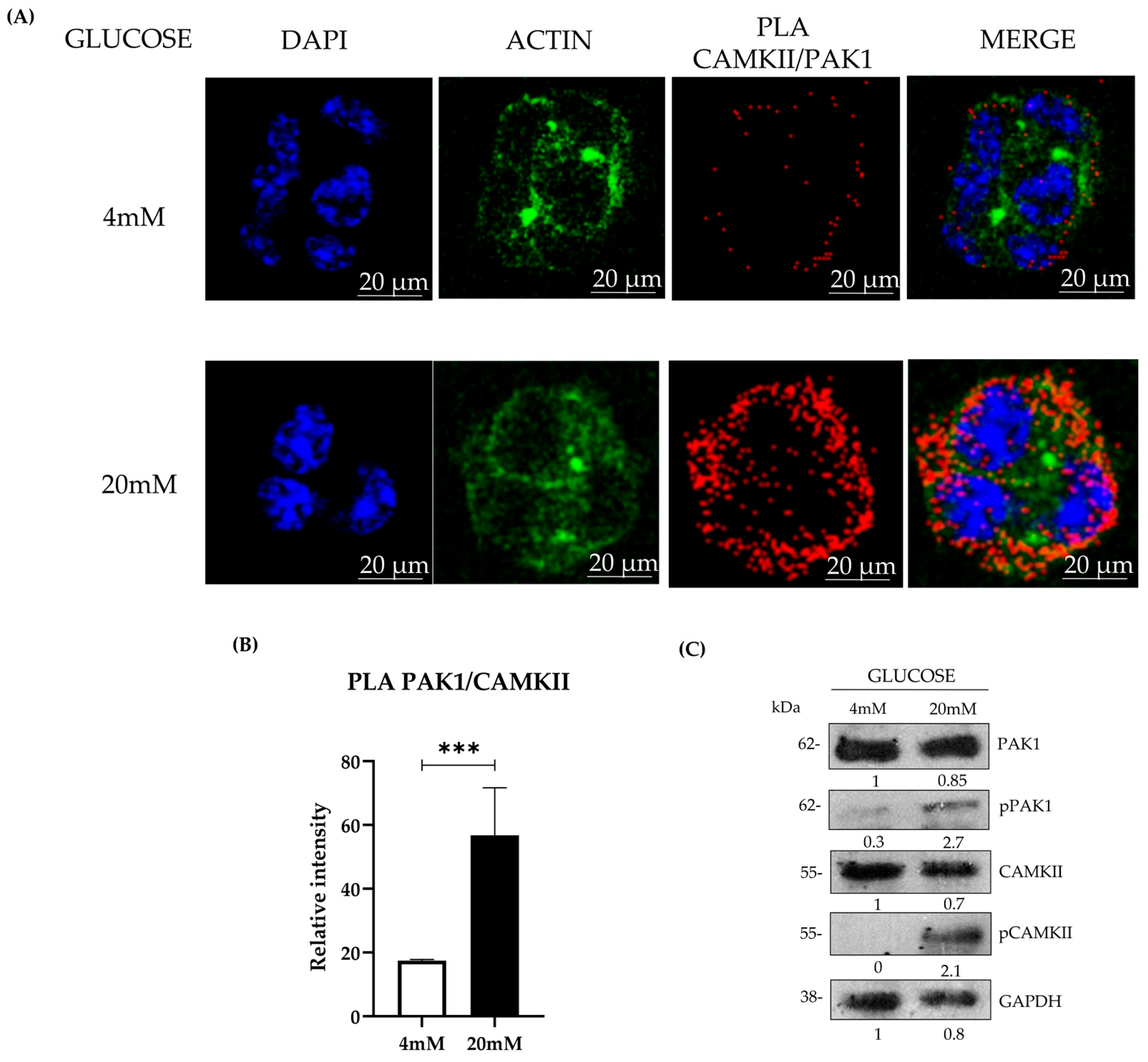
To assess Pak1-CaMKII interactions, Duolink® In Situ Detection Reagents (Sigma-Aldrich) were used per the manufacturer’s instructions. Beta-TC-6 cells (5000 per well) were disposed in 16-well chamber slides (Nunc Lab-Tek, Thermo Scientific, Waltham, MA, USA) previously treated with poly-L-lysine (Sigma-Aldrich) to promote cell adhesion and maintained overnight in high-glucose DMEM supplemented with 15% FBS at 37 °C in a humidified 5% CO2 incubator. Cells were then stimulated following the glucose-stimulated insulin secretion (GSIS) protocol. After glucose stimulation, cells were fixed, permeabilized, and blocked as described above. Cells were incubated overnight at 4 °C with anti-Pak1 and anti-CaMKII primary antibodies. The following day, samples were incubated with species-specific PLA probes (anti-rabbit PLUS and anti-mouse MINUS, Olink Bioscience, Sigma-Aldrich) for 1 h at 37 °C, followed by ligation and amplification steps per the manufacturer’s protocol. For each experimental condition, at least 100 cells per field were analyzed from five randomly selected fields, and a minimum of 300 total cells were counted per condition. All experiments were conducted in three independent biological replicates (n = 3) to ensure reproducibility. Fluorescent signals were visualized using a Leica TCS SP8 confocal microscope, and PLA-positive signals were quantified using MetaMorph software version 7.8.0.0 (Molecular Devices, San Jose, CA, USA).
2.4. Enzyme-Linked ImmunoSorbent Assay
Insulin secretion was quantified using a Mouse Insulin ELISA Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Absorbance was measured at 450 nm using a BioTek Epoch2 microplate reader (BioTek, Winooski, VT, USA). Insulin concentrations were normalized to total protein content, determined via quantification of total insulin per well using ELISA.
2.5. Western Blotting
Beta-TC-6 cells (50,000 per well) were preincubated with inhibitors for 4 h in high-glucose DMEM (15% FBS) and subsequently stimulated following the glucose-stimulated insulin secretion (GSIS) protocol. After stimulation, cells were lysed in RIPA buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% SDS, 1% sodium deoxycholate) supplemented with protease (Sigma-Aldrich) and phosphatase inhibitors (PhosSTOP, Roche, Basel, Switzerland). Proteins (30 μg) were separated via SDS-PAGE and transferred onto Immobilon-P PVDF membranes (Millipore, Burlington, MA, USA). Membranes were blocked in 5% nonfat dried milk or 1% BSA in TBS-T, incubated with primary antibodies overnight at 4 °C, followed by HRP-conjugated secondary antibodies for 1 h at room temperature. Protein bands were visualized using the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Waltham, MA, USA).
2.6. Animal Model of T2DM and Immunohistochemistry
All animal experiments were conducted following the ethical guidelines established by the Institutional Animal Care and Use Committee (IACUC) of Facultad de Estudios Superiores Iztacala, UNAM, with approval code CE/FESI/012022/1472. Male C57BL/6 mice (8 weeks old, n = 5 per group) were used to establish a murine model of Type 2 Diabetes Mellitus (T2DM). Mice in the wild-type (WT) control group were maintained on a normal chow diet, while those in the prediabetic (PD) group were fed a high-fat diet (HFD, 60% kcal from fat, 4.9 kcal/g) for 8 weeks to induce a prediabetic state. To further impair β-cell function and induce a T2DM model, a separate group of mice was fed an HFD for 12 weeks, followed by a single intraperitoneal injection of streptozotocin (STZ, 40 mg/kg). Diabetes onset was confirmed by fasting glucose evaluation (>200 mg/dL) and increased homeostatic model assessment of insulin resistance (HOMA-IR) values. After euthanasia, pancreata were collected, fixed with Bouin’s solution, and embedded in paraffin for histological analysis.
2.7. Immunohistochemistry
Pancreatic tissue sections were deparaffinized, rehydrated, and subjected to antigen retrieval in sodium citrate buffer (0.01 M, pH 6.0). Endogenous peroxidase activity was blocked with 3% hydrogen peroxide, and sections were incubated overnight with anti-PAK1 (1:750) and anti-CAMKII (1:1000) antibodies. Visualization was performed using DAB substrate, counterstained with hematoxylin, and analyzed using ImageJ software version 1.53t (NIH, Bethesda, MD, USA).
2.8. Bioinformatic Analysis of RNA-Sequencing Data from Human Pancreatic Islets
Publicly available RNA-seq data from human pancreatic islets were retrieved from the Gene Expression Omnibus (GEO) database (Accession No: GSM5009229) as reported by Wigger et al. [
29]. The dataset included samples from healthy donors (
n = 15), prediabetic individuals (
n = 11), and patients diagnosed with Type 2 Diabetes Mellitus (T2DM,
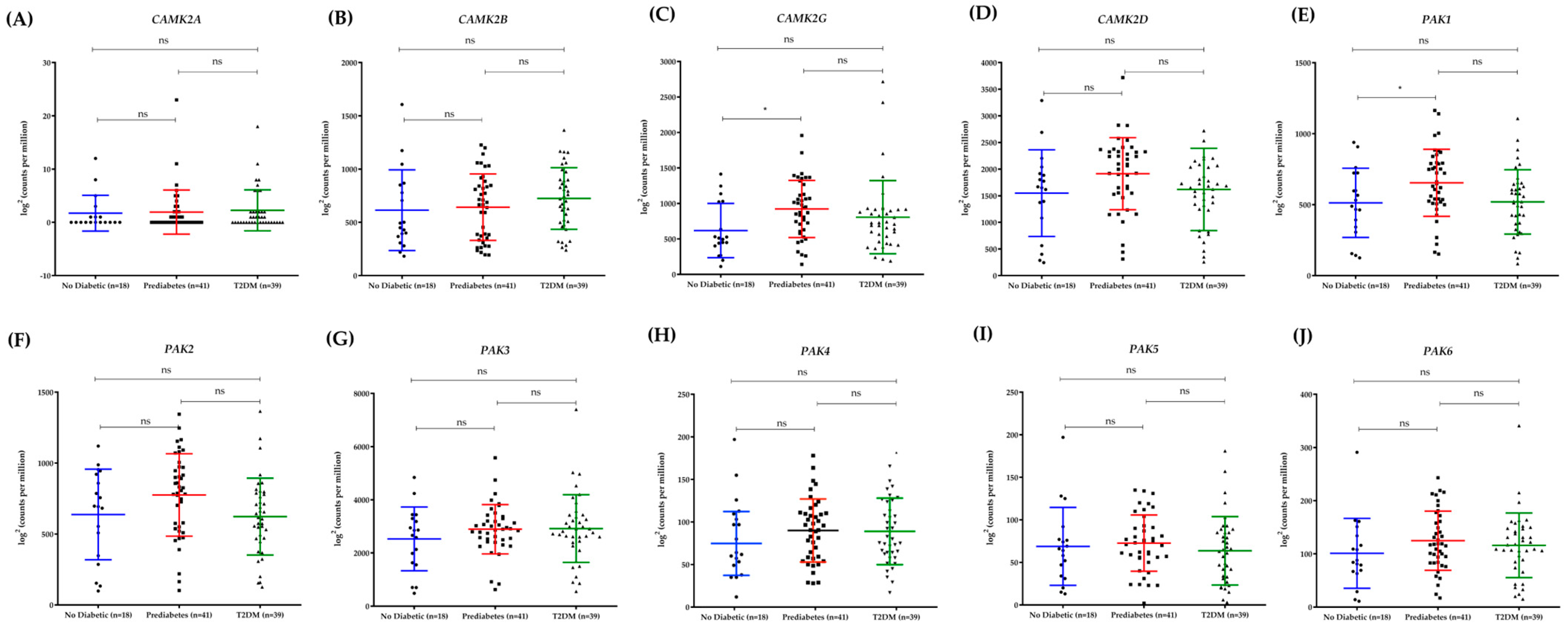
n = 39). Differential gene expression analysis was performed using DESeq2 (R Bioconductor) to assess transcriptional changes associated with pancreatic β-cell function and diabetes progression. Raw read counts were normalized using the Trimmed Mean of M-values (TMM) method, and transcript abundances were expressed as transcripts per million (TPM) and reads per kilobase per million mapped reads (RPKM). Principal component analysis (PCA) was conducted to visualize sample clustering and assess batch effects. The analysis included the Pak kinase family genes (
PAK1, ENSG00000149269;
PAK2, ENSG00000180370;
PAK3, ENSG00000077232;
PAK4, ENSG00000163902;
PAK5, ENSG00000101349;
PAK6, ENSG00000137843) and the CAMKII isoforms (
CAMK2A, ENSG00000070831;
CAMK2B, ENSG00000152492;
CAMK2G, ENSG00000157388;
CAMK2D, ENSG00000182484). To further contextualize these findings, we examined the expression of genes critical to pancreatic β-cell function, glucose metabolism, and insulin signaling, including
INS (ENSG00000254647),
INSR (ENSG00000171105),
MAFA (ENSG00000133048),
PDX1 (ENSG00000139515),
GCK (ENSG00000106633),
NEUROD1 (ENSG00000100461),
FOXO1 (ENSG00000150907), and
PKM (ENSG00000067225). Gene expression levels were compared across non-diabetic, prediabetic, and T2DM groups to assess progressive transcriptional changes.
2.9. Statistical Analysis
Data are presented as mean ± standard error of the mean (SEM). Statistical significance was determined using one-way ANOVA followed by Dunnett’s post hoc test for multiple comparisons, or Student’s t-test for pairwise comparisons, as appropriate. Prior to statistical testing, normality was assessed using the Shapiro-Wilk test, and homogeneity of variances was evaluated with Levene’s test to ensure the validity of parametric analyses. For non-normally distributed data, the Kruskal-Wallis test followed by Dunn’s post hoc test was used instead of ANOVA. To evaluate the correlation between Pak1 and CaMKII colocalization, Pearson’s correlation coefficient (r2) was used for normally distributed data, whereas Spearman’s rank correlation (ρ) was applied when normality assumptions were not met. The statistical significance of the correlation was assessed for both tests. A p < 0.05 was considered statistically significant. All results shown are representative of three independent biological experiments (n = 3 per condition), with technical triplicates performed for each experimental group. Statistical analyses were conducted using GraphPad Prism Software version 9 (San Diego, CA, USA).
4. Discussion
Type 2 Diabetes Mellitus (T2DM) is characterized by progressive β-cell dysfunction, leading to impaired insulin secretion and chronic hyperglycemia [
30]. While numerous molecular pathways governing insulin exocytosis have been extensively studied, emerging evidence suggests that kinases such as PAK1 and CAMKII may play pivotal roles in β-cell adaptation and failure during metabolic stress [
15,
31]. Our findings highlight a glucose-dependent interaction between PAK1 and CAMKII, positioning these kinases as potential modulators of insulin secretion. Furthermore, the observed biphasic expression pattern of PAK1 and CAMKII in murine and human pancreatic islets suggests a transient compensatory role in early diabetes, followed by dysregulation in advanced disease stages.
Our data reveal a strong colocalization and interaction between PAK1 and CAMKII in pancreatic β-cells, which is significantly enhanced under hyperglycemic conditions. The increase in PLA signals under high glucose suggests a direct functional interplay between these kinases, potentially coordinating cytoskeletal remodeling and vesicle trafficking during insulin secretion [
15]. Compared to other proteins involved in insulin granule mobilization, PAK1 shares functional similarities with Cdc42 and Rac1, which regulate actin polymerization and vesicle translocation [
5]. CAMKII, on the other hand, operates in a complementary manner to synaptotagmins and Rab3, which modulate Ca
2+-dependent vesicle priming and fusion with the plasma membrane [
23]. This suggests that PAK1 and CAMKII act as integrators between cytoskeletal remodeling and Ca
2+ signaling, a mechanism distinct from SNARE proteins that primarily facilitate vesicle docking.
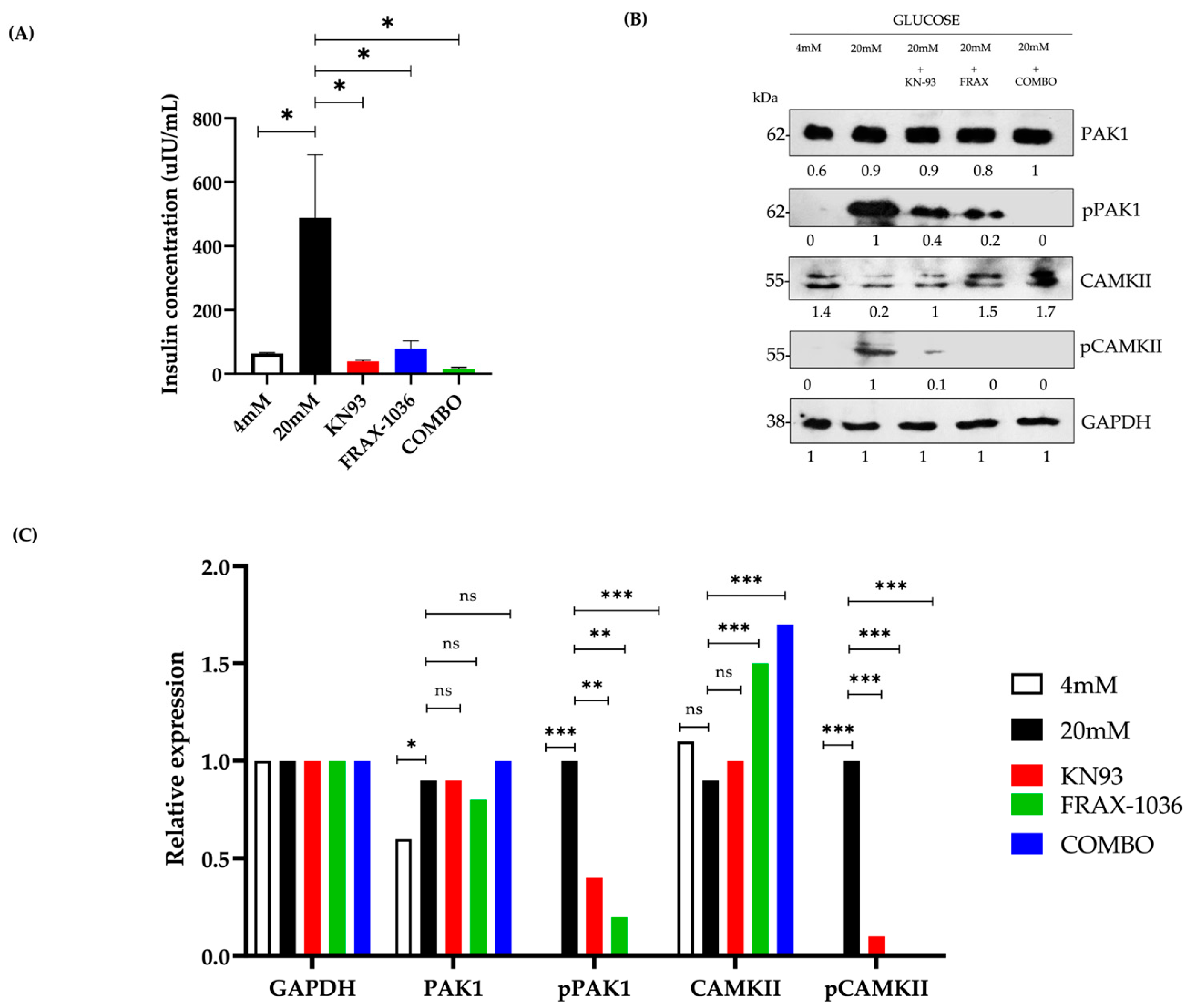
Further supporting this notion, our western blot analyses indicate that glucose stimulation leads to the phosphorylation of both PAK1 and CAMKII, correlating with increased insulin secretion. The inhibition of either kinase significantly blunted glucose-stimulated insulin release, reinforcing their critical roles in β-cell function. Quantitative analysis demonstrated that individual inhibition of CaMKII (KN-93) or PAK1 (FRAX-1036) led to a ~75% and ~70% reduction in insulin secretion, respectively (
p < 0.01). However, dual inhibition resulted in an ~90% suppression of insulin release (
p < 0.001), a reduction greater than the expected additive effect. This suggests a synergistic relationship between these kinases in β-cell insulin secretion, rather than a simple parallel contribution of independent pathways (
Figure 3A). These results align with previous findings showing that Pak1 knockout mice develop glucose intolerance and defective insulin secretion due to impaired cytoskeletal reorganization and granule trafficking [
18]. Likewise, pharmacological inhibition of CaMKII has been associated with attenuated first-phase insulin secretion and β-cell dysfunction [
32]. Our study extends these findings by providing direct evidence that simultaneous inhibition of PAK1 and CAMKII leads to a more severe defect in insulin secretion than single inhibition, reinforcing the concept that both kinases work in a coordinated manner to regulate β-cell physiology. Further investigation is required to determine whether this synergy involves direct interaction between the kinases or a shared downstream effector controlling insulin granule exocytosis. Interestingly, we also observed that individual inhibition of either PAK1 or CAMKII reduced the phosphorylation levels of both kinases, suggesting a bidirectional regulatory relationship rather than off-target effects. While FRAX-1036 and KN93 are reported to be selective for PAK1 and CAMKII respectively, their effects on the phosphorylation state of the other kinase may reflect functional interdependence within a shared signaling network.
The immunohistochemical analysis of murine pancreatic islets revealed a striking biphasic expression pattern of PAK1 and CAMKII during diabetes progression. In prediabetic mice, expression levels were significantly elevated compared to wild-type controls, suggesting an adaptive response to increased insulin demand during the hyperinsulinemic phase [
33,
34]. However, in T2DM mice, both kinases were markedly downregulated, correlating with β-cell failure and reduced insulin secretion. This pattern was mirrored in our analysis of human pancreatic islets, where PAK1 and CAMKII expression peaked in prediabetic individuals before declining in T2DM patients. These findings align with previous reports demonstrating that β-cell compensation during early diabetes involves transient upregulation of key signaling pathways, which eventually become dysfunctional as metabolic stress progresses. While most functional studies on PAK1 and CAMKII in insulin secretion have been conducted in murine models and insulinoma cell lines, our RNA-Seq analysis of human pancreatic islets demonstrated that PAK1 is differentially expressed in prediabetes and T2DM, suggesting that its regulation is also relevant in humans. The biphasic expression pattern of PAK1 and CAMKII observed in both murine and human pancreatic islets suggests a temporal shift in β-cell signaling during the course of diabetes progression. The initial upregulation in prediabetes may reflect a compensatory adaptation to increased insulin demand, where both kinases are activated to enhance cytoskeletal remodeling and calcium-dependent secretion. This phase likely corresponds to the hyperinsulinemic stage seen in early T2DM. However, the marked downregulation observed in overt T2DM may signal β-cell exhaustion, where chronic metabolic stress, glucotoxicity, and inflammation impair kinase expression and signaling capacity. Such a transition from overcompensation to failure mirrors broader models of β-cell decompensation and supports the hypothesis that PAK1 and CAMKII are dynamic regulators of β-cell resilience. Understanding this biphasic behavior could help identify biomarkers for early detection of β-cell decline or targets for therapeutic intervention before irreversible failure occurs. It is important to note that the RNA-seq data reflects basal transcriptional changes across metabolic states, whereas our experiments in Beta-TC-6 cells evaluated dynamic, glucose-induced phosphorylation events under acute stimulation. The observed lack of change in total protein levels despite increased phosphorylation is consistent with the post-translational regulation typical of kinase signaling. Therefore, the divergence between RNA and protein phosphorylation data does not indicate a contradiction, but rather highlights the distinct regulatory layers—transcriptional versus post-translational—captured in each model. However, the functional role of PAK1 in human β-cells remains largely unexplored. Future studies should aim to validate these findings in primary human islets and evaluate whether PAK1 modulation can restore insulin secretion in T2DM. Additionally, the interplay between PAK1, CAMKII, and other key β-cell regulators such as GLP-1 signaling warrants further investigation to determine its therapeutic potential. [
29,
35].
The observed decline in PAK1 and CAMKII expression in T2DM may be attributed to chronic hyperglycemia-induced β-cell exhaustion. Sustained metabolic stress has been shown to impair kinase signaling pathways, leading to defects in insulin secretion and β-cell apoptosis [
36]. Additionally, oxidative stress and glucotoxicity have been implicated in the degradation of cytoskeletal proteins and calcium signaling components, which may further contribute to the observed downregulation of these kinases [
37]. Understanding the molecular mechanisms underlying this transition from compensation to decompensation could provide valuable insights into β-cell failure in T2DM.
Given the critical roles of PAK1 and CAMKII in insulin secretion, their modulation may represent a novel therapeutic avenue for preserving β-cell function in T2DM. Previous studies have explored the potential of kinase activators in enhancing insulin secretion, with promising results in preclinical models [
38,
39]. Pharmacological activation of PAK1 has been shown to improve β-cell survival and insulin release under metabolic stress conditions [
18]. Likewise, selective CAMKII modulators have been proposed to restore β-cell calcium homeostasis and insulin granule mobilization in diabetic models [
23]. However, chronic CAMKII hyperactivation has been linked to β-cell apoptosis and oxidative stress, underscoring the need for precise therapeutic modulation [
40]. From a therapeutic perspective, targeted modulation of PAK1 and CAMKII could enhance insulin secretion at multiple levels. Activation of PAK1 may promote second-phase insulin release by facilitating cytoskeletal remodeling and vesicle mobilization, while controlled activation of CAMKII could improve first-phase insulin secretion through enhanced calcium-dependent exocytosis. In preclinical models, PAK1 activation has been shown to increase β-cell survival and insulin content under stress conditions, and CAMKII modulation has improved glucose-stimulated insulin secretion by restoring calcium dynamics. However, because excessive CAMKII activity has also been linked to oxidative stress and β-cell apoptosis, future therapies would need to fine-tune its activity rather than globally increase it. Selective activators or allosteric modulators of PAK1 and CAMKII, possibly in combination with existing incretin-based therapies, could offer a novel strategy to preserve β-cell function and delay the progression of T2DM. Future studies should investigate the feasibility of targeting these kinases with selective agonists or inhibitors to fine-tune their activity and restore β-cell function in T2DM.
Additionally, the potential of PAK1 and CAMKII as biomarkers for β-cell resilience warrants further exploration. Their early upregulation in prediabetes suggests that they may serve as indicators of β-cell compensatory capacity, helping to identify individuals at risk of disease progression before β-cell failure becomes irreversible. Longitudinal studies analyzing kinase expression in relation to metabolic parameters could provide valuable predictive insights into diabetes progression.
While our study provides compelling evidence for the functional interplay between PAK1 and CAMKII in β-cell physiology, several limitations should be acknowledged. First, although we demonstrated kinase colocalization and interaction using PLA and immunofluorescence, further biochemical analyses such as co-immunoprecipitation are needed to confirm direct protein-protein interactions. Second, our study primarily relies on pharmacological inhibition, which may have off-target effects; future investigations using β-cell-specific knockout models for PAK1 and CAMKII would provide more definitive insights. Finally, while our human islet analysis highlights the clinical relevance of these kinases, functional studies in human β-cells are necessary to validate their roles in insulin secretion and diabetes pathophysiology.
Future research should explore the molecular mechanisms underlying PAK1-mediated CAMKII activation, particularly the downstream targets involved in β-cell survival and insulin exocytosis. Additionally, studies investigating the effects of targeted kinase modulation in in vivo diabetes models could pave the way for novel therapeutic strategies aimed at preserving β-cell function in T2DM.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}