Abstract
Background: Tuberculosis (TB) and diabetes mellitus (DM) comorbidity (TB-DM) presents a significant global health challenge, with diabetes increasing susceptibility to TB, worsening clinical outcomes, and impairing immune responses. Among these dysfunctions, macrophages—the primary immune cells responsible for pathogen recognition, phagocytosis, and bacterial clearance—exhibit profound alterations in TB-DM. However, the complex interplay between metabolic dysregulation, immune impairment, and macrophage dysfunction remains poorly defined. Objective: This scoping review systematically maps the literature on macrophage dysfunction in TB-DM, identifying key immunological impairments affecting phagocytosis, cytokine production, antigen presentation, macrophage polarisation, reactive oxygen species (ROS) and nitric oxide (NO) regulation, and chronic inflammation. Methods: A systematic search was conducted in PubMed, Web of Science, and Embase, covering studies from 2014 to 2024. Inclusion criteria focused on human studies investigating macrophage-specific mechanisms in TB-DM. Data extraction and synthesis were performed using Covidence, with findings grouped into key immunological themes. Results: A total of 44 studies were included, revealing significant impairments in macrophage function in TB-DM. Findings indicate reduced NO production, variable ROS dysregulation, altered M1/M2 polarisation, defective antigen presentation, and chronic inflammation. Elevated IL-10 and VEGF were associated with immune suppression and granuloma destabilisation, while eicosanoids (PGE2, LXA4) contributed to sustained inflammation. Conclusions: Macrophage dysfunction emerges as a central driver of immune failure in TB-DM, creating a self-perpetuating cycle of inflammation, immune exhaustion, and bacterial persistence. Understanding these mechanisms is essential for developing biomarker-driven diagnostics, host-directed therapies, targeted immunomodulation, and improving TB outcomes in diabetic populations. Future research should explore macrophage-targeted interventions to enhance immune function and mitigate TB-DM burden.
1. Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis (M. tb), remains a leading cause of death from infectious diseases worldwide, with its burden disproportionately affecting low- and middle-income countries [1]. Despite significant advances in TB control, the disease persists as a major global health challenge, particularly in populations with predisposing conditions. Among these, diabetes mellitus (DM) has emerged as a significant risk factor for TB, not only increasing susceptibility to infection but also worsening clinical outcomes, including higher relapse rates, delayed sputum conversion, and increased mortality [2]. The global prevalence of DM is rising rapidly, affecting over 537 million adults in 2021, with projections exceeding 700 million by 2045 [3]. As DM prevalence continues to increase, its convergence with TB, commonly referred to as TB-DM comorbidity, represents a growing public health crisis, particularly in regions where both conditions are highly prevalent.
The immunological consequences of TB-DM comorbidity remain incompletely understood, but it is evident that DM profoundly alters the host immune response, impairing the body’s ability to control M. tb [4]. Macrophages, the primary immune cells involved in pathogen recognition, phagocytosis, and bacterial clearance, are particularly vulnerable to dysfunction in the context of DM. These cells serve a dual role as both the first line of defence and as the primary intracellular reservoir for M. tb, making their function critical to disease progression. However, in TB-DM, macrophage phagocytosis, cytokine production, antigen presentation, and polarisation are disrupted, leading to reduced bacterial clearance, chronic inflammation, and persistent infection [4,5].
Although individual aspects of macrophage dysfunction in TB-DM have been well documented—including reduced nitric oxide (NO) production, variable reactive oxygen species (ROS) regulation, altered polarisation, and cytokine imbalances—how these mechanisms interact to drive disease progression remains poorly defined. Existing research often examines these pathways in isolation, failing to capture the complex interplay between metabolic dysfunction and TB immunopathogenesis. Variability in findings across studies, particularly regarding ROS production and cytokine dynamics, further underscores the need for a comprehensive synthesis of the available evidence.
This scoping review systematically maps the literature on macrophage dysfunction in TB-DM, focusing on key macrophage-mediated immune processes: phagocytosis, cytokine production, antigen presentation, polarisation, the regulation of ROS and NO, and chronic inflammation. Unlike systematic reviews, which aim to establish causal relationships, this scoping review takes a broader approach to synthesise evidence across diverse study designs, identify knowledge gaps, and provide an updated understanding of macrophage dysfunction in TB-DM. By integrating findings from the past decade (2014–2024), this review aims to clarify how diabetes alters macrophage function in TB, inform future research priorities, and contribute to the development of immunomodulatory therapies and improved clinical management strategies.
2. Methods
2.1. Eligibility Criteria
The eligibility criteria included original research articles investigating macrophage function in human populations with TB and DM, compared with individuals with TB alone. Studies were required to focus on macrophage-specific mechanisms, including phagocytosis, cytokine production, antigen presentation, macrophage polarisation, ROS and NO production, and chronic inflammation. Articles published between 2014 and 2024 were included to ensure relevance to current research. Only studies published in English were considered due to resource limitations for translation. Reviews, meta-analyses, editorials, opinion pieces, and studies involving animal models or unrelated comorbidities were excluded to ensure that the synthesis focused solely on new research relevant to macrophage dysfunction in TB-DM.
2.2. Information Sources
The primary sources of evidence were the bibliographic databases PubMed, Web of Science, and Embase. These databases were chosen for their extensive coverage of biomedical research, multidisciplinary scope, and inclusion of high-quality, peer-reviewed studies. Searches were conducted for studies published between 1 January 2014 and 31 December 2024, with the final search completed in January 2025. The reference lists of the included studies were manually reviewed to identify additional relevant articles. The Covidence platform was utilised to streamline the screening, selection, and data extraction processes [6].
A systematic search strategy was developed and refined for each database to ensure thorough and reproducible results. Boolean operators and controlled vocabulary terms (e.g., MeSH, Emtree) were used to capture relevant studies. The search strategies for each database are outlined below:
PubMed: (“Tuberculosis”[MeSH] OR “TB” OR “tuberculous”) AND (“Diabetes Mellitus”[MeSH] OR “diabetes” OR “Type 1 Diabetes” OR “Type 2 Diabetes” OR “T1DM” OR “T2DM”) AND (“Macrophages”[MeSH] OR “macrophage function” OR “cytokines” OR “antigen presentation” OR “immune dysregulation” OR “reactive oxygen species” OR “ROS” OR “nitric oxide” OR “NO”). Filters were applied to include studies published between 1 January 2014 and 31 December 2024 and limited to articles written in English. PubMed was selected for its broad coverage of biomedical and clinical research.
Web of Science: TS = (“tuberculosis” OR “TB” OR “tuberculous”) AND TS = (“diabetes mellitus” OR “diabetes” OR “Type 1 Diabetes” OR “Type 2 Diabetes” OR “T1DM” OR “T2DM”) AND TS = (“macrophage function” OR “cytokines” OR “antigen presentation” OR “immune dysregulation” OR “reactive oxygen species” OR “ROS” OR “nitric oxide” OR “NO”). Filters were applied to restrict results to studies published in English between 1 January 2014 and 31 December 2024. Web of Science was selected for its cross-disciplinary scope and citation-tracking features.
Embase: (‘tuberculosis’/exp OR ‘tuberculosis’ OR ‘TB’ OR ‘tuberculous’) AND (‘diabetes mellitus’/exp OR ‘diabetes mellitus’ OR ‘diabetes’ OR ‘Type 1 Diabetes’ OR ‘Type 2 Diabetes’ OR ‘T1DM’ OR ‘T2DM’) AND (‘macrophages’/exp OR ‘macrophage function’ OR ‘cytokines’ OR ‘antigen presentation’ OR ‘immune dysregulation’ OR ‘reactive oxygen species’ OR ‘ROS’ OR ‘nitric oxide’ OR ‘NO’). Filters limited the search to articles published in English between 1 January 2014 and 31 December 2024. Embase was chosen for its inclusion of biomedical and pharmacological research.
Selection of Sources of Evidence
The selection process was conducted using the Covidence platform. Two reviewers independently screened titles and abstracts for potential eligibility. Articles meeting the inclusion criteria underwent a full-text review, with any disagreements resolved through discussion or consultation with a third reviewer. A PRISMA-ScR [7] flow diagram was used to document the selection process, including the number of records identified, screened, included, and excluded, with reasons for exclusion.
2.3. Data Charting and Extraction
Data were charted using a standardised extraction form within the Covidence platform, capturing study characteristics (e.g., authors, year, and study design), population details (e.g., TB, TB-DM, and latent or active TB), and macrophage-specific outcomes. Two reviewers independently performed data extraction to ensure accuracy and consistency, resolving discrepancies through discussion or consultation with a third reviewer.
The extracted variables included macrophage-specific outcomes, such as phagocytosis efficacy, cytokine profiles (e.g., interleukin (IL)-10, IFN-γ, and TNF-α), antigen presentation markers (e.g., major histocompatibility complex (MHC) class II (MHC-II) expression), macrophage polarisation markers (e.g., M1/M2), and levels of ROS and NO. Where necessary, data were grouped to enhance clarity in reporting and synthesis.
2.4. Critical Appraisal of Individual Sources of Evidence
The critical appraisal of individual studies was not performed, as the purpose of this scoping review was to map the evidence and identify gaps rather than assess study quality. This approach aligned with the exploratory nature of scoping reviews, allowing for the inclusion of a broader range of evidence.
2.5. Synthesis of Results
The data were synthesised descriptively, with findings grouped according to the predefined themes of macrophage dysfunction. Results were presented narratively, supported by tables summarising the study characteristics and key outcomes. The synthesis integrated evidence across studies to highlight the common mechanisms of macrophage dysregulation in TB-DM and identify areas for further research. Covidence facilitated the organisation and synthesis of the data, ensuring a clear and systematic presentation.
3. Results
3.1. Selection of Sources of Evidence
Overview: Although formal quality assessment was not conducted, studies were rigorously selected to ensure relevance and reliability. The systematic approach to study selection strengthened the review’s focus on macrophage dysfunction in TB-DM comorbidity, providing a solid foundation for synthesis.
The initial database search identified 822 studies. After the removal of 300 duplicates, 522 studies remained for screening. The titles and abstracts of these studies were screened independently by two reviewers, excluding 444 studies that did not meet the inclusion criteria. The remaining 78 studies underwent full-text review, with 34 excluded for reasons such as non-macrophage focus, lack of original data, or failure to meet thematic relevance. Ultimately, 44 studies were included in the review. All stages of the review process were documented using a PRISMA-ScR flow diagram (Figure 1), providing a clear representation of the study selection process.
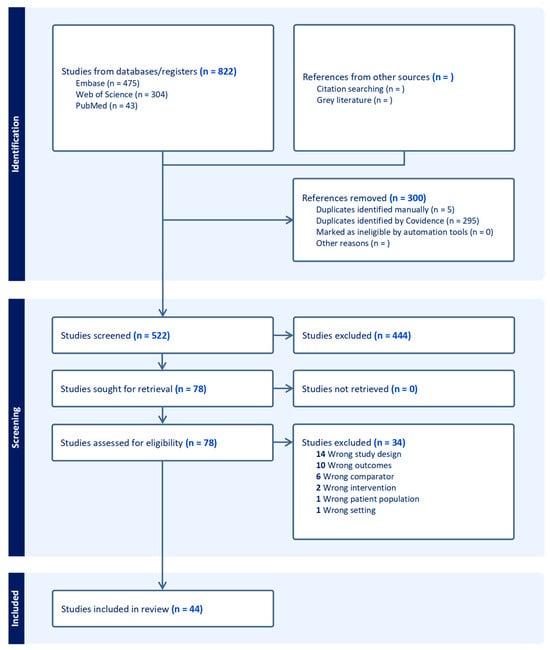
Figure 1.
PRISMA flow diagram. This flow diagram outlines the study selection process for the scoping review. A total of 822 studies were identified from three databases (Embase, Web of Science, and PubMed). After removing 300 duplicates, 522 studies were screened, with 444 excluded based on the title and abstract review. Seventy-eight full-text studies were assessed for eligibility, with thirty-four excluded for reasons such as study design, outcomes, comparator, intervention, patient population, or setting. Ultimately, 44 studies were included in the final review.
3.2. Characteristics of Sources of Evidence
The included studies encompass diverse designs and outcomes, reflecting a comprehensive examination of macrophage dysfunction in TB-DM. The wide geographic scope and range of publication years further ensure the findings’ generalisability and relevance.
The 44 included studies encompassed a range of study designs, including observational studies, cross-sectional analyses, and immunophenotyping investigations, with a focus on macrophage dysfunction in the context of TB and TB-DM. These studies reported on various macrophage-specific outcomes. Upon review of these outcomes, five key and reciprocally influenced themes emerged: altered cytokine signalling profiles (e.g., IL-10, IFN-γ, and TNF-α), the production of ROS and NO, altered macrophage polarisation (M1/M2 profiles), impaired antigen presentation markers (e.g., MHC-II expression) with reduced phagocytosis efficacy and cellular exhaustion, and chronic inflammation and systemic dysfunction. The included studies were published between 2014 and 2024 and represented research conducted across diverse settings.
While this review includes studies from a wide range of geographic settings—including South and Southeast Asia, Latin America, and Sub-Saharan Africa—there was limited consistency in reporting population-specific variables, making it difficult to compare findings by region. However, immune responses may vary significantly by geographic and ethnic background due to differences in host genetics, co-infections, environmental exposures, and healthcare access. The Supplementary Materials (Supplemental Table S1) provide a detailed table summarising the study characteristics, including authorship, year of publication, study design, population, and key findings. The citations for all the included studies are listed in the Reference Section.
3.2.1. Cytokines in TB-DM Comorbidity
Cytokine dysregulation is a key driver of immune dysfunction in TB-DM comorbidity. Hyperglycaemia and associated metabolic dysfunction alter the balance between pro-inflammatory cytokines (e.g., TNF-α, IL-6, and IFN-γ) and anti-inflammatory cytokines (e.g., IL-10 and TGF-β), creating a paradoxical immune state characterised by excessive inflammation and suppressed immune resolution [5,8,9,10]. These imbalances disrupt granuloma formation and maintenance [11], compromising the host’s ability to contain M. tb and promoting bacterial dissemination (Figure 2).
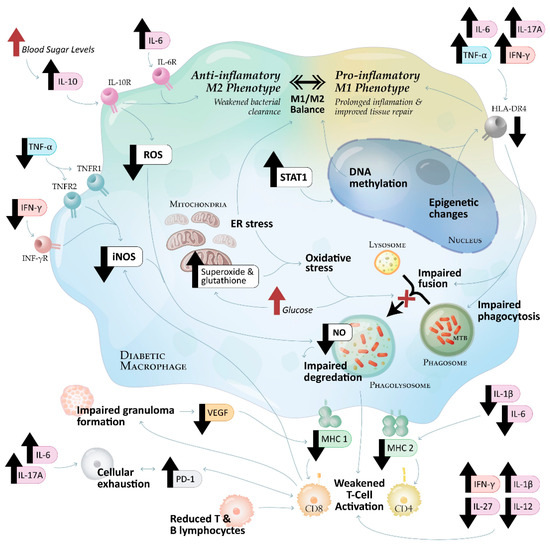
Figure 2.
Macrophage dysfunction in tuberculosis–diabetes mellitus (TB-DM) comorbidity: key immune dysregulation pathways. Immune dysregulation in tuberculosis (TB) and diabetes (DM), highlighting key cytokine and metabolic interactions impairing macrophage function and T-cell activation. Reduced TNF-α (via TNFR1/TNFR2) and IFN-γ (via IFN-γR) signalling leads to diminished inducible nitric oxide synthase (iNOS) expression, reducing nitric oxide (NO) production and impairing phagolysosomal degradation. Hyperglycaemia increases IL-10, signalling through IL-10R to promote M2 macrophage polarisation and suppress reactive oxygen species (ROS), further reducing NO production and bacterial clearance. IL-6 (via IL-6R) also contributes to M2 differentiation.
Cytokines directly influence macrophage bactericidal activity, ROS and NO production [12,13], antigen presentation [14], macrophage polarisation [12], and chronic inflammation [15]. The disruption of this intricate coordination impairs the immune system’s capacity to effectively control M. tb, resulting in bacterial persistence [16,17], enhanced tissue damage [9,15], and immune exhaustion [18]. Cytokine dysregulation in TB-DM not only disrupts immune responses at the site of M. tb infection, such as within granulomas, but also triggers widespread systemic inflammation [19,20]. This systemic inflammation exacerbates immune dysfunction and causes tissue damage in areas beyond the localised infection, further impairing the body’s ability to control the disease [15,21].
While cytokine dysregulation is a well-documented phenomenon in TB-DM, some studies highlight variability in cytokine profiles depending on factors such as glycaemic control, disease stage, and study methodologies [9,15,21]. For example, although reduced TNF-α and IFN-γ levels are commonly reported, some studies have observed persistently elevated TNF-α levels, suggesting that macrophage dysfunction may arise from impaired responsiveness to cytokines rather than their outright suppression [15,19]. Similarly, IL-10 levels have been shown to fluctuate, with higher levels associated with poor glycaemic control, underscoring the heterogeneity of immune responses in TB-DM [9,22].
Thus, cytokine dysregulation serves as a pivotal mediator of macrophage dysfunction and immune imbalance in TB-DM. The following sections will examine how cytokine-driven imbalances and other contributing factors influence key immune processes, including ROS/NO production, macrophage polarisation, antigen presentation, and chronic inflammation.
3.2.2. Impacts of Reactive Oxygen Species and Nitric Oxide in TB-DM Comorbidity
Overview
ROS and NO are essential components of the macrophage-mediated immune response against M. tb. NO is a key effector molecule that disrupts bacterial metabolism, while ROS induces oxidative damage to bacterial DNA, proteins, and membranes, assisting in pathogen clearance [13]. However, these molecules must be tightly regulated, as excessive production can lead to oxidative stress and tissue damage, while insufficient production compromises bacterial killing and immune control [9,13]. Disruptions in ROS and NO production due to cytokine imbalances and metabolic dysfunction are summarised in Figure 2.
Cytokines such as interferon-gamma (IFN-γ) and tumour necrosis factor-alpha (TNF-α) play a critical role in the regulation of NO and ROS by activating macrophages and upregulating inducible nitric oxide synthase (iNOS) [9,13,23]. However, in TB-DM comorbidity, this regulation is disrupted due to hyperglycaemia-induced metabolic changes, cytokine imbalances, and epigenetic modifications, leading to immune dysfunction and exacerbated oxidative stress [13,19]. The following sections summarise these findings and their implications for macrophage function.
Key Findings
- Reduced Nitric Oxide Production
A consistent finding across studies was the likely suppression of NO production, impairing macrophage bactericidal activity. While only one study [13] directly measured NO reduction, several studies reported mechanisms that would lead to NO suppression, including the downregulation of iNOS, reduced IFN-γ and TNF-α levels, and increased oxidative stress markers [14,23,24,25] (Figure 2).
Epigenetic modifications and metabolic dysregulation further compounded NO suppression in TB-DM comorbidity. Panda et al. [13] reported that chronic hyperglycaemia led to a reduction in NO production, impairing macrophage-mediated bacterial clearance. This effect was particularly pronounced in individuals with poorly controlled diabetes, where persistent hyperglycaemia reinforced inflammatory dysfunction (Figure 2). Although Kumar et al. [15] described persistent inflammation in TB-DM, further research is needed to confirm whether hyperglycaemia directly induces the DNA methylation of the inducible nitric oxide synthase (iNOS) promoter. Additionally, Fernández et al. [22] found that cortisol dysregulation in TB-DM contributed to immune suppression by impairing pro-inflammatory cytokine signalling, which could indirectly reduce NO production. Collectively, these factors contribute to a significant impairment in NO-dependent bacterial killing, rendering TB-DM patients more vulnerable to disease progression (Figure 2).
- Dysregulated Reactive Oxygen Species Production
Unlike NO, ROS production demonstrated greater variability across studies, with findings indicating both excessive and insufficient ROS production, depending on disease stage and metabolic status. Several studies [13,19] reported that hyperglycaemia-induced oxidative stress resulted in excessive ROS production, contributing to tissue damage and immune dysfunction. Elevated glucose levels in diabetes are known to increase mitochondrial superoxide production and alter glutathione peroxidase activity, thereby contributing to oxidative stress and exacerbating inflammatory damage [26]. While ROS overproduction is initially part of the immune response against M. tb, sustained oxidative stress damages host tissues, promoting immune exhaustion and ineffective pathogen clearance.
Notably, chronic oxidative stress in TB-DM is further amplified by dysregulated inflammatory signalling via TRAF6-mediated pathways and prolonged ER stress, both of which contribute to sustained ROS generation and immune dysfunction [27,28]. TRAF6 activation can enhance ROS generation through NADPH oxidase stimulation, while prolonged ER stress disrupts redox homeostasis, increasing oxidative damage [19]. These mechanisms create a self-perpetuating feedback loop where excessive ROS stimulates pro-inflammatory cytokine secretion, damages mitochondrial function, and prolongs immune activation, ultimately worsening disease progression in TB-DM [29]. The interplay between oxidative stress, ER stress, and macrophage dysfunction is illustrated in Figure 2.
In contrast, other studies observed reduced ROS production in TB-DM, particularly in individuals with poorly controlled diabetes and prolonged hyperglycaemia [25]. Advanced metabolic dysfunction was linked to immune dysfunction and a defective oxidative burst—a rapid release of ROS by phagocytes, crucial for intracellular bacterial killing—resulting in weakened immune responses [21]. Additionally, elevated levels of IL-10, an anti-inflammatory cytokine, were associated with ROS suppression, further reducing oxidative burst capacity and impairing pathogen clearance [25].
These findings suggest that ROS dysregulation in TB-DM is dynamic, with early-stage hyperglycaemia driving excessive ROS production, whereas prolonged metabolic dysfunction leads to ROS deficiency and impaired bacterial killing [13,19]. The variability in ROS findings may reflect differences in disease progression, glycaemic control, and immune-modulating factors across study populations. A comprehensive breakdown of study-specific findings and conflicting results is provided in Supplemental Table S2.
3.2.3. Dysregulation of Macrophage Polarisation in TB-DM
Overview
Dysregulated macrophage polarisation is a hallmark of TB-DM comorbidity [30]. The imbalance between the pro-inflammatory M1 phenotype and the anti-inflammatory M2 phenotype impairs immune function, leading to chronic inflammation, tissue damage, and reduced bacterial clearance. Findings from 29 studies highlight cytokine imbalances, oxidative stress, and molecular dysregulation as key drivers that disrupt macrophage polarisation (Figure 2). Key macrophage polarisation observations and their impacts are summarised in Table 1.

Table 1.
Overview of macrophage dysregulation in tuberculosis–diabetes mellitus comorbidity by thematic group. Key observations related to immune dysregulation in tuberculosis–diabetes mellitus (TB-DM) comorbidity, focusing on macrophage function. This table highlights how specific mechanisms affect macrophage activity for each theme, contributing to immune dysfunction and disease progression. References are included to provide evidence for each observation. See Supplemental Table S1 for detailed descriptions and additional references.
Key Findings
- Dysregulated Macrophage Polarisation in TB-DM
In TB-DM, macrophages are skewed toward the pro-inflammatory M1 phenotype, which initially supports bacterial killing. However, chronic metabolic dysfunction sustains this M1 dominance, leading to prolonged inflammation, impaired tissue repair, and suboptimal pathogen clearance. Elevated levels of pro-inflammatory cytokines, including IFN-γ, TNF-α, IL-6, IL-17A, and IL-1β, reinforce M1 dominance [5,8,12,14,20,24,31,33,34].
In TB-DM, the excessive activation of the STAT1 signalling pathway, usually critical for M1 polarisation and antimicrobial responses [44], leads to immune dysregulation in TB-DM. While STAT1 activation enhances pathogen clearance in early infection, persistent overactivation reinforces inflammatory cytokine production and limits macrophage plasticity, preventing adaptive immune responses [44]. This leads to an impaired resolution of infection, excessive tissue damage, and a failure to effectively transition toward an anti-inflammatory M2 phenotype that is necessary for healing [15,39].
In TB-DM, macrophages are either locked in a sustained M1-dominant state or undergo an incomplete transition to a dysfunctional M2 phenotype, impairing immune resolution. Cytokine imbalances play a central role in this dysfunction. Elevated IL-6 and IL-10 levels [9,34,36] promote anti-inflammatory states that suppress M1 activity, while reduced IL-22, crucial for antimicrobial defence, weakens pathogen control [45,46].
When the M2 transition occurs, it is often incomplete or maladaptive. Elevated IL-10 and VEGF levels suggest an M2-like state that prioritises tissue remodelling and angiogenesis over bacterial clearance, enabling pathogen persistence [9,13,36]. Oxidative stress further impairs macrophage function, as excessive ROS damages host tissues, while reduced NO weakens bacterial clearance by M1 macrophages [13,15,24]. Epigenetic and molecular dysregulation, including DNA methylation and ER stress, impair pathways essential for balanced M1/M2 activity [19,39].
Finally, pathological angiogenesis driven by VEGF exacerbates polarisation dysfunction, disrupting granuloma integrity and facilitating chronic infection [8,46]. Simultaneous elevations of IL-13 and pro-inflammatory cytokines further create a dysregulated immune landscape that undermines the balance between M1 and M2 states [40].
Overall, macrophages in TB-DM exhibit dysregulated polarisation characterised by persistent M1 dominance or maladaptive M2 activity, contributing to chronic inflammation, tissue damage, and bacterial survival. Table 1 outlines the overarching impacts of polarisation imbalances in TB-DM.
Conflicting Findings
Most studies report M1 dominance and an incomplete or maladaptive M2 transition in TB-DM, but some findings indicate variability in polarisation dynamics. Fernández et al. [34] observed that IL-10 and IL-6 promoted M2 polarisation in certain contexts, suggesting a potential compensatory anti-inflammatory response. In contrast, Kumar et al. [41] noted that despite the presence of IL-10, macrophages remained locked in an M1-dominant state, highlighting a possible resistance to M2 transition. Additionally, some studies suggest that VEGF-driven angiogenesis can push macrophages toward an M2-like phenotype that prioritises tissue remodelling over bacterial clearance [13], while others report a failure to transition fully, resulting in an ineffective immune response [36]. These differences may stem from variations in disease stage, glycaemic control, or study methodologies.
These discrepancies highlight the context-dependent nature of macrophage polarisation in TB-DM, suggesting that glycaemic control, disease stage, and inflammatory signalling networks may differentially shape polarisation outcomes across patient populations.
3.2.4. Impaired Antigen Presentation in TB-Diabetes Mellitus
Overview
Phagocytosis is a critical initial step in the immune defence against M. tb, involving macrophages and dendritic cells engulfing the bacterium into a phagosome, which subsequently fuses with a lysosome to form a phagolysosome. Within this compartment, M. tb is degraded into peptides that are presented on MHC molecules, facilitating the activation of T cells. Specifically, MHC-II molecules present these peptides to CD4+ T cells, which release cytokines such as IFN-γ and TNF-α to enhance macrophage bactericidal activity and maintain granuloma stability—a key feature of immune containment [47]. Simultaneously, intracellular M. tb antigens are processed for presentation via MHC-I molecules, activating CD8+ T cells to mediate the cytotoxic killing of infected macrophages [47]. This dual mechanism ensures granuloma integrity, limits bacterial dissemination, and maintains immune control [48].
However, in individuals with TB-DM, this finely tuned immune system is disrupted. MHC class I and II expression are dysregulated, impairing antigen presentation to CD8+ and CD4+ T cells. This weakens T-cell activation, reducing IFN-γ and TNF-α secretion, which are essential for macrophage activation and granuloma maintenance [8,35]. The disrupted antigen presentation pathways, including impaired phagosome–lysosome fusion, reduced MHC expression, and weakened T-cell responses, are illustrated in Figure 2.
Cytokine imbalances in TB-DM disrupt antigen presentation. Elevated TNF-α and IL-1β promote chronic inflammation but impair antigen-presenting cell function, while decreased IL-12 and IL-27 reduce T-cell priming, weakening pathogen-specific immunity [9,49]. These disruptions in antigen presentation and immune activation allow M. tb to persist, leading to disease progression and compromising the host’s ability to contain the infection [23,36].
Key Findings
- Defective Phagocytosis and Antigen Processing
Phagocytosis, the first line of defence against M. tb, is significantly impaired in TB-DM due to chronic hyperglycaemia and oxidative stress, which disrupt phagosome–lysosome fusion and limit bacterial degradation and antigen processing [8,23]. The failure of phagosome–lysosome fusion, a key mechanism contributing to antigen presentation defects, is highlighted in Figure 2.
The resulting oxidative stress and dysregulation of key bactericidal molecules, such as ROS and NO, further impair macrophage function by weakening bacterial clearance and damaging cellular components [9]. These early defects reduce the availability of bacterial peptides for MHC molecule loading, disrupting downstream antigen presentation and T-cell activation.
Beyond metabolic dysfunction, epigenetic changes in TB-DM further impair macrophage function by disrupting phagocytosis and antigen presentation. DNA methylation reduces the expression of HLA-DR and key phagocytic receptors (e.g., Fcγ receptors and complement receptors), limiting bacterial recognition and uptake [19]. Additionally, STAT1 hyperactivation in TB-DM disrupts cytoskeletal remodelling and vesicle trafficking, delaying phagosome maturation and impairing bacterial clearance [39]. This sustains high IFN-γ and TNF-α levels, paradoxically inhibiting macrophage plasticity and reinforcing a cycle of defective phagocytosis and chronic inflammation [39].
- 2.
- MHC Dysregulation and Impaired T-Cell Activation
MHC Class II Impairment (CD4+ T cell dysfunction)
Antigen presentation via MHC-II is critical for CD4+ T cell activation, which facilitates macrophage activation through IFN-γ and TNF-α secretion. However, TB-DM is associated with reduced MHC-II expression due to cytokine imbalances. While TNF-α is essential for upregulating MHC-II expression on macrophages, its levels are often dysregulated in TB-DM, leading to ineffective macrophage activation [34]. Even when TNF-α is elevated, chronic inflammation and cytokine antagonism reduce its functional impact [15,19].
Moreover, low levels of IL-1β and IL-6 further impair MHC-II expression [23], while elevated IL-10 suppresses antigen presentation, allowing M. tb to evade immune detection [9]. Together, these changes reduce CD4+ T cell activation, granuloma stability, and macrophage bactericidal capacity, contributing to persistent infection.
MHC Class I Impairment (CD8+ T cell dysfunction)
MHC-I molecules are essential for presenting intracellular M. tb antigens to CD8+ T cells, enabling the cytotoxic killing of infected macrophages. In TB-DM, VEGF-driven hypoxia within granulomas inhibits MHC-I expression, impairing CD8+ T cell activation and functionality [19]. This effect is compounded by the reduced expression of cytotoxic markers, such as perforin and granzyme B, which are critical for infected macrophage elimination [8].
The combined impairments in MHC-I and MHC-II pathways disrupt both CD4+ and CD8+ T cell responses, weakening granuloma integrity, promoting bacterial dissemination, and facilitating immune evasion (Figure 2).
- 3.
- Cellular Exhaustion
T cell exhaustion, characterised by the elevated expression of inhibitory receptors such as PD-1, is a significant contributor to immune dysfunction in TB-DM [50]. Exhausted T cells exhibit reduced responsiveness to antigens presented by MHC molecules, impairing their ability to maintain effective feedback loops with macrophages and diminishing immune coordination [36,37]. The role of PD-1 upregulation in T-cell exhaustion and immune dysfunction is illustrated in Figure 2.
This dysfunction weakens CD4+ T cell-mediated cytokine production, including IFN-γ and TNF-α, which are essential for macrophage activation and granuloma integrity [36]. Similarly, CD8+ T cell exhaustion limits cytotoxic responses against infected macrophages, enabling M. tb persistence and granuloma destabilisation [37].
Beyond exhaustion, reductions in T and B lymphocyte counts, commonly observed in TB-DM, further weaken adaptive immunity, compromising the immune system’s capacity to sustain the granuloma structure and restrict bacterial spread [31,36,37,39]. The cumulative effect of T cell dysfunction and lymphocyte depletion exacerbates defects in antigen processing and presentation, further impairing immune surveillance.
However, emerging evidence suggests that T cell exhaustion may not be irreversible. Some studies indicate that under specific conditions, granuloma function and immune responses can be restored, with PD-1 inhibitors showing promise in reversing exhaustion and enhancing the host defence against TB-DM [37].
Conflicting Studies
While many studies highlight the mechanisms of impaired antigen presentation in TB-DM, some findings challenge these conclusions. For example, while TNF-α is frequently described as dysregulated in TB-DM, some studies report that its levels remain elevated and still contribute to macrophage activation [15]. This suggests that defects in antigen presentation may arise not solely from cytokine imbalances but also from altered macrophage responsiveness, persistent inflammatory signalling, or metabolic dysfunction affecting antigen-processing pathways.
Similarly, conflicting evidence exists regarding the role of VEGF-driven hypoxia in antigen presentation. While some studies link hypoxia to reduced MHC expression and impaired T cell activation, others argue that its primary effect is on the granuloma structure rather than directly interfering with MHC-mediated antigen presentation [41]. These discrepancies suggest that the impact of VEGF and hypoxia on immune function may be context-dependent, varying based on disease stage and metabolic status.
3.2.5. Chronic Inflammation and Macrophage Dysfunction in TB-DM
Chronic inflammation is a defining feature of TB-DM, contributing to sustained immune activation and macrophage dysfunction. Persistent elevations in pro-inflammatory cytokines such as TNF-α, IL-6, and IL-17A drive immune exhaustion, impair antigen presentation, and disrupt macrophage polarisation, reinforcing bacterial persistence and tissue damage [9,15]. Additionally, increased levels of eicosanoids, including prostaglandin E2 (PGE2) and lipoxin A4 (LXA4), interfere with inflammation resolution and macrophage recruitment, creating an immune environment that favours bacterial survival [42].
VEGF-driven pathological angiogenesis further destabilises granulomas, creating an inflammatory microenvironment that promotes ongoing macrophage activation without effective bacterial clearance [41,46]. Oxidative stress exacerbates this dysfunction, with both excessive and insufficient ROS responses leading to either tissue damage or impaired bacterial killing [13,19]. This sustained inflammatory state, coupled with metabolic dysregulation, pushes macrophages toward an exhausted phenotype, reducing their bactericidal capacity and prolonging infection [24,25]. The supporting study-specific evidence is presented in Supplemental Table S2.
Synthesis of Findings
TB-DM comorbidity profoundly disrupts immune function, with macrophage dysfunction emerging as the central driver of pathogenesis. This review identifies macrophage impairment as a unifying factor linking metabolic dysfunction, immune dysregulation, and bacterial persistence in TB-DM. Cytokine dysregulation acts as a unifying factor, influencing ROS/NO production, antigen presentation, and macrophage polarisation, thereby sustaining chronic inflammation and impairing immune control of M. tb.
Persistent hyperglycaemia induces metabolic stress, suppressing NO production [19,49] and dysregulating ROS levels [13,17,25,32], leading to impaired bacterial clearance and sustained inflammation. A novel insight from this review is the stage-dependent shift in ROS dynamics, where early hyperglycaemia drives ROS overproduction, while prolonged metabolic dysfunction suppresses ROS, exacerbating bacterial persistence. This imbalance is compounded by cytokine-driven immune dysfunction, where pro-inflammatory cytokines such as TNF-α and IL-6 perpetuate inflammation, while elevated IL-10 suppresses macrophage activation and antigen presentation [9]. Dysregulated eicosanoids, including prostaglandin E2 (PGE2) and lipoxin A4 (LXA4), further interfere with inflammation resolution and disrupt macrophage recruitment, exacerbating immune dysfunction [42].
VEGF-induced hypoxia and epigenetic modifications collectively impair antigen presentation, destabilising granulomas and promoting M. tb survival [19,41]. Macrophage polarisation remains dysregulated, with persistent M1 activation amplifying inflammation and tissue damage, while incomplete M2 transitions fail to resolve inflammation or support tissue repair [13,19]. These findings suggest that macrophage dysfunction is not merely a consequence of TB-DM but an active driver of disease progression, reinforcing a vicious cycle of immune impairment and bacterial evasion.
Together, these findings demonstrate macrophage dysfunction as the critical bottleneck in TB-DM immunity, where chronic inflammation and metabolic stress drive a self-perpetuating cycle of immune impairment, bacterial survival, and tissue destruction. Targeting these interconnected dysfunctions may provide avenues for improving TB-DM treatment outcomes, particularly by modulating macrophage function and restoring immune balance.
Metabolic dysregulation induces mitochondrial superoxide accumulation, glutathione imbalance, and endoplasmic reticulum (ER) stress, exacerbating oxidative stress and impairing phagolysosome fusion. Increased STAT1 activity drives DNA methylation and epigenetic modifications, reducing HLA-DR4 expression. TNF-α, IFN-γ, IL-6, and IL-17A further suppress HLA-DR4, impairing phagosome–lysosome fusion and antigen presentation.
Reduced IL-1β and IL-6 downregulate MHC-II expression, weakening antigen presentation. T-cell activation is impaired by increased IFN-γ and IL-1β, alongside reduced IL-27 and IL-12, while persistent IL-6 and IL-17A signalling promote T-cell exhaustion via PD-1 upregulation. CD8+ T-cell dysfunction compromises granuloma integrity, reducing VEGF levels and MHC-I expression, further weakening cytotoxic responses. These immune and metabolic alterations create a permissive environment for M. tb persistence and disease progression in TB-DM.
4. Discussion
This scoping review systematically synthesises evidence on macrophage dysfunction in TB-DM, highlighting the key mechanisms driving immune dysregulation. Findings from this review establish macrophage impairment as the cornerstone of immune dysfunction in TB-DM, integrating metabolic, inflammatory, and epigenetic disruptions into a unified model of disease progression. The results demonstrate that TB-DM comorbidity not only disrupts fundamental macrophage functions—such as NO production, antigen presentation, and cytokine signalling—but also establishes a self-reinforcing cycle of chronic inflammation and immune exhaustion. Understanding these mechanisms is crucial for guiding future research and informing targeted interventions.
While existing reviews have documented the bidirectional relationship between TB and DM, demonstrating how hyperglycaemia compromises both innate and adaptive immunity [51,52,53], they primarily discuss macrophage dysfunction as part of broader innate immune impairments rather than providing a focused analysis of macrophage-specific mechanisms. This review addresses this gap by directly linking macrophage dysfunction to key immune impairments, including altered ROS/NO regulation, eicosanoid-mediated inflammation, and VEGF-induced antigen presentation deficits. Furthermore, it challenges the prevailing assumption that TB-DM is solely driven by ROS overproduction [54,55], highlighting a stage-dependent shift, where early hyperglycaemia leads to excessive ROS, but prolonged metabolic dysfunction eventually suppresses ROS, further impairing bacterial clearance [13,19].
Importantly, inconsistencies in reported immune findings may stem from heterogeneity in patient populations and experimental conditions. For example, cytokine profiles can be influenced by the TB disease stage, background inflammatory status, and levels of glycaemic control. Similarly, the immune phenotype of macrophages may differ based on their tissue of origin or the presence of comorbid conditions. In vitro studies often use standardised stimuli that may not fully capture the complexity of in vivo environments, potentially limiting generalisability. A more harmonised approach to study design and patient characterisation—including the consistent reporting of HbA1c, TB status, and demographic variables—would greatly improve the comparability of future studies and help explain immune variability in TB-DM. A notable contribution of this review is the identification of eicosanoid disruption as a driver of immune dysfunction in TB-DM, an aspect that has received comparatively less attention in prior studies. Elevated PGE2 and lipoxin A4 impair inflammation resolution and macrophage recruitment, facilitating bacterial persistence [15,19]. This dysregulated inflammatory response is further compounded by epigenetic modifications, such as the DNA methylation of iNOS promoters, which suppress NO production and impair macrophage function, ultimately reducing bacterial clearance [14]. Together, these findings highlight the multifaceted nature of immune dysfunction in TB-DM and underscore the need for a more targeted immunomodulatory approach to treatment, moving beyond glycaemic control alone.
Macrophage dysfunction in TB-DM also has significant implications for diagnostics and clinical management. A key translational impact of these findings is the link between antigen presentation deficits and the reduced sensitivity of tuberculin skin tests (TSTs) and interferon-gamma release assays (IGRAs) in TB-DM patients [16,38]. Alternative biomarkers, such as heme oxygenase-1 and VEGF-A, may provide more reliable indicators of TB susceptibility and disease progression [24,34,41].
Beyond mechanistic insights, this review underscores the clinical relevance of macrophage dysfunction in TB-DM, particularly how cytokine dysregulation, oxidative stress, and impaired immune clearance contribute to persistent bacillary burden, granuloma instability, and poor treatment outcomes [9,13,15,20,25,34,40]. These immune impairments likely explain the 1.7-fold increased risk of TB treatment failure and the 53% higher risk of relapse observed in TB-DM patients [56,57], reinforcing the urgent need for macrophage-targeted therapies.
Although many of the studies included are preclinical, their findings offer valuable direction for translational research. Several macrophage-associated markers, including VEGF-A, IL-10, and heme oxygenase-1 [24,34,41], show potential as biomarkers for TB susceptibility, immune dysfunction, or treatment monitoring in diabetic populations. Additionally, mechanisms such as PD-1-mediated T-cell exhaustion and ROS/NO imbalance highlight the potential therapeutic targets for host-directed therapy [36,37]. Integrating these mechanistic insights into translational research pipelines may support the development of targeted immunomodulatory approaches and precision diagnostics for TB-DM.
5. Limitations
Despite providing a comprehensive synthesis of macrophage dysfunction in TB-DM, this review has several limitations.
First, significant heterogeneity exists across studies, particularly in study design, patient populations, and glycaemic control assessments, making direct comparisons challenging. Many studies remain preclinical, limiting the immediate translation of findings into clinical practice.
Second, this review focuses exclusively on macrophage dysfunction, excluding the roles of other immune cells such as T cells, neutrophils, and dendritic cells, as well as autophagy, which are all known contributors to TB-DM pathogenesis. While autophagy is a key immune regulatory mechanism, its role in TB-DM remains unclear, with conflicting evidence on whether hyperglycaemia enhances or suppresses autophagy [58]. While a macrophage-centred lens allowed for thematic depth, it is important to recognise that these mechanisms occur within a broader immunological context involving complex cellular crosstalk and signalling networks. Future research should integrate broader systems immunology approaches to examine multi-cellular immune interactions.
The heterogeneity in study design, population characteristics, and assessment of glycaemic control presents a limitation in drawing definitive comparisons and weakens the generalisability of individual findings. Nevertheless, the emergence of consistent immunological patterns across these varied studies strengthens the conclusion that macrophage dysfunction is a central feature of TB-DM. Future research should prioritise standardised methodologies and reporting practices to improve comparability and support robust synthesis.
Despite these limitations, this review provides a strong foundation for future investigations, identifying key pathways of immune dysfunction that may inform targeted therapeutic interventions. Addressing these gaps will require translational studies linking immune dysfunction to treatment outcomes and exploring novel therapeutic approaches.
6. Future Directions
Optimising glycaemic control should be a central component of TB-DM management. Poor glycaemic control is linked to increased inflammation, immune exhaustion, and impaired macrophage function, all of which contribute to disease progression [59]. Clinical trials assessing the impact of intensive glycaemic management on TB outcomes could provide valuable insights into whether tighter glucose regulation improves bacterial clearance and reduces TB relapse rates. Future research should explore whether specific glycaemic targets, combined with TB therapy, enhance immune function and reduce disease severity. Additionally, the potential role of glucose-lowering agents with immunomodulatory properties, such as GLP-1 receptor agonists, warrants further investigation in TB-DM.
However, given the complex immune dysfunction associated with TB-DM, glycaemic control alone may not be sufficient to fully restore host immune responses. A key research priority is the development of host-directed therapies (HDTs) to enhance immune function and reduce TB severity in diabetic individuals. Programmed death (PD)-1 inhibitors, which reverse T-cell exhaustion, have shown promise in enhancing pathogen clearance and immune control in TB-DM [60,61]. Given their success in chronic infections and cancer, further investigation into their therapeutic role in TB-DM is warranted [62]. Additionally, metformin, a widely used antidiabetic drug, has demonstrated immunomodulatory effects, including enhanced autophagy, reduced inflammation, and improved macrophage function [63]. Integrating metformin as an HDT in TB-DM treatment regimens may be particularly beneficial for patients with poor glycaemic control, improving both immune function and clinical outcomes [64].
Refining TB diagnostics for diabetic populations is another critical area for future research. The reduced sensitivity of TST and IGRA in TB-DM due to impaired antigen presentation and T-cell exhaustion highlights the need for alternative screening methods. Integrating immune biomarkers, metabolomic profiling, and epigenetic signatures could improve diagnostic accuracy and provide more reliable indicators of disease progression. Identifying the immune correlates of protection in TB-DM will be essential for optimising risk stratification and tailoring treatment strategies.
Future research should also prioritise longitudinal studies to establish the relationship between macrophage dysfunction, TB progression, and treatment failure in TB-DM patients. Longitudinal studies are essential to understanding whether macrophage impairments resolve with improved glycaemic control or persist despite treatment and how they correlate with clinical outcomes such as relapse or treatment failure. Additionally, systems biology approaches, including single-cell transcriptomics, metabolic profiling, and epigenetic mapping, should be used to capture the full scope of immune dysfunction and identify potential therapeutic targets. Understanding these mechanisms in greater detail will enable the development of precision-based interventions for TB-DM management.
To enhance clinical translation, future studies should focus on validating macrophage-associated immune markers as diagnostic or prognostic tools. Longitudinal clinical studies linking these biomarkers to treatment outcomes and TB relapse risk in diabetic populations are particularly needed. Furthermore, mechanistic findings from preclinical research—such as epigenetic modulation of iNOS or the role of eicosanoids in immune suppression—should inform the design of host-directed therapies. Bridging basic immunology with clinical implementation will be critical to improving outcomes for individuals with TB-DM.
While this review was intentionally focused on macrophages, future work should explore how macrophage dysfunction interfaces with broader immune responses. The interaction between macrophages and T cells, particularly CD4+ and CD8+ subsets, is essential for maintaining granuloma integrity and controlling M. tb. In TB-DM, dysfunction in these networks, including impaired antigen presentation and T-cell exhaustion, likely compounds immune failure. Additionally, innate immune mechanisms such as autophagy—which modulates bacterial clearance and inflammation—may be disrupted in TB-DM but remain underexplored. Integrating macrophage findings with data from systems immunology and multi-cellular studies will be essential to fully characterise TB-DM immunopathogenesis.
7. Conclusions
This scoping review enhances our understanding of macrophage dysfunction as a central driver of TB-DM pathogenesis. It identifies key immune impairments—including ROS dysregulation, the epigenetic suppression of NO, and eicosanoid-driven immune dysfunction—that collectively compromise bacterial clearance and drive disease progression. These findings underscore how metabolic stress and immune dysregulation interact, leading to persistent infection, treatment failure, and higher relapse rates.
This review identifies key knowledge gaps by linking macrophage-specific dysfunctions to diagnostic and therapeutic limitations in TB-DM. Understanding the immune correlates of protection is crucial for developing precision medicine strategies, including risk stratification tools, targeted immunotherapies, and biomarker-driven diagnostics. Addressing these translational gaps will facilitate patient-specific interventions, ultimately improving treatment success rates and reducing TB-DM burden worldwide.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/diabetology6050035/s1. Supplemental Table S1: Summary of studies exploring the interplay between tuberculosis (TB) and diabetes mellitus (DM) in relation to macrophage function and immune modulation. This table provides an overview of 44 studies investigating the impact of diabetes mellitus (DM) on tuberculosis (TB) pathogenesis, with a specific focus on macrophage dysfunction and immune responses. Columns include the study number, authorship, year of publication, title of the paper, study design, population studied, and key findings relevant to TB-DM comorbidity. The findings highlight altered immune profiles, cytokine dysregulation, and impaired macrophage activity as critical elements in the TB-DM nexus. * The studies included in this table examine immune responses and disease progression in various groups, including TB-DM patients (those with both active tuberculosis and diabetes), LTBI-DM patients (individuals with latent tuberculosis infection and diabetes), and TB-PDM patients (those with pulmonary TB and prediabetes). Some studies also focus on T1D patients (individuals with type 1 diabetes) or analyse specific biological samples, such as macrophages derived from TB-DM patients or plasma from TB-DM patients, to assess immune markers and inflammatory pathways. Supplemental Table S2. Immune dysregulation in tuberculosis—diabetes mellitus (TB-DM) comorbidity: observations, macrophage impacts, and thematic integration. Key observations related to immune dysregulation in tuberculosis (TB) and diabetes mellitus (DM) comorbidity, with a focus on macrophage function. Observations are categorised by their effects on macrophage activity, corresponding immunological themes, and supporting references. Themes include reactive oxygen species and nitric oxide, impaired phagocytosis, dysregulation of macrophage polarisation, antigen presentation, altered cytokine production, and chronic inflammation. Refs. [65,66,67,68,69,70,71,72,73,74] are cited in Supplementary Materials.
Author Contributions
S.E.B. conceived the study, designed the methodology, conducted the literature search, screened articles, performed data synthesis and analysis, and wrote the manuscript. A.S. screened articles, reviewed included studies, and contributed to manuscript revisions. A.M. screened articles, reviewed included studies, and contributed to manuscript revisions. J.I. screened articles, reviewed included studies, and contributed to manuscript revisions. S.L. screened articles, reviewed included studies, and contributed to manuscript revisions. J.B.G. screened articles, reviewed included studies, and contributed to manuscript revisions. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data were generated or analysed in this study. This manuscript is a scoping review of the published literature, and all data supporting the reported results are derived from publicly available sources, as cited in the references.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| TB | Tuberculosis |
| DM | Diabetes Mellitus |
| TB-DM | Tuberculosis–Diabetes Mellitus Comorbidity |
| M. tb | Mycobacterium Tuberculosis |
| NO | Nitric Oxide |
| ROS | Reactive Oxygen Species |
| MHC | Major Histocompatibility Complex |
| IFN-γ | Interferon-Gamma |
| TNF-α | Tumour Necrosis Factor-Alpha |
| IL | Interleukin |
| VEGF | Vascular Endothelial Growth Factor |
| PGE2 | Prostaglandin E2 |
| LXA4 | Lipoxin A4 |
| iNOS | Inducible Nitric Oxide Synthase |
| PD-1 | Programmed Death-1 |
| PRISMA | Preferred Reporting Items for Systematic Reviews and Meta-Analyses |
References
- Global Tuberculosis Report 2024; WHO, Ed.; World Health Organization: Geneva, Switzerland, 2024. [Google Scholar]
- Baker, M.A.; Harries, A.D.; Jeon, C.Y.; Hart, J.E.; Kapur, A.; Lönnroth, K.; Ottmani, S.E.; Goonesekera, S.D.; Murray, M.B. The impact of diabetes on tuberculosis treatment outcomes: A systematic review. BMC Med. 2011, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- GBD 2021 Diabetes Collaborators. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the Global Burden of Disease Study 2021. Lancet 2023, 402, 203–234. [Google Scholar] [CrossRef]
- Berbudi, A.; Rahmadika, N.; Tjahjadi, A.I.; Ruslami, R. Type 2 Diabetes and its Impact on the Immune System. Curr. Diabetes Rev. 2020, 16, 442–449. [Google Scholar]
- Pavan Kumar, N.; Nair, D.; Banurekha, V.V.; Dolla, C.; Kumaran, P.; Sridhar, R.; Babu, S. Type 2 diabetes mellitus coincident with pulmonary or latent tuberculosis results in modulation of adipocytokines. Cytokine 2016, 79, 74–81. [Google Scholar] [CrossRef]
- Covidence Systematic Review Software; V.H. Innovation: Melbourne, Australia, 2025.
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.J.; Horsley, T.; Weeks, L.; et al. PRISMA Extension for Scoping Reviews (PRISMA-ScR): Checklist and Explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef]
- Kumar, N.P.; Moideen, K.; Dolla, C.; Kumaran, P.; Babu, S. Prediabetes is associated with the modulation of antigen-specific Th1/Tc1 and Th17/Tc17 responses in latent Mycobacterium tuberculosis infection. PLoS ONE 2017, 12, e0178000. [Google Scholar] [CrossRef]
- Singh, K.; Hussain, T.; Gupta, B.; Pati, S. A Comparative Investigation on Cytokine Expression in Pulmonary Tuberculosis and Comorbidity with Type 2 Diabetes Mellitus. Int. J. Mycobacteriol. 2024, 13, 165–170. [Google Scholar] [CrossRef]
- Kumar, N.P.; Moideen, K.; George, P.J.; Dolla, C.; Kumaran, P.; Babu, S. Coincident diabetes mellitus modulates Th1-, Th2-, and Th17-cell responses in latent tuberculosis in an IL-10- and TGF-beta-dependent manner. Eur. J. Immunol. 2016, 46, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Ferlita, S.; Yegiazaryan, A.; Noori, N.; Lal, G.; Nguyen, T.; To, K.; Venketaraman, V. Type 2 Diabetes Mellitus and Altered Immune System Leading to Susceptibility to Pathogens, Especially Mycobacterium tuberculosis. J. Clin. Med. 2019, 8, 2219. [Google Scholar] [CrossRef]
- Panda, S.; Arora, A.; Luthra, K.; Mohan, A.; Vikram, N.K.; Kumar Gupta, N.; Singh, A. Hyperglycemia modulates M1/M2 macrophage polarization in chronic diabetic patients with pulmonary tuberculosis infection. Immunobiology 2024, 229, 152787. [Google Scholar] [CrossRef]
- Panda, S.; Seelan, D.M.; Faisal, S.; Arora, A.; Luthra, K.; Palanichamy, J.K.; Mohan, A.; Vikram, N.K.; Gupta, N.K.; Ramakrishnan, L.; et al. Chronic hyperglycemia drives alterations in macrophage effector function in pulmonary tuberculosis. Eur. J. Immunol. 2022, 52, 1595–1609. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, N.; Martinez, A.G.R.; Garcia-Hernandez, M.H.; Hernandez-Pando, R.; Castaneda-Delgado, J.E.; Lugo-Villarino, G.; Cougoule, C.; Neyrolles, O.; Rivas-Santiago, B.; Valtierra-Alvarado, M.A.; et al. Type-2 diabetes alters the basal phenotype of human macrophages and diminishes their capacity to respond, internalise, and control Mycobacterium tuberculosis. Mem. Inst. Oswaldo Cruz 2018, 113, e170326. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.P.; Fukutani, K.F.; Shruthi, B.S.; Alves, T.; Silveira-Mattos, P.S.; Rocha, M.S.; West, K.; Natarajan, M.; Viswanathan, V.; Babu, S.; et al. Persistent inflammation during anti-tuberculosis treatment with diabetes comorbidity. eLife 2019, 8, e46477. [Google Scholar] [CrossRef] [PubMed]
- Dasan, B.; Rajamanickam, A.; Pandiarajan, A.N.; Shanmugam, S.; Nott, S.; Babu, S. Immunological mechanisms of tuberculosis susceptibility in TB-infected individuals with type 2 diabetes mellitus: Insights from mycobacterial growth inhibition assay and cytokine analysis. Clin. Microbiol. 2025, 13, e01445-24. [Google Scholar] [CrossRef]
- Leniseptaria, A.A.; Saraswati, I.; Pakaya, D. Comparative studies evaluating macrophage activity of pulmonary tuberculosis patients with and without diabetes mellitus (tb-dm and non tb-dm). Bangladesh J. Med. Sci. 2018, 17, 107–111. [Google Scholar] [CrossRef]
- Wei, R.; Li, P.; Xue, Y.; Liu, Y.; Gong, W.; Zhao, W. Impact of Diabetes Mellitus on the Immunity of Tuberculosis Patients: A Retrospective, Cross-Sectional Study. Risk Manag. Healthc. Policy 2022, 15, 611–627. [Google Scholar] [CrossRef]
- Prada-Medina, C.A.; Fukutani, K.F.; Pavan Kumar, N.; Gil-Santana, L.; Babu, S.; Lichtenstein, F.; West, K.; Sivakumar, S.; Menon, P.A.; Viswanathan, V.; et al. Systems Immunology of Diabetes-Tuberculosis Comorbidity Reveals Signatures of Disease Complications. Sci. Rep. 2017, 7, 1999. [Google Scholar] [CrossRef]
- Mily, A.; Sarker, P.; Taznin, I.; Hossain, D.; Haq, M.A.; Kamal, S.M.M.; Agerberth, B.; Brighenti, S.; Raqib, R. Slow radiological improvement and persistent low-grade inflammation after chemotherapy in tuberculosis patients with type 2 diabetes. BMC Infect. Dis. 2020, 20, 933. [Google Scholar] [CrossRef]
- Kathamuthu, G.R.; Pavan Kumar, N.; Moideen, K.; Dolla, C.; Kumaran, P.; Babu, S. Multi-Dimensionality Immunophenotyping Analyses of MAIT Cells Expressing Th1/Th17 Cytokines and Cytotoxic Markers in Latent Tuberculosis Diabetes Comorbidity. Pathogens 2022, 11, 87. [Google Scholar] [CrossRef]
- Fernandez, R.D.V.; Diaz, A.; Bongiovanni, B.; Gallucci, G.; Bertola, D.; Gardenez, W.; Lioi, S.; Bertolin, Y.; Galliano, R.; Bay, M.L.; et al. Evidence for a More Disrupted Immune-Endocrine Relation and Cortisol Immunologic Influences in the Context of Tuberculosis and Type 2 Diabetes Comorbidity. Front. Endocrinol. 2020, 11, 126. [Google Scholar] [CrossRef]
- Aravindhan, V.; Bobhate, A.; Sathishkumar, K.; Patil, A.; Kumpatla, S.; Viswanathan, V. Unique Reciprocal Association Seen Between Latent Tuberculosis Infection and Diabetes Is Due to Immunoendocrine Modulation (DM-LTB-1). Front. Microbiol. 2022, 13, 884374. [Google Scholar] [CrossRef] [PubMed]
- Andrade, B.B.; Kumar, N.P.; Sridhar, R.; Banurekha, V.V.; Jawahar, M.S.; Nutman, T.B.; Sher, A.; Babu, S. Heightened plasma levels of heme oxygenase-1 and tissue inhibitor of metalloproteinase-4 as well as elevated peripheral neutrophil counts are associated with TB-diabetes comorbidity. Chest 2014, 145, 1244–1254. [Google Scholar] [CrossRef]
- Chao, W.C.; Yen, C.L.; Wu, Y.H.; Chen, S.Y.; Hsieh, C.Y.; Chang, T.C.; Ou, H.Y.; Shieh, C.C. Increased resistin may suppress reactive oxygen species production and inflammasome activation in type 2 diabetic patients with pulmonary tuberculosis infection. Microbes Infect. 2015, 17, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Iacobini, C.; Vitale, M.; Pesce, C.; Pugliese, G.; Menini, S. Diabetic Complications and Oxidative Stress: A 20-Year Voyage Back in Time and Back to the Future. Antioxidants 2021, 10, 727. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Dainichi, T.; Matsumoto, R.; Mostafa, A.; Kabashima, K. Immune Control by TRAF6-Mediated Pathways of Epithelial Cells in the EIME (Epithelial Immune Microenvironment). Front. Immunol. 2019, 10, 1107. [Google Scholar] [CrossRef]
- Silwal, P.; Kim, J.K.; Kim, Y.J.; Jo, E.K. Mitochondrial Reactive Oxygen Species: Double-Edged Weapon in Host Defense and Pathological Inflammation During Infection. Front. Immunol. 2020, 11, 1649. [Google Scholar] [CrossRef]
- Martinez, N.; Kornfeld, H. Diabetes and immunity to tuberculosis. Eur. J. Immunol. 2014, 44, 617–626. [Google Scholar] [CrossRef]
- Kumar, N.P.; Banurekha, V.V.; Nair, D.; Kumaran, P.; Dolla, C.K.; Babu, S. Type 2 diabetes—Tuberculosis co-morbidity is associated with diminished circulating levels of IL-20 subfamily of cytokines. Tuberculosis 2015, 95, 707–712. [Google Scholar] [CrossRef]
- Kumar, N.P.; Moideen, K.; George, P.J.; Dolla, C.; Kumaran, P.; Babu, S. Impaired Cytokine but Enhanced Cytotoxic Marker Expression in Mycobacterium tuberculosis-Induced CD8+ T Cells in Individuals with Type 2 Diabetes and Latent Mycobacterium tuberculosis Infection. J. Infect. Dis. 2016, 213, 866–870. [Google Scholar] [CrossRef]
- Rajamanickam, A.; Kothandaraman, S.P.; Kumar, N.P.; Viswanathan, V.; Shanmugam, S.; Hissar, S.; Nott, S.; Kornfeld, H.; Babu, S. Cytokine and chemokine profiles in pulmonary tuberculosis with pre-diabetes. Front. Immunol. 2024, 15, 1447161. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, R.; Diaz, A.; D’Attilio, L.; Bongiovanni, B.; Santucci, N.; Bertola, D.; Besedovsky, H.; Del Rey, A.; Bay, M.L.; Bottasso, O. An adverse immune-endocrine profile in patients with tuberculosis and type 2 diabetes. Tuberculosis 2016, 101, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Kathamuthu, G.R.; Kumar, N.P.; Moideen, K.; Menon, P.A.; Babu, S. Decreased Frequencies of Gamma/Delta T Cells Expressing Th1/Th17 Cytokine, Cytotoxic, and Immune Markers in Latent Tuberculosis-Diabetes/Pre-Diabetes Comorbidity. Front. Cell Infect. Microbiol. 2021, 11, 756854. [Google Scholar] [CrossRef]
- Kathamuthu, G.R.; Kumar, N.P.; Moideen, K.; Menon, P.A.; Babu, S. High Dimensionality Reduction and Immune Phenotyping of Natural Killer and Invariant Natural Killer Cells in Latent Tuberculosis-Diabetes Comorbidity. J. Immunol. Res. 2022, 2022, 2422790. [Google Scholar] [CrossRef]
- Ssekamatte, P.; Nabatanzi, R.; Sitenda, D.; Nakibuule, M.; Bagaya, B.S.; Kibirige, D.; Kyazze, A.P.; Kateete, D.P.; Sande, O.J.; van Crevel, R.; et al. Impaired Mycobacterium tuberculosis-specific T-cell memory phenotypes and functional profiles among adults with type 2 diabetes mellitus in Uganda. Front. Immunol. 2024, 15, 1480739. [Google Scholar] [CrossRef]
- Faurholt-Jepsen, D.; Aabye, M.G.; Jensen, A.V.; Range, N.; Praygod, G.; Jeremiah, K.; Changalucha, J.; Faurholt-Jepsen, M.; Jensen, L.; Jensen, S.M.; et al. Diabetes is associated with lower tuberculosis antigen-specific interferon gamma release in Tanzanian tuberculosis patients and non-tuberculosis controls. Scand. J. Infect. Dis. 2014, 46, 384–391. [Google Scholar] [CrossRef]
- Liu, S.; Ren, W.; Yu, J.; Li, C.; Tang, S. Identification of Hub Genes Associated with Diabetes Mellitus and Tuberculosis Using Bioinformatic Analysis. Int. J. Gen. Med. 2021, 14, 4061–4072. [Google Scholar] [CrossRef]
- Masood, K.I.; Irfan, M.; Masood, Q.; Yameen, M.; Jamil, B.; Ram, N.; Rao, S.; Rottenberg, M.; Hasan, Z.; Latent, M. tuberculosis infection is associated with increased inflammatory cytokine and decreased suppressor of cytokine signalling (SOCS)-3 in the diabetic host. Scand. J. Immunol. 2022, 95, e13134. [Google Scholar] [CrossRef]
- Kumar, N.P.; Moideen, K.; Sivakumar, S.; Menon, P.A.; Viswanathan, V.; Kornfeld, H.; Babu, S. Tuberculosis-diabetes co-morbidity is characterized by heightened systemic levels of circulating angiogenic factors. J. Infect. 2017, 74, 10–21. [Google Scholar] [CrossRef]
- Pavan Kumar, N.; Moideen, K.; Nancy, A.; Viswanathan, V.; Shruthi, B.S.; Shanmugam, S.; Hissar, S.; Kornfeld, H.; Babu, S. Plasma Eicosanoid Levels in Tuberculosis and Tuberculosis-Diabetes Co-morbidity Are Associated with Lung Pathology and Bacterial Burden. Front. Cell Infect. Microbiol. 2019, 9, 335. [Google Scholar] [CrossRef]
- Chen, H.; Su, L.; Bao, J.; Zhang, K.; Li, Y.; Mao, E. The impact of pulmonary tuberculosis on immunological and metabolic features of diabetic patients. Front. Immunol. 2022, 13, 973991. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Kumar, N.P.; Moideen, K.; Dhakshinraj, S.D.; Banurekha, V.V.; Nair, D.; Dolla, C.; Kumaran, P.; Babu, S. Profiling leucocyte subsets in tuberculosis-diabetes co-morbidity. Immunology 2015, 146, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Tong, F.; Lai, J.; Lu, Z.; Bao, Z.; Cao, J. Clinical features, immunologic parameter and treatment outcome of Chinese tuberculosis patients with or without DM. Front. Med. 2024, 11, 1386124. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.V.; Boom, W.H. Regulation of antigen presentation by Mycobacterium tuberculosis: A role for Toll-like receptors. Nat. Rev. Microbiol. 2010, 8, 296–307. [Google Scholar] [CrossRef]
- Flynn, J.L.; Chan, J. Immunology of tuberculosis. Annu. Rev. Immunol. 2001, 19, 93–129. [Google Scholar] [CrossRef]
- Aravindhan, V.; Bobhate, A.; Sathishkumar, K.; Viswanathan, V. Serum levels of novel anti-inflammatory cytokine Interleukin-38 in diabetes patients infected with latent tuberculosis (DM-LTB-3). J. Diabetes Complicat. 2022, 36, 108133. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Ye, Z.; Li, L.; Yang, L.; Zhuang, L.; Aspatwar, A.; Wang, L.; Gong, W. Impact of diabetes mellitus on tuberculosis prevention, diagnosis, and treatment from an immunologic perspective. Exploration 2024, 4, 20230138. [Google Scholar] [CrossRef]
- Kumar Nathella, P.; Babu, S. Influence of diabetes mellitus on immunity to human tuberculosis. Immunology 2017, 152, 13–24. [Google Scholar] [CrossRef]
- Selvan, P.; Nagesh, N.J.; Vajravelu, L.K.; Akram, C.S.; Karniha, B. A Bidirectional Immunological Relationship between Diabetes Mellitus and Tuberculosis: A Narrative Review. J. Clin. Diagn. Res. 2024, 18, DE01–DE05. [Google Scholar] [CrossRef]
- Navasardyan, I.; Yeganyan, S.; Nguyen, H.; Vaghashia, P.; Subbian, S.; Venketaraman, V. Role of Oxidative Stress in Tuberculosis Meningitis Infection in Diabetics. Biomedicines 2023, 11, 2568. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Huangfu, P.; Ugarte-Gil, C.; Golub, J.; Pearson, F.; Critchley, J. The effects of diabetes on tuberculosis treatment outcomes: An updated systematic review and meta-analysis. Int. J. Tuberc. Lung Dis. 2019, 23, 783–796. [Google Scholar] [CrossRef]
- Rehman, A.U.; Khattak, M.; Mushtaq, U.; Latif, M.; Ahmad, I.; Rasool, M.F.; Shakeel, S.; Hayat, K.; Hussain, R.; Alhazmi, G.A.; et al. The impact of diabetes mellitus on the emergence of multi-drug resistant tuberculosis and treatment failure in TB-diabetes comorbid patients: A systematic review and meta-analysis. Front. Public Health 2023, 11, 1244450. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Mukhopadhyay, M.; Bhattacharyya, M.; Karmakar, P. Is autophagy associated with diabetes mellitus and its complications? A review. EXCLI J. 2018, 17, 709–720. [Google Scholar]
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. Eur. Cardiol. 2019, 14, 50–59. [Google Scholar] [CrossRef]
- Kamboj, D.; Gupta, P.; Basil, M.V.; Mohan, A.; Guleria, R.; Bhatnagar, A.; Mehta, G.; Kumar, P.; Saurabh, A.; Deepak, R.; et al. Improved Mycobacterium tuberculosis clearance after the restoration of IFN-γ(+) TNF-α(+) CD4(+) T cells: Impact of PD-1 inhibition in active tuberculosis patients. Eur. J. Immunol. 2020, 50, 736–747. [Google Scholar] [CrossRef]
- Zhao, L.; Fan, K.; Sun, X.; Li, W.; Qin, F.; Shi, L.; Gao, F.; Zheng, C. Host-directed therapy against mycobacterium tuberculosis infections with diabetes mellitus. Front. Immunol. 2023, 14, 1305325. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Ahmed, R. CD4 T-cell immunotherapy for chronic viral infections and cancer. Immunotherapy 2013, 5, 975–987. [Google Scholar] [CrossRef]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55.e6. [Google Scholar] [CrossRef] [PubMed]
- Naicker, N.; Sigal, A.; Naidoo, K. Metformin as Host-Directed Therapy for TB Treatment: Scoping Review. Front. Microbiol. 2020, 11, 435. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, Y.; Zhang, Q.; Gao, Y. Association between serum globulins and diabetes mellitus in American latent tuberculosis infection patients: A cross-sectional study. Medicine 2024, 103, e39949. [Google Scholar] [CrossRef]
- He, X.; Wang, Y.; Yang, Y.; He, Q.; Sun, L.; Jin, J. Quantitative proteomics reveals plasma protein profile and potential pathways in pulmonary tuberculosis patients with and without diabetes. Tuberculosis 2023, 143, 102424. [Google Scholar] [CrossRef]
- Janssen, A.W.M.; Stienstra, R.; Jaeger, M.; van Gool, A.J.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P.; Tack, C.J. Understanding the increased risk of infections in diabetes: Innate and adaptive immune responses in type 1 diabetes. Metabolism 2021, 121, 154795. [Google Scholar] [CrossRef]
- Kumar, N.P.; Sridhar, R.; Nair, D.; Banurekha, V.V.; Nutman, T.B.; Babu, S. Type 2 diabetes mellitus is associated with altered CD8(+) T and natural killer cell function in pulmonary tuberculosis. Immunology 2015, 144, 677–686. [Google Scholar] [CrossRef]
- Kumar, N.P.; Banurekha, V.V.; Nair, D.; Sridhar, R.; Kornfeld, H.; Nutman, T.B.; Babu, S. Coincident pre-diabetes is associated with dysregulated cytokine responses in pulmonary tuberculosis. PLoS ONE 2014, 9, e112108. [Google Scholar] [CrossRef]
- Kumar, N.P.; Moideen, K.; Bhootra, Y.; Nancy, A.; Viswanathan, V.; Shruthi, B.S.; Sivakumar, S.; Natarajan, M.; Kornfeld, H.; Babu, S. Elevated circulating levels of monocyte activation markers among tuberculosis patients with diabetes co-morbidity. Immunology 2019, 156, 249–258. [Google Scholar] [CrossRef]
- Lachmandas, E.; Thiem, K.; van den Heuvel, C.; Hijmans, A.; de Galan, B.E.; Tack, C.J.; Netea, M.G.; van Crevel, R.; van Diepen, J.A. Patients with type 1 diabetes mellitus have impaired IL-1beta production in response to Mycobacterium tuberculosis. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 371–380. [Google Scholar] [CrossRef]
- Sanchez-Jimenez, R.; Ceron, E.; Bernal-Alcantara, D.; Castillejos-Lopez, M.; Gonzalez-Trujano, E.; Negrete-Garcia, M.C.; Alvarado-Vasquez, N. Association between IL-15 and insulin plasmatic concentrations in patients with pulmonary tuberculosis and type 2 diabetes. Tuberculosis 2018, 111, 114–120. [Google Scholar] [CrossRef]
- Valtierra-Alvarado, M.A.; Lugo-Villarino, G.; Duenas-Arteaga, F.; Gonzalez-Contreras, B.E.; Lugo-Sanchez, A.; Castaneda-Delgado, J.E.; González-Amaro, R.; Venegas Gurrola, O.A.; González Valadez, A.D.; Enciso-Moreno, J.A.; et al. Impact of type 2 diabetes on the capacity of human macrophages infected with Mycobacterium tuberculosis to modulate monocyte differentiation through a bystander effect. Immunol. Cell Biol. 2021, 99, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ma, A.; Han, X.; Chan, L.; Liang, H.; Litifu, A.; Xue, F. T Cell Profile was Altered in Pulmonary Tuberculosis Patients with Type 2 Diabetes. Med. Sci. Monit. 2018, 24, 636–642. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).