



Do T2DM and Hyperglycaemia Affect the Expression Levels of the Regulating Enzymes of Cellular O-GlcNAcylation in Human Saphenous Vein Smooth Muscle Cells?

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Isolation of HSVSMCs
2.2.2. Cell Culture
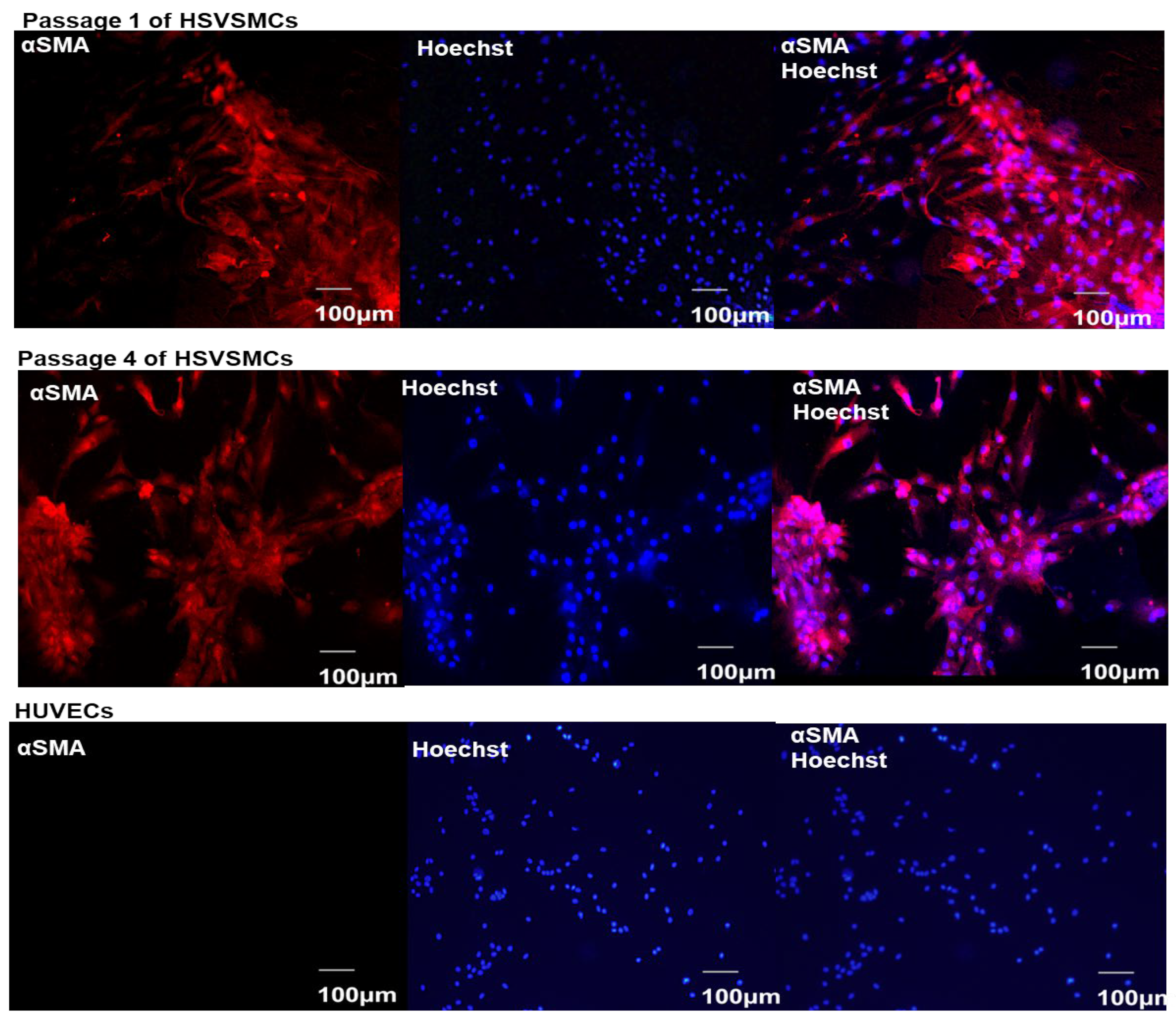
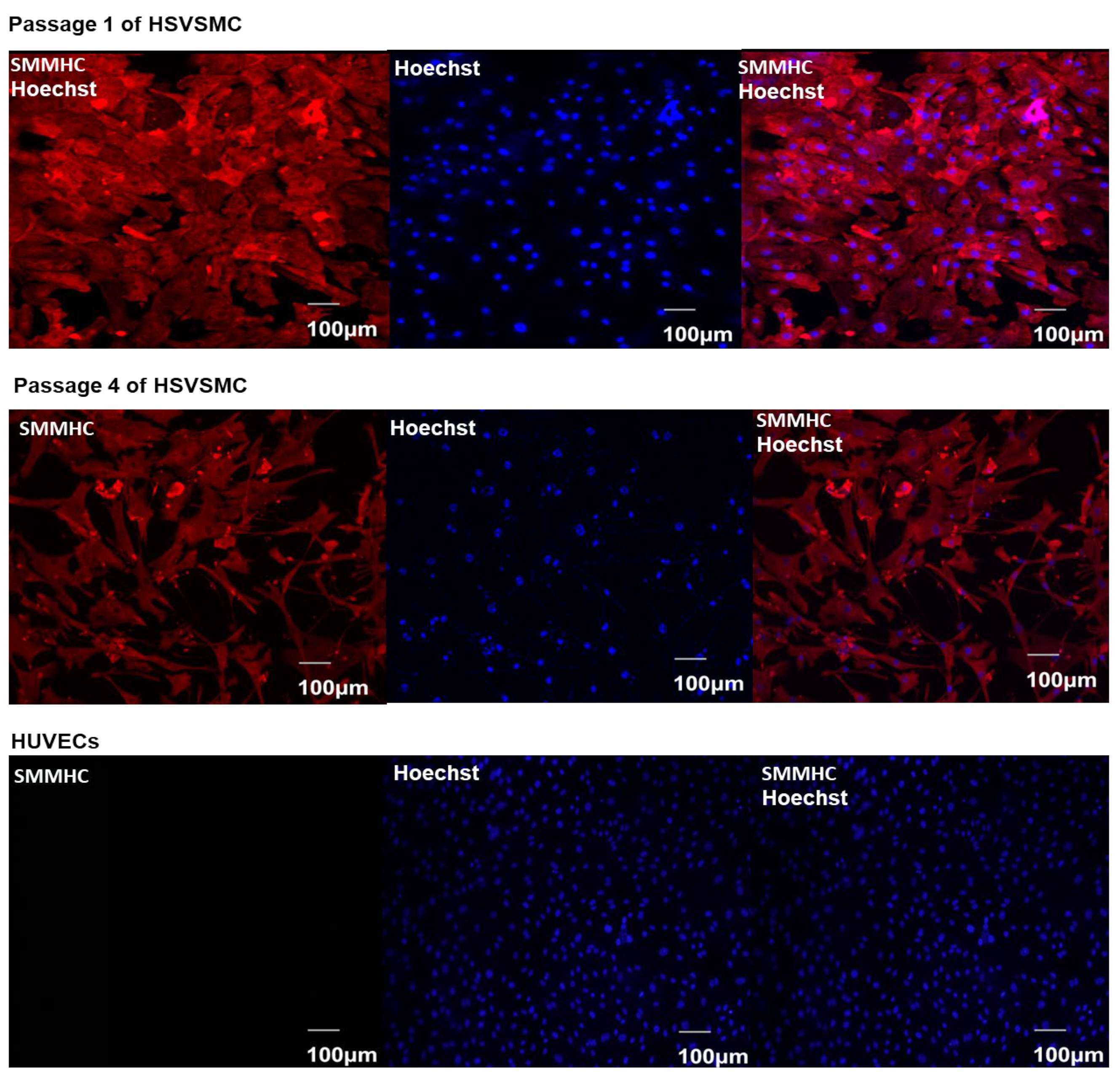
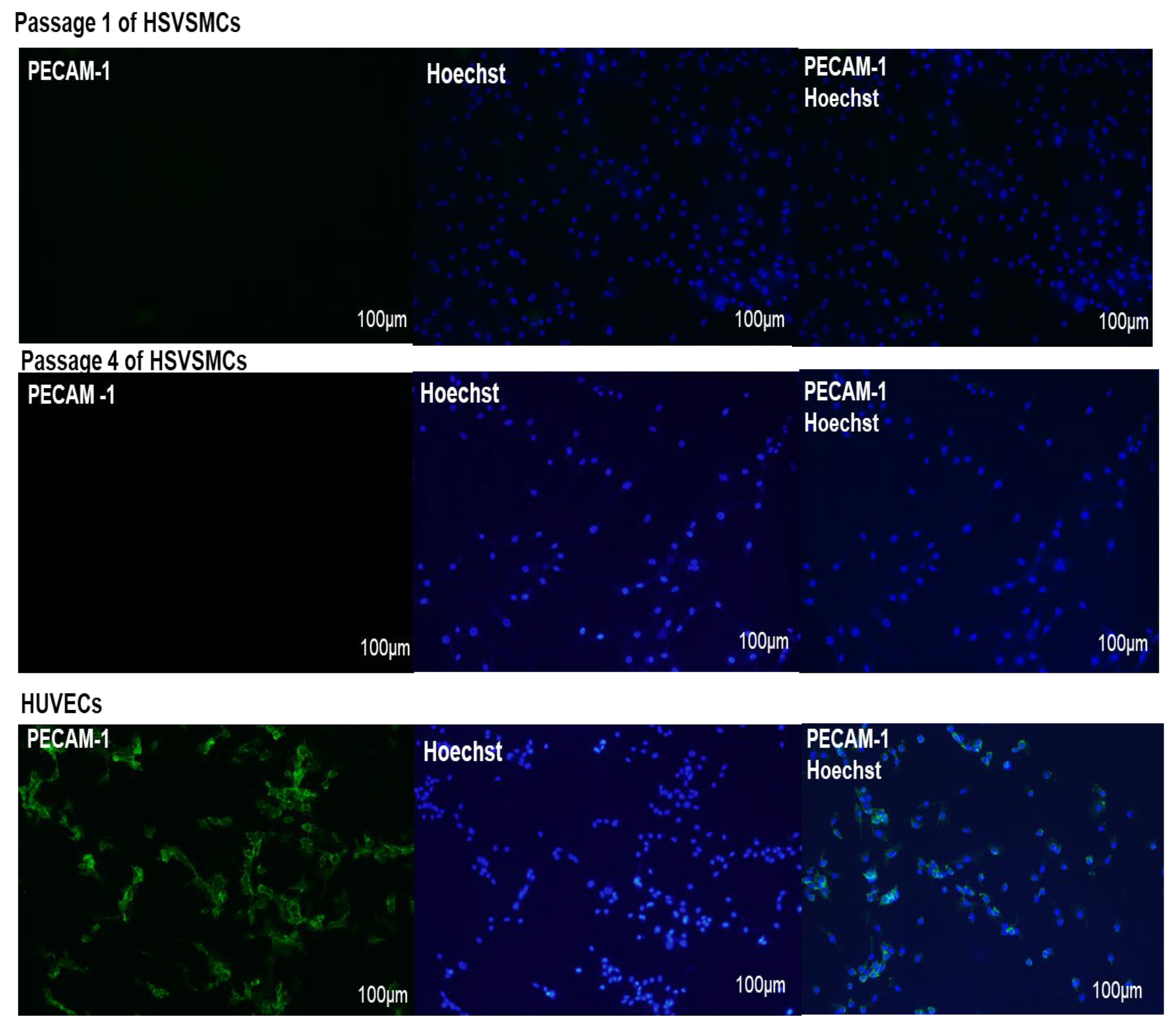
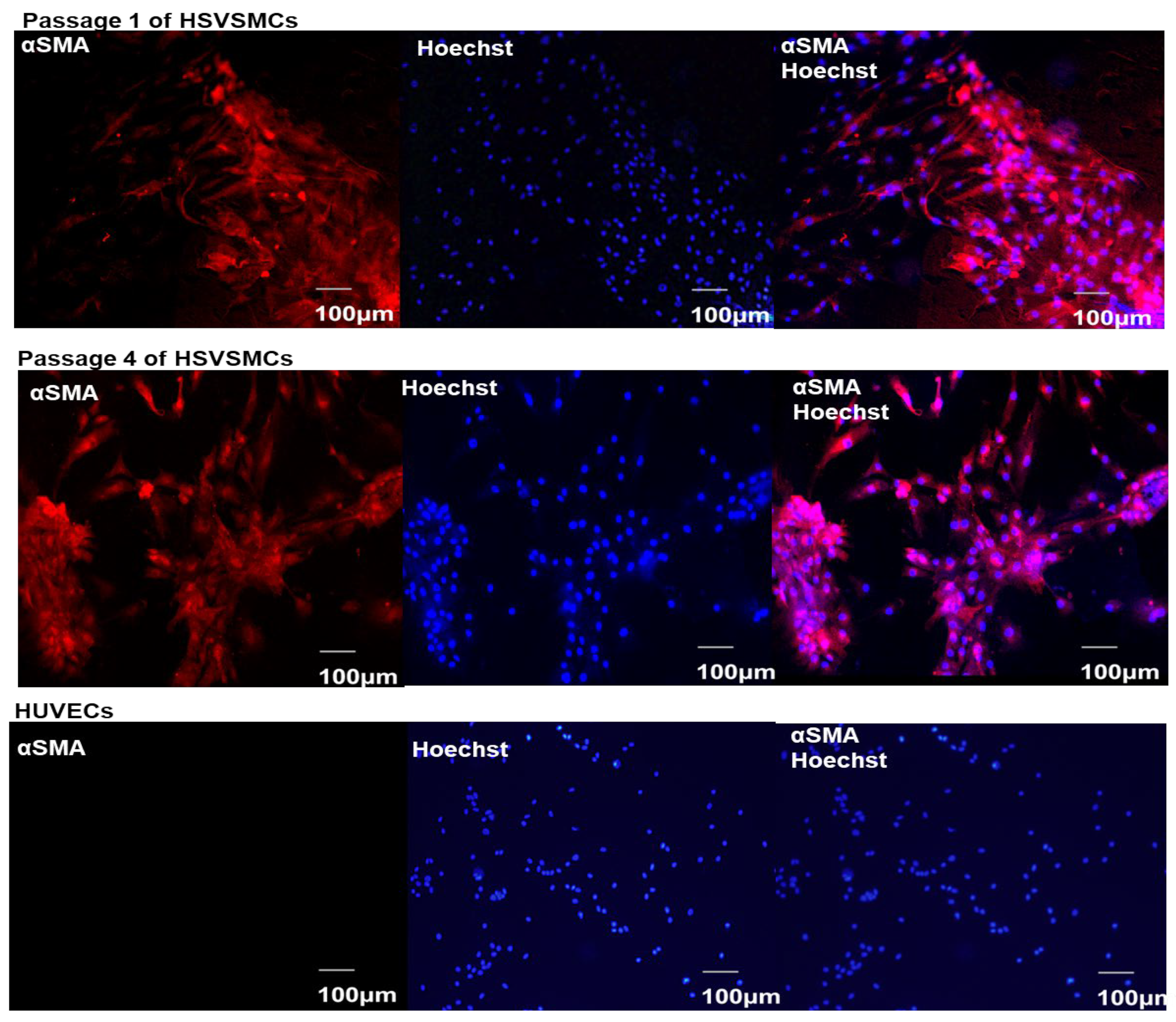
2.2.3. Characterisation of HSVSMCs by Immunofluorescence Microscopy
2.2.4. Generation of Cell Lysates
2.2.5. Transfection of HEK 293T Cells
2.2.6. SDS-PAGE and Immunoblotting
2.2.7. Statistical Analysis
3. Results
3.1. Characterisation of Isolated HSVSMCs
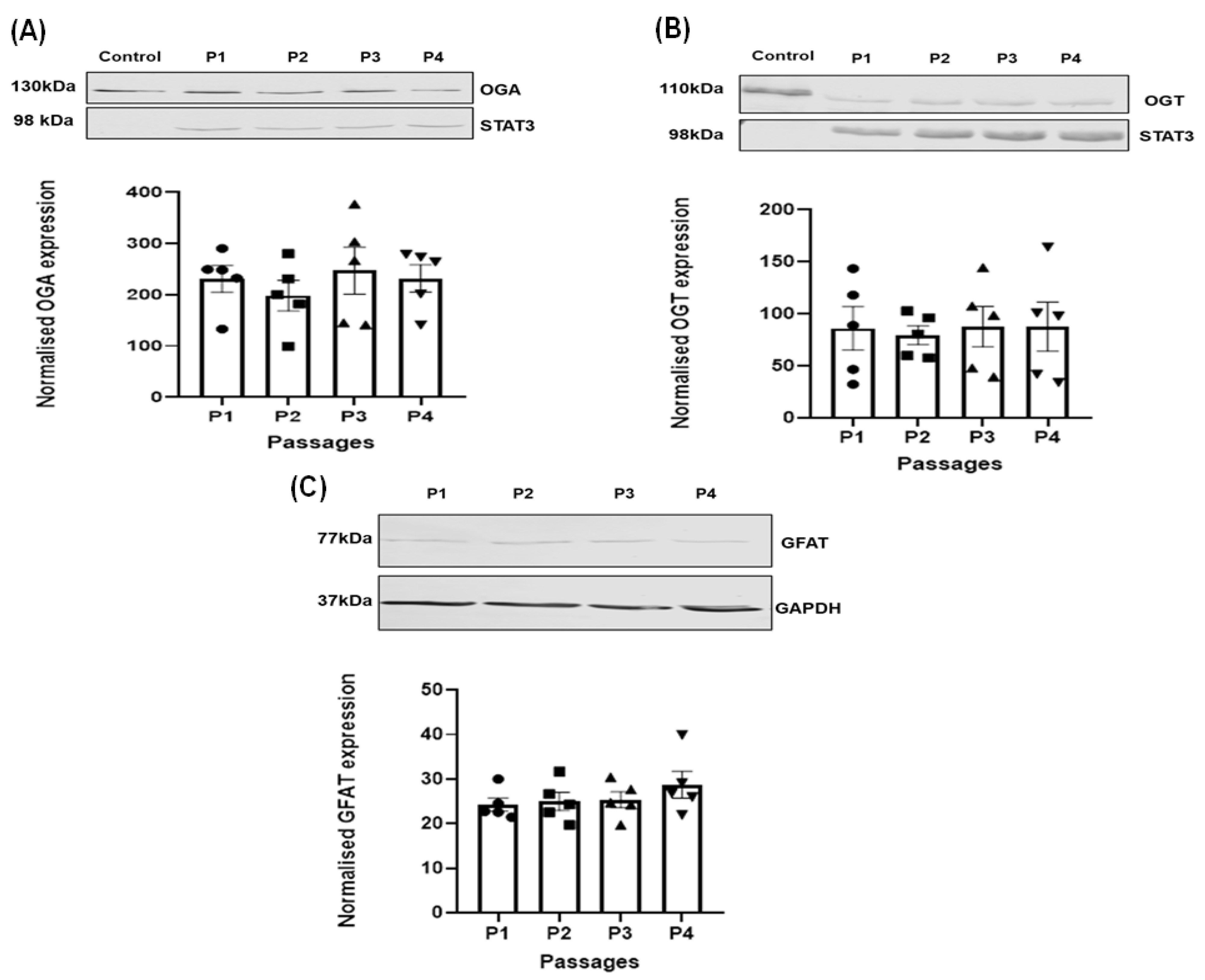
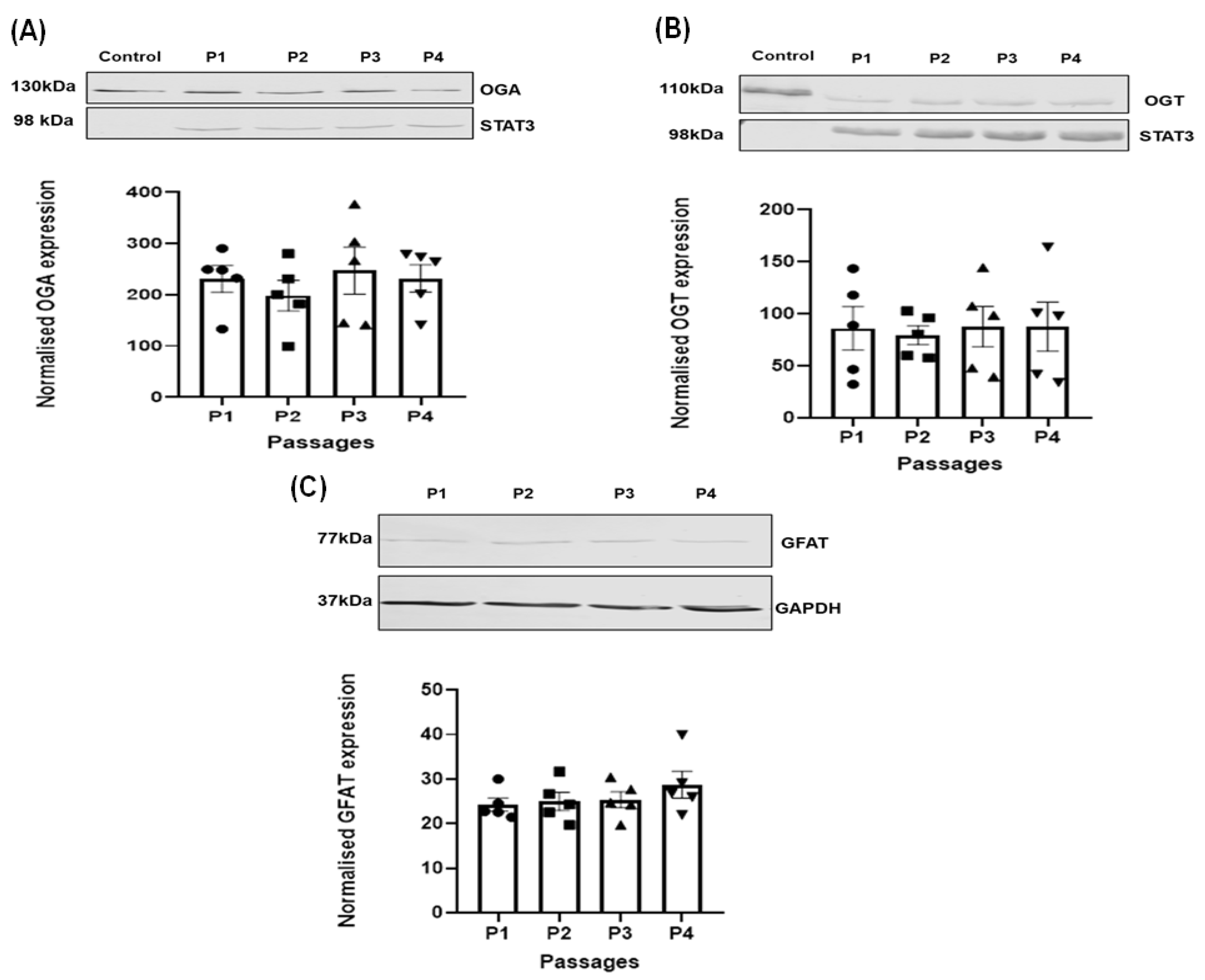
3.2. Effect of Passage on the Expression of the Regulating Enzymes of O-GlcNAcylation in HSVSMC
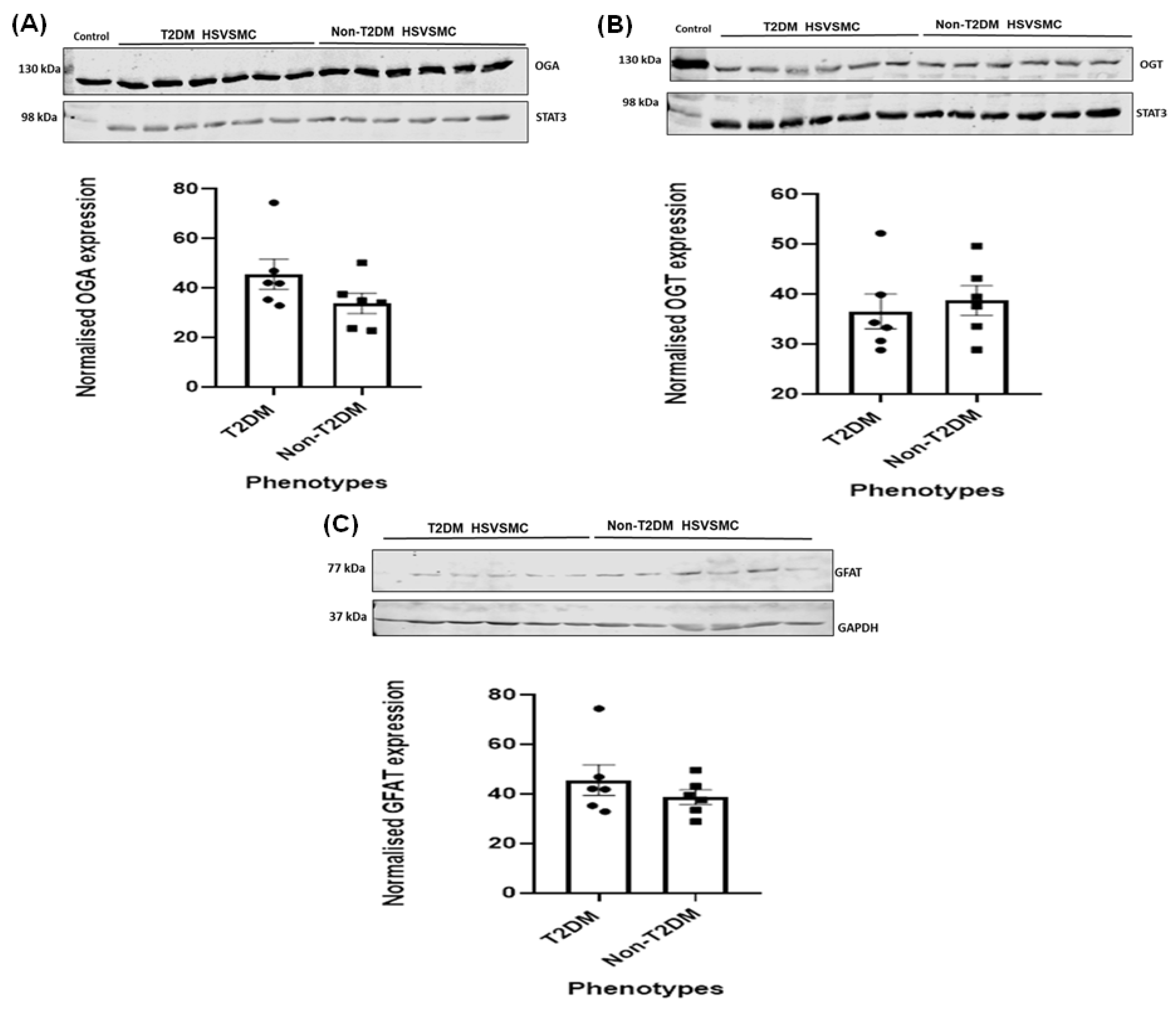
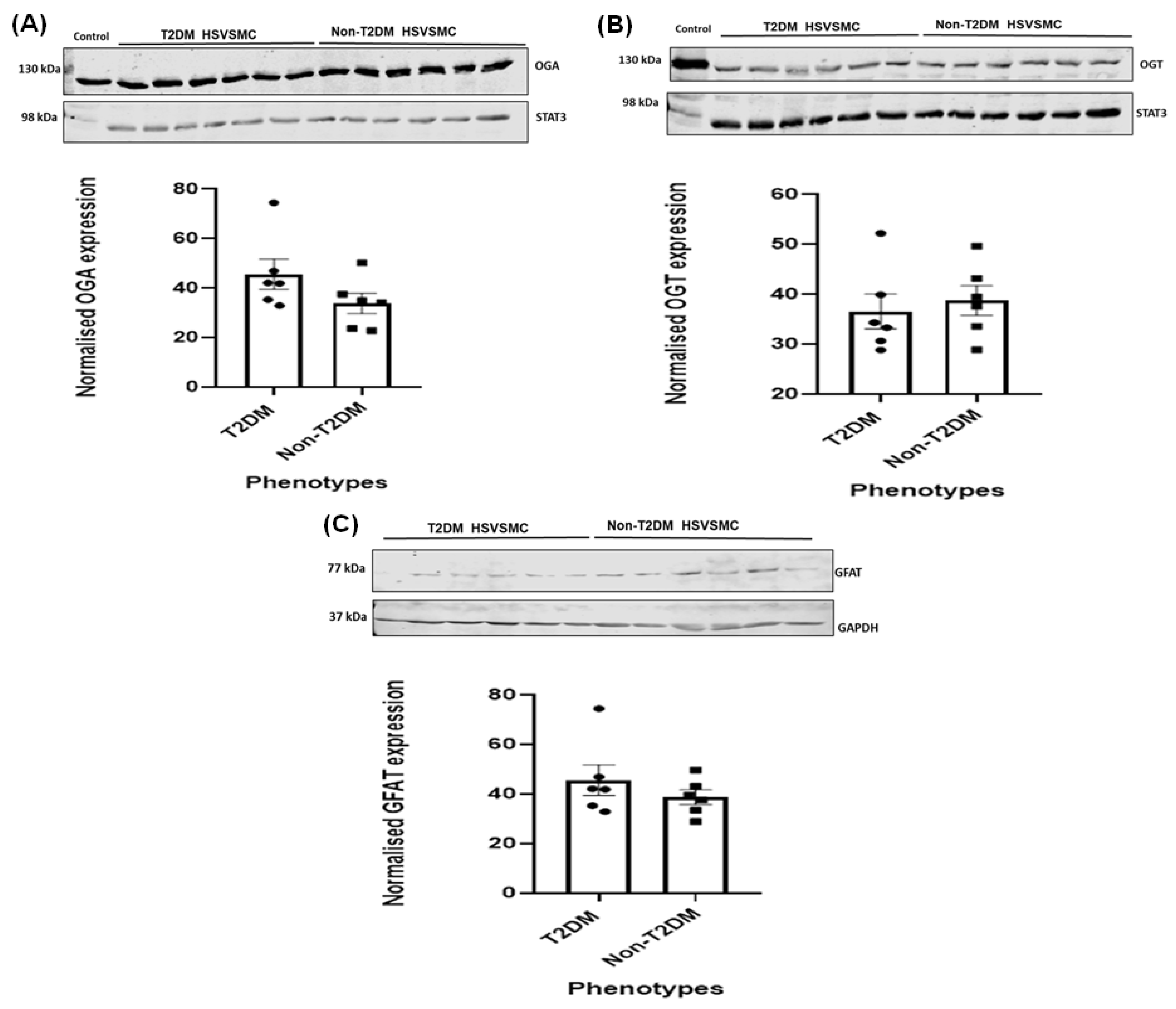
3.3. Comparison of OGA, OGT, and GFAT Expression Levels in HSVSMCs from T2DM versus Control Patients
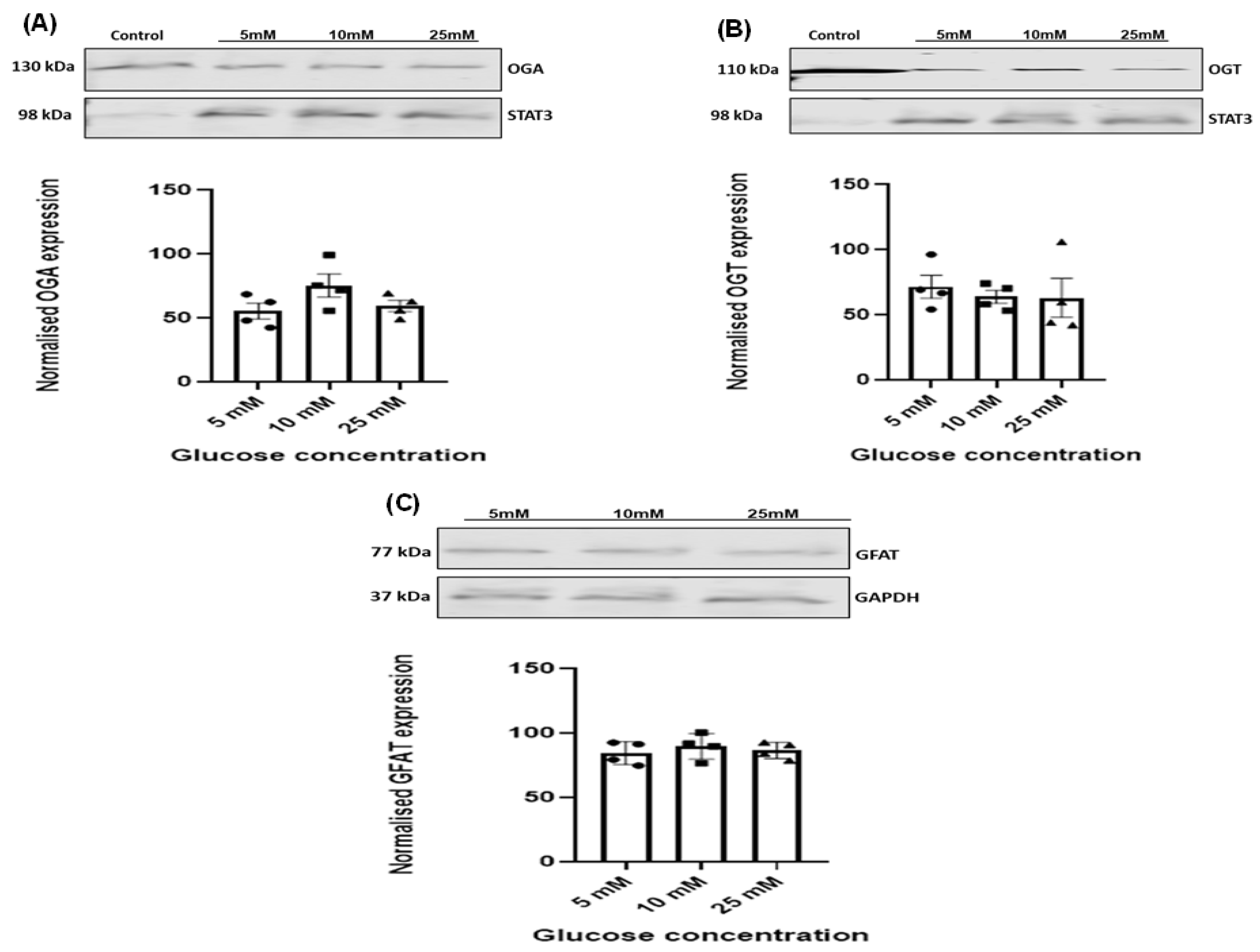
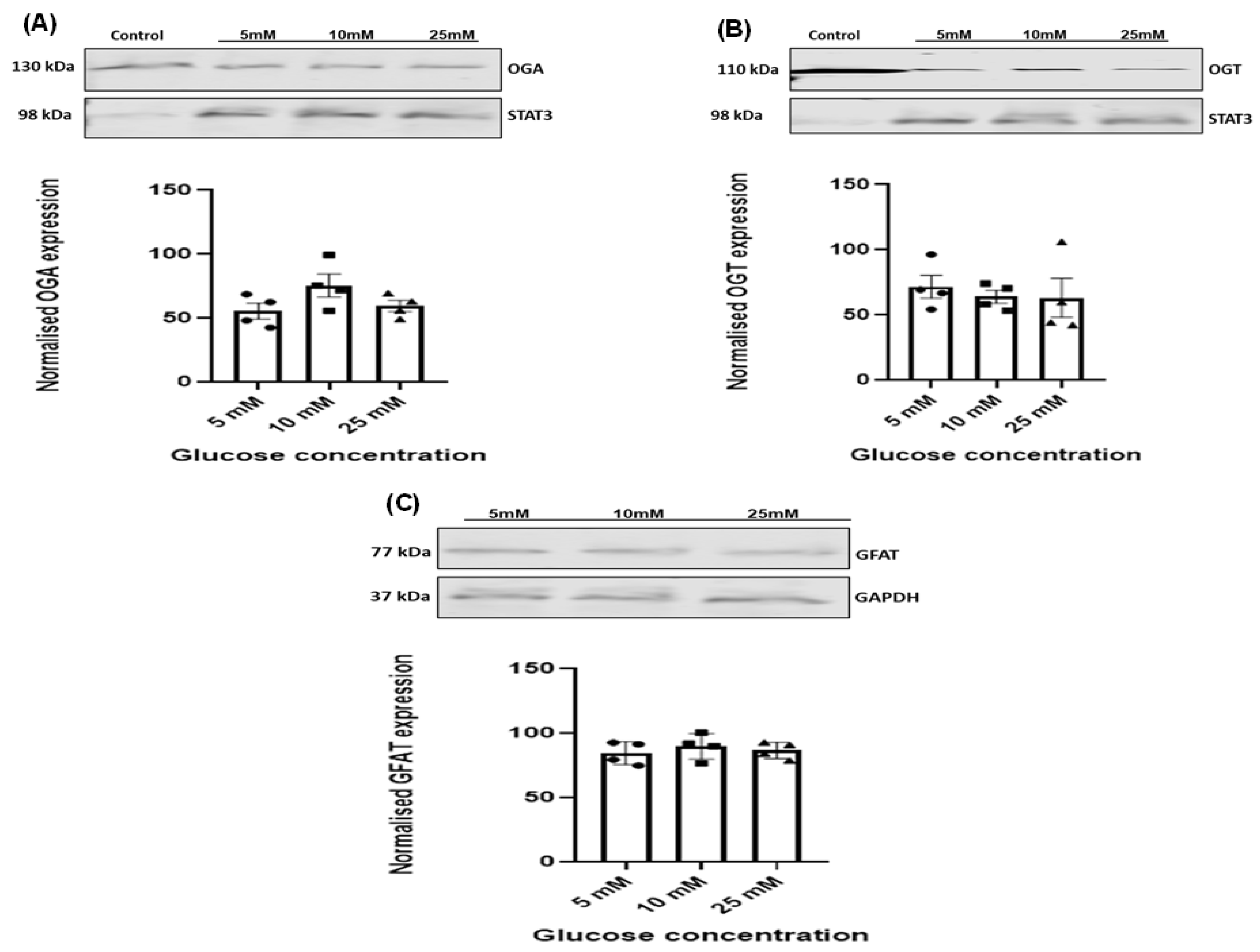
3.4. Effect of Increasing Glucose Concentrations on OGT, OGA, and GFAT Expression Levels in HSVSMCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W.; Housley, M.P.; Slawson, C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 2007, 446, 1017–1022. [Google Scholar] [CrossRef]
- Hart, G.W.; Haltiwanger, R.S.; Holt, G.D.; Kelly, W.G. Glycosylation in the nucleus and cytoplasm. Annu. Rev. Biochem. 1989, 58, 841–874. [Google Scholar] [CrossRef]
- Bolanle, I.O.; Riches-Suman, K.; Williamson, R.; Palmer, T.M. Emerging roles of protein O-GlcNAcylation in cardiovascular diseases: Insights and novel therapeutic targets. Pharmacol. Res. 2021, 165, 105467. [Google Scholar] [CrossRef] [PubMed]
- Bolanle, I.O.; Palmer, T.M. Targeting protein O-GlcNAcylation, a link between type 2 diabetes mellitus and inflammatory disease. Cells 2022, 11, 705. [Google Scholar] [CrossRef] [PubMed]
- Wells, L.; Vosseller, K.; Hart, G.W. Glycosylation of nucleocytoplasmic proteins: Signal transduction and O-GlcNAc. Science 2001, 291, 2376–2378. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A., III; Peters, E.C.; Driggers, E.M.; Hsieh-Wilson, L.C. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 2012, 337, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Slawson, C.; Hart, G.W. O-GlcNAc signalling: Implications for cancer cell biology. Nat. Rev. Cancer 2011, 11, 678–684. [Google Scholar] [CrossRef]
- Ruan, H.B.; Singh, J.P.; Li, M.D.; Wu, J.; Yang, X. Cracking the O-GlcNAc code in metabolism. Trends Endocrinol. Metab. 2013, 24, 301–309. [Google Scholar] [CrossRef]
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The emerging link between O-GlcNAc and Alzheimer disease. J. Biol. Chem. 2014, 289, 34472–34481. [Google Scholar] [CrossRef]
- Bond, M.R.; Hanover, J.A. O-GlcNAc cycling: A link between metabolism and chronic disease. Annu. Rev. Nutr. 2013, 33, 205–229. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, R.M.; Silva, J.F.; Alves, J.V.; Dias, T.B.; Rassi, D.M.; Garcia, L.V.; Lobato, N.D.; Tostes, R.C. Increased O-GlcNAcylation of endothelial nitric oxide synthase compromises the anti-contractile properties of perivascular adipose tissue in metabolic syndrome. Front. Physiol. 2018, 9, 341. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, M.B.; Nam, Y.; Jiang, J.; Sliz, P.; Walker, S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 2011, 469, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Bolanle, I.O.; Riches-Suman, K.; Loubani, M.; Williamson, R.; Palmer, T.M. Revascularisation of type 2 diabetics with coronary artery disease: Insights and therapeutic targeting of O-GlcNAcylation. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Masaki, N.; Feng, B.; Bretón-Romero, R.; Inagaki, E.; Weisbrod, R.M.; Fetterman, J.L.; Hamburg, N.M. O-GlcNAcylation Mediates Glucose-Induced Alterations in Endothelial Cell Phenotype in Human Diabetes Mellitus. J. Am. Heart Assoc. 2020, 9, e014046. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos-dos-Santos, A.; de Queiroz, R.M.; da Costa Rodrigues, B.; Todeschini, A.R.; Dias, W.B. Hyperglycemia and aberrant O-GlcNAcylation: Contributions to tumor progression. J. Bioenerg. Biomembr. 2018, 50, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.R.; Hanover, J.A. A little sugar goes a long way: The cell biology of O-GlcNAc. J. Cell Biol. 2015, 208, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Zucker, D.; Passamani, E.; Peduzzi, P.; Takaro, T.; Fisher, L.D.; Kennedy, J.W.; Davis, K.; Killip, T.; Norris, R.; et al. Effect of coronary artery bypass graft surgery on survival: Overview of 10-year results from randomised trials by the Coronary Artery Bypass Graft Surgery Trialists Collaboration. Lancet 1994, 344, 563–570. [Google Scholar] [CrossRef]
- National Health Service (NHS) Inform. Coronary Heart Bypass Graft. 2020. Available online: https://www.nhsinform.scot/tests-and-treatments/surgical-procedures/coronary-artery-bypass-graft (accessed on 25 June 2023).
- Motwani, J.G.; Topol, E.J. Aortocoronary saphenous vein graft disease: Pathogenesis, predisposition, and prevention. Circulation 1998, 97, 916–931. [Google Scholar] [CrossRef]
- De Vries, M.R.; Simons, K.H.; Jukema, J.W.; Braun, J.; Quax, P.H. Vein graft failure: From pathophysiology to clinical outcomes. Nat. Rev. Cardiol. 2016, 13, 451–470. [Google Scholar] [CrossRef]
- Riches, K.; Alshanwani, A.R.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Wood, I.C.; Turner, N.A.; Porter, K.E. Elevated expression levels of miR-143/5 in saphenous vein smooth muscle cells from patients with Type 2 diabetes drive persistent changes in phenotype and function. J. Mol. Cell. Cardiol. 2014, 74, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.C.; Reis, A.; Noll, T. Preparation of endothelial cells from microand macrovascular origin. In Practical Methods in Cardiovascular Research; Springer: Berlin/Heidelberg, Germany, 2005; pp. 610–629. [Google Scholar]
- Madi, H.A.; Riches, K.; Warburton, P.; O’Regan, D.J.; Turner, N.A.; Porter, K.E. Inherent differences in morphology, proliferation, and migration in saphenous vein smooth muscle cells cultured from nondiabetic and Type 2 diabetic patients. Am. J. Physiol.-Cell Physiol. 2009, 297, C1307–C1317. [Google Scholar] [CrossRef] [PubMed]
- McKavanagh, P.; Yanagawa, B.; Zawadowski, G.; Cheema, A. Management and prevention of saphenous vein graft failure: A review. Cardiol. Ther. 2017, 6, 203–223. [Google Scholar] [CrossRef] [PubMed]
- Campeau, L.J.; Lesperance, J.; Hermann, J.; Corbara, F.; Grondin, C.M.; Bourassa, M.G. Loss of the improvement of angina between 1 and 7 years after aortocoronary bypass surgery: Correlations with changes in vein grafts and in coronary arteries. Circulation 1979, 60, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Riches-Suman, K.; Hussain, A. Identifying and targeting the molecular signature of smooth muscle cells undergoing early vascular ageing. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2022, 1868, 166403. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Miano, J.M.; Cserjesi, P.; Ligon, K.L.; Periasamy, M.; Olson, E.N. Smooth muscle myosin heavy chain exclusively marks the smooth muscle lineage during mouse embryogenesis. Circ. Res. 1994, 75, 803–812. [Google Scholar] [CrossRef]
- Woodfin, A.; Voisin, M.B.; Nourshargh, S. PECAM-1: A multi-functional molecule in inflammation and vascular biology. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2514–2523. [Google Scholar] [CrossRef]
- Weissberg, P.L.; Cary, N.R.; Shanahan, C.M. Gene expression and vascular smooth muscle cell phenotype. Blood Press. Suppl. 1995, 2, 68–73. [Google Scholar]
- Clempus, R.E.; Sorescu, D.; Dikalova, A.E.; Pounkova, L.; Jo, P.; Sorescu, G.P.; Lasseègue, B.; Griendling, K.K. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 42–48. [Google Scholar] [CrossRef]
- Mouriaux, F.; Zaniolo, K.; Bergeron, M.A.; Weidmann, C.; De La Fouchardiere, A.; Fournier, F.; Droit, A.; Morcos, M.W.; Landreville, S.; Guérin, S.L. Effects of long-term serial passaging on the characteristics and properties of cell lines derived from uveal melanoma primary tumors. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5288–5301. [Google Scholar] [CrossRef]
- Neumann, E.; Riepl, B.; Knedla, A.; Lefèvre, S.; Tarner, I.H.; Grifka, J.; Steinmeyer, J.; Schölmerich, J.; Gay, S.; Müller-Ladner, U. Cell culture and passaging alters gene expression pattern and proliferation rate in rheumatoid arthritis synovial fibroblasts. Arthritis Res. Ther. 2010, 12, R83. [Google Scholar] [CrossRef]
- Frid, M.G.; Shekhonin, B.V.; Koteliansky, V.E.; Glukhova, M.A. Phenotypic changes of human smooth muscle cells during development: Late expression of heavy caldesmon and calponin. Dev. Biol. 1992, 153, 185–193. [Google Scholar] [CrossRef]
- Gimona, M.; Herzog, M.; Vandekerckhove, J.; Small, J.V. Smooth muscle specific expression of calponin. FEBS Lett. 1990, 274, 159–162. [Google Scholar]
- Ortiz-Munoz, G.; Martin-Ventura, J.L.; Hernandez-Vargas, P.; Mallavia, B.; Lopez-Parra, V.; Lopez-Franco, O.; Munoz-Garcia, B.; Fernandez-Vizarra, P.; Ortega, L.; Egido, J.; et al. Suppressors of cytokine signaling modulate JAK/STAT-mediated cell responses during atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 525–531. [Google Scholar] [CrossRef]
- Xiang, S.; Dong, N.G.; Liu, J.P.; Wang, Y.; Shi, J.W.; Wei, Z.J.; Hu, X.J.; Gong, L. Inhibitory effects of suppressor of cytokine signaling 3 on inflammatory cytokine expression and migration and proliferation of IL-6/IFN-γ-induced vascular smooth muscle cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2013, 33, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Liu, J.; Dong, N.; Shi, J.; Xiao, Y.; Wang, Y.; Hu, X.; Gong, L.; Wang, W. Suppressor of cytokine signaling 3 is a negative regulator for neointimal hyperplasia of vein graft stenosis. J. Vasc. Res. 2014, 51, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Uhrin, P.; Mocan, A.; Waltenberger, B.; Breuss, J.M.; Tewari, D.; Mihaly-Bison, J.; Huminiecki, Ł.; Starzyński, R.R.; Tzvetkov, N.T.; et al. Vascular smooth muscle cell proliferation as a therapeutic target. Part 1: Molecular targets and pathways. Biotechnol. Adv. 2018, 36, 1586–1607. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, B.D.; Love, D.C.; Hanover, J.A. Recombinant O-GlcNAc transferase isoforms: Identification of O-GlcNAcase, yes tyrosine kinase, and tau as isoform-specific substrates. Glycobiology 2006, 16, 415–421. [Google Scholar] [CrossRef]
- Liu, C.; Dong, W.; Li, J.; Kong, Y.; Ren, X. O-GlcNAc Modification and Its Role in Diabetic Retinopathy. Metabolites 2022, 12, 725. [Google Scholar] [CrossRef]
- Ma, J.; Hart, G.W. Protein O-GlcNAcylation in diabetes and diabetic complications. Expert Rev. Proteom. 2013, 10, 365–380. [Google Scholar] [CrossRef]
- Umapathi, P.; Mesubi, O.O.; Banerjee, P.S.; Abrol, N.; Wang, Q.; Luczak, E.D.; Wu, Y.; Granger, J.M.; Wei, A.C.; Reyes Gaido, O.E.; et al. Excessive O-GlcNAcylation causes heart failure and sudden death. Circulation 2021, 143, 1687–1703. [Google Scholar] [CrossRef]
- Chatham, J.C.; Zhang, J.; Wende, A.R. Role of O-linked N-acetylglucosamine protein modification in cellular (patho) physiology. Physiol. Rev. 2021, 101, 427–493. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.V.; Andreadou, I.; Hausenloy, D.J.; Bøtker, H.E. The role of O-GlcNAcylation for protection against ischemia-reperfusion injury. Int. J. Mol. Sci. 2019, 20, 404. [Google Scholar] [CrossRef]
- Park, K.; Saudek, C.D.; Hart, G.W. Increased expression of β-N-acetylglucosaminidase in erythrocytes from individuals with pre-diabetes and diabetes. Diabetes 2010, 59, 1845–1850. [Google Scholar] [CrossRef]
- Akimoto, Y.; Hart, G.W.; Wells, L.; Vosseller, K.; Yamamoto, K.; Munetomo, E.; Ohara-Imaizumi, M.; Nishiwaki, C.; Nagamatsu, S.; Hirano, H.; et al. Elevation of the post-translational modification of proteins by O-linked N-acetylglucosamine leads to deterioration of the glucose-stimulated insulin secretion in the pancreas of diabetic Goto–Kakizaki rats. Glycobiology 2007, 17, 127–140. [Google Scholar] [CrossRef] [PubMed]
- McClain, D.A.; Lubas, W.A.; Cooksey, R.C.; Hazel, M.; Parker, G.J.; Love, D.C.; Hanover, J.A. Altered glycan-dependent signaling induces insulin resistance and hyperleptinemia. Proc. Natl. Acad. Sci. USA 2002, 99, 10695–10699. [Google Scholar] [CrossRef]
- Yang, X.; Ongusaha, P.P.; Miles, P.D.; Havstad, J.C.; Zhang, F.; So, W.V.; Kudlow, J.E.; Michell, R.H.; Olefsky, J.M.; Field, S.J.; et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 2008, 451, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Soesanto, Y.; Luo, B.; Parker, G.; Jones, D.; Cooksey, R.C.; McClain, D.A. Pleiotropic and age-dependent effects of decreased protein modification by O-linked N-acetylglucosamine on pancreatic β-cell function and vascularization. J. Biol. Chem. 2011, 286, 26118–26126. [Google Scholar] [CrossRef]
- Dentin, R.; Hedrick, S.; Xie, J.; Yates, J., III; Montminy, M. Hepatic glucose sensing via the CREB coactivator CRTC2. Science 2008, 319, 1402–1405. [Google Scholar] [CrossRef]
- Yki-Järvinen, H.; Virkamäki, A.; Daniels, M.C.; McClain, D.; Gottschalk, W.K. Insulin and glucosamine infusions increase O-linked N-acetyl-glucosamine in skeletal muscle proteins in vivo. Metabolism 1998, 47, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.A.; Weinstein, M.L.; Lindenmayer, G.E.; Buse, M.G. Effects of diabetes and hyperglycemia on the hexosamine synthesis pathway in rat muscle and liver. Diabetes 1995, 44, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; Gratton, J.P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Montagnani, M.; Chen, H.; Barr, V.A.; Quon, M.J. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser1179. J. Biol. Chem. 2001, 276, 30392–30398. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | T2DM | Non-T2DM | |
|---|---|---|---|
| 1 | Number of samples | 17 | 38 |
| 2 | Age range | 56–80 | 48–84 |
| 3 | Sex | Male—15 Female—2 | Male—37 Female—1 |
| 4 | Ethnicity | White British—17 | White British—37 Asian—1 |
| 5 | Reason for surgery | CAD | CAD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolanle, I.O.; Durham, G.A.; Hobkirk, J.P.; Loubani, M.; Sturmey, R.G.; Palmer, T.M. Do T2DM and Hyperglycaemia Affect the Expression Levels of the Regulating Enzymes of Cellular O-GlcNAcylation in Human Saphenous Vein Smooth Muscle Cells? Diabetology 2024, 5, 162-177. https://doi.org/10.3390/diabetology5020013
Bolanle IO, Durham GA, Hobkirk JP, Loubani M, Sturmey RG, Palmer TM. Do T2DM and Hyperglycaemia Affect the Expression Levels of the Regulating Enzymes of Cellular O-GlcNAcylation in Human Saphenous Vein Smooth Muscle Cells? Diabetology. 2024; 5(2):162-177. https://doi.org/10.3390/diabetology5020013
Chicago/Turabian StyleBolanle, Israel O., Gillian A. Durham, James P. Hobkirk, Mahmoud Loubani, Roger G. Sturmey, and Timothy M. Palmer. 2024. "Do T2DM and Hyperglycaemia Affect the Expression Levels of the Regulating Enzymes of Cellular O-GlcNAcylation in Human Saphenous Vein Smooth Muscle Cells?" Diabetology 5, no. 2: 162-177. https://doi.org/10.3390/diabetology5020013
APA StyleBolanle, I. O., Durham, G. A., Hobkirk, J. P., Loubani, M., Sturmey, R. G., & Palmer, T. M. (2024). Do T2DM and Hyperglycaemia Affect the Expression Levels of the Regulating Enzymes of Cellular O-GlcNAcylation in Human Saphenous Vein Smooth Muscle Cells? Diabetology, 5(2), 162-177. https://doi.org/10.3390/diabetology5020013