
Recent Advances in Monitoring Microbial Toxins in Food Samples by HPLC-Based Techniques: A Review

 , , , and
, , , and 
Abstract
1. Introduction
- Direct contact can occur due to contaminated hands of food handlers or infected animals and contaminated surfaces;
- Cross-contamination can occur due to pathogens transferred from raw foods or utensils used without proper cleaning, as well as to improper storage;
- Water contamination can occur by irrigating crops with contaminated water or using contaminated water during food processing, washing, or cooking;
- Airborne transmission can occur by spreading pathogens through the air in dust particles, water droplets, or poorly maintained or contaminated ventilation systems.
- Soil contamination can be caused by naturally present pathogens or by using untreated manure as fertilizers;
- Human factors, such as poor hygiene or sickness of food handlers, which can transmit pathogens to food through coughing, sneezing, or direct contact;Pests, which can include insects and rodents;
- Contaminated ingredients that could become contaminated during the supply chain.
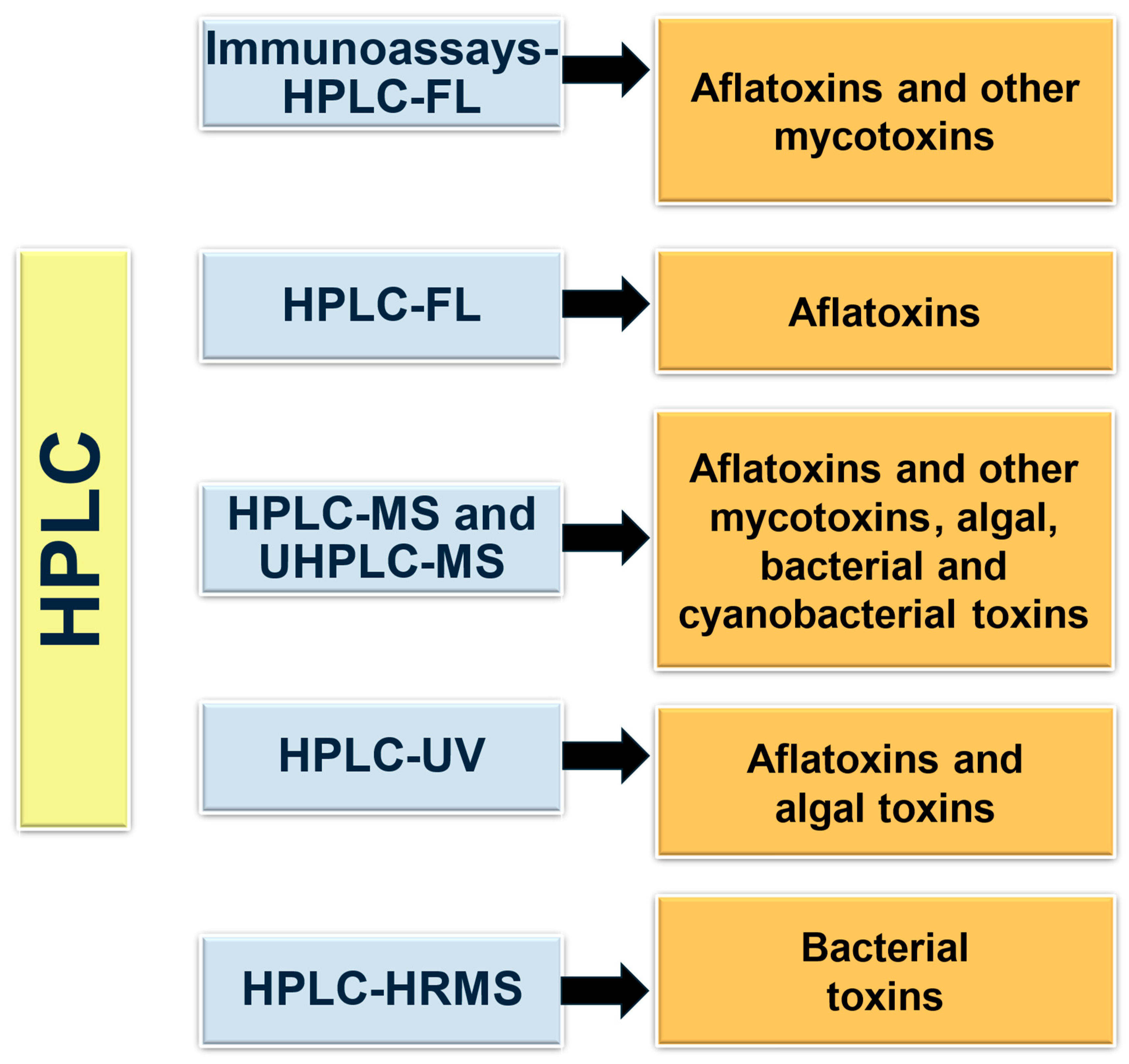
2. HPLC-Based Methodologies for Microbial Toxin Detection in Food Samples
2.1. Aflatoxins and Other Mycotoxins
2.1.1. HPLC-UV
2.1.2. HPLC-FD
2.1.3. Novel Technologies: HPLC-MS/MS, LC-HRMS, and UPLC-MS/MS
2.2. Bacterial Toxins
2.2.1. HPLC-UV and HPLC-HRMS
2.2.2. UHPLC-MS/MS and Nano-LC-MS/MS
2.3. Cyanotoxins and Harmful Algal Blooms Toxins
2.3.1. HPLC-UV
2.3.2. HPLC-MS and UHPLC-MS
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Strategy for Food Safety 2022–2030. 2022. Available online: https://iris.who.int/handle/10665/364638 (accessed on 13 July 2024).
- Newland & Newland LLP. What Is the Difference between Foodborne Illness and Food Poisoning? 2016. Available online: https://ask.usda.gov/s/article/What-is-the-difference-between-food-poisoning-and-foodborne-illness (accessed on 4 February 2024).
- Tatini, S.R. Thermal stability of enterotoxins in food. J. Food Prot. 1976, 39, 432–438. [Google Scholar] [CrossRef]
- Bruslin, L. General Microbiology, 1st ed.; Oregon State University: Corvallis, OR, USA, 2019. [Google Scholar] [CrossRef]
- Tang, X.; Zuo, J.; Yang, C.; Jiang, J.; Zhang, Q.; Ping, J.; Li, P. Current trends in biosensors for biotoxins (mycotoxins, marine toxins, and bacterial food toxins): Principles, application, and perspective. TrAC Trends Anal. Chem. 2023, 165, 117144. [Google Scholar] [CrossRef]
- Ray, B. Fundamental Food Microbiology, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Tokio Food Safety Information Center. What are Mycotoxins? 2024. Available online: https://www.hokeniryo.metro.tokyo.lg.jp/shokuhin/eng/kabi/kabidoqa.html#:~:text=%EF%BC%91%20Mycotoxins%20are%20resistant%20to,times%20(under%2060%20minutes) (accessed on 6 April 2024).
- Wakenell, P. Management and medicine of backyard poultry. In Current Therapy in Avian Medicine and Surgery; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 550–565. [Google Scholar] [CrossRef]
- Bennett, J.W.; Klich, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.M.; Qamar, F.; Saifi, M.; Abdin, M.Z. Natural inhibitors: A sustainable way to combat aflatoxins. Front. Microbiol. 2022, 13, 993834. [Google Scholar] [CrossRef]
- Ghazaei, C. Advances in the Study of Bacterial Toxins, Their Roles and Mechanisms in Pathogenesis. Malays. J. Med. Sci. 2022, 29, 4–17. [Google Scholar] [CrossRef]
- Czura, A.W.; Campus, E. Chapter 15—Microbial mechanisms of pathogenicity. In Microbiology: An Introduction, 13th ed.; Tortora, G., Funke, B., Case, C., Eds.; Pearson: Boston, MA, USA, 2019; pp. 423–444. [Google Scholar]
- Kumar, S. Textbook of Microbiology, 1st ed.; Jaypee Brothers Medical Publishers: New Delhi, India, 2012. [Google Scholar]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef]
- Wells, M.L.; Karlson, B.; Wulff, A.; Kudela, R.; Trick, C.; Asnaghi, V.; Berdalet, E.; Cochlan, W.; Davidson, K.; De Rijcke, M. Future HAB science: Directions and challenges in a changing climate. Harmful Algae 2020, 91, 101632. [Google Scholar] [CrossRef]
- Otero, P.; Silva, M. Emerging Marine Biotoxins in European Waters: Potential Risks and Analytical Challenges. Mar. Drugs 2022, 20, 199. [Google Scholar] [CrossRef]
- Young, N.; Sharpe, R.A.; Barciela, R.; Nichols, G.; Davidson, K.; Berdalet, E.; Fleming, L.E. Marine harmful algal blooms and human health: A systematic scoping review. Harmful Algae 2020, 98, 101901. [Google Scholar] [CrossRef]
- Lassudrie, M.; Hegaret, H.; Wikfors, G.H.; da Silva, P.M. Effects of marine harmful algal blooms on bivalve cellular immunity and infectious diseases: A review. Dev. Comp. Immunol. 2020, 108, 103660. [Google Scholar] [CrossRef]
- Backer, L.C.; Manassaram-Baptiste, D.; LePrell, R.; Bolton, B. Cyanobacteria and algae blooms: Review of health and environmental data from the harmful algal bloom-related illness surveillance system (HABISS) 2007–2011. Toxins 2015, 7, 1048–1064. [Google Scholar] [CrossRef] [PubMed]
- Cantoral-Uriza, E.A.; Asencio-Martínez, A.D.; Aboal-Sanjurjo, M. Cianotoxinas: Efectos ambientales y sanitarios. Medidas de prevención. Hidrobiológica 2017, 27, 241–251. [Google Scholar]
- Oshiro, N.; Gago-Martínez, A.; Tubaro, A. Chemistry, Toxicology and Etiology of Marine Biotoxins. J. Mar. Sci. Eng. 2024, 12, 236. [Google Scholar] [CrossRef]
- Nowruzi, B.; Porzani, S.J. Toxic compounds produced by cyanobacteria belonging to several species of the order Nostocales: A review. J. Appl. Toxicol. 2021, 41, 510–548. [Google Scholar] [CrossRef] [PubMed]
- Falfushynska, H.; Kasianchuk, N.; Siemens, E.; Henao, E.; Rzymski, P. A review of common cyanotoxins and their effects on fish. Toxics 2023, 11, 118. [Google Scholar] [CrossRef]
- Kamala, K.; Kumar, V.P. Chapter 1. Food products and food contamination. In Microbial Contamination and Food Degradation; Academic Press: Cambridge, MA, USA, 2018; pp. 1–19. [Google Scholar]
- European Parliament. Directive (EC) 2002/32/EC of the European Parliament and the Council of 7 May 2002 on Undesirable Substances in Animal Feed. L140. 2002. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CONSLEG:2002L0032:20061020:EN:PDF (accessed on 1 January 2024).
- Ministério da Saúde. PORTARIA N° 2914. 2011. Available online: https://bvsms.saude.gov.br/bvs/publicacoes/portaria_2914_2011.pdf (accessed on 5 February 2024).
- National Health and Medical Research Council; National Resource Management Ministerial Council. Australian Drinking Water Guidelines, Paper 6 National Water Quality Management Strategy. 2011. Available online: https://www.nhmrc.gov.au/sites/default/files/documents/reports/aust-drinking-water-guidelines.pdf (accessed on 10 February 2024).
- Gazzetta Ufficiale. Attuazione Della Direttiva 98/83/CE Relativa Alla Qualità Delle Acque Destinate Al Consumo Umano, Decreto Legislativo 2 Febbraio. 2001. Available online: https://www.physico.eu/pdf/direttiva-europea-98-83-ce.pdf (accessed on 12 February 2024).
- Governement Gazette of Republic of South Africa. Water Services Act. Available online: https://www.fao.org/faolex/results/details/en/c/LEX-FAOC015982/ (accessed on 28 February 2024).
- GB 5749-2006; Standards of Drinking Water Quality. Standardization Administration of China. Ministry of Health of China: Beijing, China, 2007.
- Boletín Oficial del Estado. Real Decreto 140/2003, de 7 de Febrero, Criterios Sanitarios de la Calidad del Agua de Consumo Humano, 7228. 2003. Available online: https://www.boe.es/buscar/act.php?id=BOE-A-2003-3596 (accessed on 6 February 2024).
- Ando, M. New regulation of tap water quality and standard methods in Japan. Jpn. J. Toxicol. Environ. Health 1994, 40, 317–327. [Google Scholar] [CrossRef]
- Picardo, M.; Filatova, D.; Nunez, O.; Farré, M. Recent advances in the detection of natural toxins in freshwater environments. TrAC Trends Anal. Chem. 2019, 112, 75–86. [Google Scholar] [CrossRef]
- Sheth, U.V. Detection of Toxin Genes by PCR Based Methods. In Biosafety Assessment of Probiotic Potential. Methods and Protocols in Food Science; Dwivedi, M.K., Amaresan, N., Sankaranarayanan, A., Begum, R., Eds.; Humana: New York, NY, USA, 2022; pp. 107–121. [Google Scholar] [CrossRef]
- Rhiannon, D. The promise of toxicogenomics for genetic toxicology: Past, present and future. Mutagenesis 2020, 35, 153–159. [Google Scholar]
- Scott, R.P.W. Essential oils. In Encyclopedia of Analytical Science, 2nd ed.; Worsfold, P., Townshend, A., Poole, C., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2005; pp. 554–561. [Google Scholar]
- Romero-Sánchez, I.; Ramírez-García, L.; Gracia-Lor, E.; Madrid-Albarrán, Y. Simultaneous determination of aflatoxins B1, B2, G1 and G2 in commercial rices using immunoaffinity column clean-up and HPLC-MS/MS. Food Chem. 2022, 395, 133611. [Google Scholar] [CrossRef]
- Li, C.; Chu, S.; Tan, S.; Yin, X.; Jiang, Y.; Dai, X.; Gong, X.; Fang, X.; Tian, D. Towards higher sensitivity of mass spectrometry: A perspective from the mass analyzers. Front. Chem. 2021, 9, 813359. [Google Scholar] [CrossRef]
- Otero, C.; Arredondo, C.; Echeverriá-Vega, A.; Gordillo-Fuenzalida, F. Penicillium spp. Mycotoxins found in food and feed and their health effects. World Mycotoxin J. 2020, 13, 323–343. [Google Scholar] [CrossRef]
- Algammal, A.M.; Elsayed, M.E.; Hashem, H.R.; Ramadan, H.; Sheraba, N.S.; El-Diasty, E.M.; Abbas, S.M.; Hetta, H.F. Molecular and HPLC-based approaches for detection of aflatoxin B1 and ochratoxin A released from toxigenic Aspergillus species in processed meat. BMC Microbiol. 2021, 21, 82. [Google Scholar] [CrossRef] [PubMed]
- Maggira, M.; Sakaridis, I.; Ioannidou, M.; Samouris, G. Comparative Evaluation of Three Commercial Elisa Kits Used for the Detection of Aflatoxins B1, B2, G1, and G2 in Feedstuffs and Comparison with an HPLC Method. Vet. Sci. 2022, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Schrenk, D.; Bignami, M.; Bodin, L.; Chipman, J.K.; del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.; Leblanc, J.C.; Nebbia, C.S.; et al. Risk assessment of aflatoxins in food. EFSA J. 2020, 18, e06040. [Google Scholar] [CrossRef] [PubMed]
- Kabak, B. Aflatoxins in foodstuffs: Occurrence and risk assessment in Turkey. J. Food Compos. Anal. 2021, 96, 103734. [Google Scholar] [CrossRef]
- Awuchi, C.G.; Amagwula, I.O.; Priya, P.; Kumar, R.; Yezdani, U.; Gayoor Khan, M. Aflatoxins In Foods And Feeds: A Review on Health Implications, Detection, and Control. Env. Pharmacol. Life Sci. 2020, 9, 149–155. [Google Scholar]
- Benkerroum, N. Chronic and acute toxicities of aflatoxins: Mechanisms of action. Int. J. Environ. Res. Public Health 2020, 17, 423. [Google Scholar] [CrossRef]
- Omar, S.S.; Haddad, M.A.; Parisi, S. Validation of HPLC and Enzyme-Linked Immunosorbent Assay (ELISA) techniques for detection and quantification of aflatoxins in different food samples. Foods 2020, 9, 661. [Google Scholar] [CrossRef]
- The Commission of the European Communities. COMMISSION REGULATION (EC) No 1881/2006: Maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Union 2006. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2006:364:0005:0024:EN:PDF (accessed on 18 February 2024).
- Sarmento-Amoras, E.; Pena-Costa, A.L. Aflatoxins: A Brief Review of their Chemical Properties, Toxicological Effects and Control Measures. Arch. Ecotoxicol. 2020, 2, 43–46. [Google Scholar] [CrossRef]
- Schoenau, E.A. Elements of method design. Am. Chem. Soc. Symp. Ser. 2019, 1300, 3–16. [Google Scholar] [CrossRef]
- Uzeh, R.E.; Adebowale, E.T. Aflatoxigenic fungi and aflatoxins in locally processed peanut butter in Lagos, Nigeria. J. Microbiol. Biotechnol. Food Sci. 2021, 10, e3546. [Google Scholar] [CrossRef]
- Alilou, S.; Amirzehni, M.; Eslami, P.A. A simple fluorometric method for rapid screening of aflatoxins after their extraction by magnetic MOF-808/graphene oxide composite and their discrimination by HPLC. Talanta 2021, 235, 122709. [Google Scholar] [CrossRef] [PubMed]
- Palma, P.; Godoy, M.; Vidal, M.; Rivera, A.; Calderón, R. Adaptation, optimization, and validation of a sensitive and robust method for the quantification of total aflatoxins (B1, B2, G1, and G2) in the spice merkén by HPLC-FLD with post-column derivatization. Microchem. J. 2022, 178, 107342. [Google Scholar] [CrossRef]
- Shuib, N.S.; Saad, B. In-syringe dispersive micro-solid phase extraction method for the HPLC-fluorescence determination of aflatoxins in milk. Food Control 2022, 132, 108510. [Google Scholar] [CrossRef]
- Maggira, M.; Ioannidou, M.; Sakaridis, I.; Samouris, G. Determination of aflatoxin m1 in raw milk using an hplc-fl method in comparison with commercial elisa kits—Application in raw milk samples from various regions of greece. Vet. Sci. 2021, 8, 46. [Google Scholar] [CrossRef]
- Turksoy, S.; Kabak, B. Determination of aflatoxins and ochratoxin A in wheat from different regions of Turkey by HPLC with fluorescence detection. Acta Aliment. 2020, 49, 118–124. [Google Scholar] [CrossRef]
- Food and Agriculture Organization. Food Control (Maximum Levels of Aflatoxins in Food) Regulations. 2011. Available online: https://faolex.fao.org/docs/pdf/BOT196888.pdf (accessed on 2 February 2024).
- Shen, M.H.; Singh, R.K. Determining aflatoxins in raw peanuts using immunoaffinity column as sample clean-up method followed by normal-phase HPLC-FLD analysis. Food Control 2022, 139, 109065. [Google Scholar] [CrossRef]
- Dhanshetty, M.; Thorat, P.; Banerjee, K. High-Throughput Analysis of Aflatoxins in Cereals, Nuts, and Processed Products Involving Automated Immunoaffinity Cleanup and Inline HPLC-Fluorescence Detection. J. AOAC Int. 2021, 104, 1526–1532. [Google Scholar] [CrossRef]
- Aliakbarzadeh, G.; Mahmoudi-Meymand, M.; Mazaheri, M. Verification of a standard method based on immunoaffinity column cleanup and HPLC-FLD analysis for determination of aflatoxins in peanut kernels. Food Control 2023, 152, 109820. [Google Scholar] [CrossRef]
- Wang, L.J.; Chen, Z.W.; Ma, T.Z.; Qing, J.; Liu, F.; Xu, Z.; Jiao, Y.; Luo, S.H.; Cheng, Y.H.; Ding, L. A novel magnetic metal–organic framework absorbent for rapid detection of aflatoxins B1, B2, G1 and G2 in rice by HPLC-MS/MS. Anal. Methods 2022, 14, 2522–2530. [Google Scholar] [CrossRef]
- Jayasinghe, G.D.T.M.; Domínguez-González, R.; Bermejo-Barrera, P.; Moreda-Piñeiro, A. Miniaturized vortex assisted-dispersive molecularly imprinted polymer micro-solid phase extraction and HPLC-MS/MS for assessing trace aflatoxins in cultured fish. Anal. Methods 2020, 12, 4351–4362. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Pan, A.; Zhang, C.; Guo, M.; Lou, C.; Zhang, J.; Wu, H.; Wang, X. Fast extraction of aflatoxins, ochratoxins and enniatins from maize with magnetic covalent organic framework prior to HPLC-MS/MS detection. Food Chem. 2023, 404, 134464. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Mehta, A. Rapid and sensitive detection of mycotoxins by advanced and emerging analytical methods: A review. Food Sci. Nutr. 2020, 8, 2183–2204. [Google Scholar] [CrossRef] [PubMed]
- Miklós, G.; Angeli, C.; Ambrus, Á.; Nagy, A.; Kardos, V.; Zentai, A.; Kerekes, K.; Farkas, Z.; Jóźwiak, Á.; Bartók, T. Detection of aflatoxins in different matrices and food-chain positions. Front. Microbiol. 2020, 11, 1916. [Google Scholar] [CrossRef]
- Center for Food Safety and Applied Nutrition Office of Regulatory Affairs. Compliance Policy Guide Sec 570.375 Aflatoxins in Peanuts and Peanut Products (FDA-2021-D-0244). 2021. Available online: https://www.fda.gov/media/72073/download (accessed on 10 February 2024).
- European Commision Regulation. Commision Regulation (EC) No 1881/2006 of 19 December 2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs; European Commission: Brussels, Belgium, 2006. [Google Scholar]
- Wu, J.; Ali, S.; Ouyang, Q.; Wang, L.; Rong, Y.; Chen, Q. Highly specific and sensitive detection of aflatoxin B1 in food based on upconversion nanoparticles-black phosphorus nanosheets aptasensor. Microchem. J. 2021, 171, 106847. [Google Scholar] [CrossRef]
- De Girolamo, A.; Lippolis, V.; Pascale, M. Overview of recent liquid chromatography mass spectrometry-based methods for natural toxins detection in food products. Toxins 2022, 14, 328. [Google Scholar] [CrossRef]
- Calleri, E.; Marrubini, G.; Brusotti, G.; Massolini, G.; Caccialanza, G. Development and integration of an immunoaffinity monolithic disk for the on-line solid-phase extraction and HPLC determination with fluorescence detection of aflatoxin B1 in aqueous solutions. J. Pharm. Biomed. Anal. 2007, 44, 396–403. [Google Scholar] [CrossRef]
- Wang, Y.; Chu, L.; Qu, J.; Ding, B.; Kang, X. A novel sample pretreatment of nanofiber-packed solid-phase extraction of aflatoxin B1, B2, G1 and G2 in foods and simultaneous determination with HPLC. Food Chem. 2024, 436, 137699. [Google Scholar] [CrossRef]
- Tang, Y.Y.; Lin, H.Y.; Chen, Y.C.; Su, W.T.; Wang, S.C.; Chiueh, L.C.; Shin, Y.C. Development of a quantitative multi-mycotoxin method in rice, maize, wheat and peanut using UPLC-MS/MS. Food Anal. Methods 2013, 6, 727–736. [Google Scholar] [CrossRef]
- Wu, Y.; Ye, J.; Xuan, Z.; Li, L.; Wang, H.; Wang, S.; Liu, H.; Wang, S. Development and validation of a rapid and efficient method for simultaneous determination of mycotoxins in coix seed using one-step extraction and UHPLC-HRMS. Food Addit. Contam. Part A 2021, 38, 148–159. [Google Scholar] [CrossRef]
- Wagner, A.B. Bacterial Food Poisoning. Texas A&M AgriLife 2008. Available online: https://aggie-horticulture.tamu.edu/food-technology/bacterial-food-poisoning/ (accessed on 14 February 2024).
- Aljamali, N.M.; Al Najim, M.M.; Alabbasy, A.J. Global Academic Journal of Pharmacy and Drug Research Review on Food poisoning (Types, Causes, Symptoms, Diagnosis, Treatment). Glob. Acad. J. Pharm. Drug Res. 2021, 3, 54–61. [Google Scholar] [CrossRef]
- Sospedra, I.; Marín, R.; Mañes, J.; Soriano, J.M. Rapid whole protein quantification of staphylococcal enterotoxin B by liquid chromatography. Food Chem. 2012, 133, 163–166. [Google Scholar] [CrossRef]
- Morya, S.; Amoah, A.E.D.D.; Snaebjornsson, S.O. Food poisoning hazards and their consequences over food safety. In Microorganisms for Sustainable Environment and Health; Elsevier: Amsterdam, The Netherlands, 2020; pp. 383–400. [Google Scholar]
- Liang, M.; Chen, R.; Xian, Y.; Hu, J.; Hou, X.; Wang, B.; Wu, Y.; Wang, L. Determination of bongkrekic acid and isobongkrekic acid in rice noodles by HPLC-Orbitrap HRMS technology using magnetic halloysite nanotubes. Food Chem. 2021, 344, 128682. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.; Kasper, A.; Steck, A.R.; Schier, J.G. Bongkrekic Acid—A Review of a Lesser-Known Mitochondrial Toxin. J. Med. Toxicol. 2017, 13, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Weagant, S.D.; Jinneman, K. Chapter 4A: Diarrheagenic Escherichia coli. 2020. Available online: https://www.fda.gov/food/laboratory-methods-food/bam-chapter-4a-diarrheagenic-escherichia-coli (accessed on 20 March 2024).
- Linsinger, T.P.J. Use of recovery and bias information in analytical chemistry and estimation of its uncertainty contribution. TrAC Trends Anal. Chem. 2008, 27, 916–923. [Google Scholar] [CrossRef]
- Wang, H.; Hu, L.; Chang, X.; Hu, Y.; Zhang, Y.; Zhou, P.; Cui, X. Determination of bacterial toxin toxoflavin and fervenulin in food and identification of their degradation products. Food Chem. 2023, 399, 134010. [Google Scholar] [CrossRef]
- Sospedra, I.; De Simone, C.; Soriano, J.M.; Mañes, J.; Ferranti, P.; Ritieni, A. Liquid chromatography-ultraviolet detection and quantification of heat-labile toxin produced by enterotoxigenic E. coli cultured under different conditions. Toxicon 2018, 141, 73–78. [Google Scholar] [CrossRef]
- Shah, H.N.; Saheer, E. Using nano-LC-MS/MS to investigate the toxicity of outbreak E. coli O104: H4 strain. Culture 2012, 33, 1–8. [Google Scholar]
- Nierzwicki-Bauer, S.A. Protocols for Cyanobacteria Sampling and Detection of Cyanotoxin. Thajuddin, N., Narayanan, A.S., Dhanasekaran, D., Eds.; 2023. Available online: https://www.researchgate.net/profile/Valentina-Messineo/publication/376604780_Extraction_and_Quantification_of_BMAA_from_Fish_Tissue/links/660d475bf5a5de0a9ff70d75/Extraction-and-Quantification-of-BMAA-from-Fish-Tissue.pdf (accessed on 21 May 2024).
- Sánchez-Parra, E.; Boutarfa, S.; Aboal, M. Are cyanotoxins the only toxic compound potentially present in microalgae supplements? Results from a study of ecological and non-ecological products. Toxins 2020, 12, 552. [Google Scholar] [CrossRef]
- Fontaine, J.; Duy, S.V.; Troncy, C.; Dinh, Q.T.; Simon, D.F.; Munoz, G.; Sauvé, S. Screening of multi-class cyanotoxins in algal dietary supplements marketed in North America. Algal Res. 2023, 73, 103162. [Google Scholar] [CrossRef]
- Mafra Jr, L.L.; de Souza, D.A.; Menezes, M.; Schramm, M.A.; Hoff, R. Marine biotoxins: Latest advances and challenges toward seafood safety, using Brazil as a case study. Curr. Opin. Food Sci. 2023, 53, 101078. [Google Scholar] [CrossRef]
- Akif, F.; Genten, F.; Ettoubi, E.; Blaghen, M. Marine Biotoxins: Origins, Effects, Distribution, Prevention and Treatment. Int. J. Innov. Sci. Res. Technol. 2020, 5, 928–941. [Google Scholar]
- Wang, Z.; Doucette, G.J. Determination of lipophilic marine biotoxins by liquid chromatography-tandem mass spectrometry in five shellfish species from Washington State, USA. J. Chromatogr. A 2021, 1639, 461902. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.; Zaczek-Moczydlowska, M.A.; Edwards, C.; Turner, A.D.; Miller, P.I.; Moore, H.; McKinney, A.; Lawton, L.; Campbell, K. Current trends and challenges for rapid smart diagnostics at point-of-site testing for marine toxins. Sensors 2021, 21, 2499. [Google Scholar] [CrossRef] [PubMed]
- Government of Canada. Health Canada’s Maximum Levels for Chemical Contaminants in Foods. 2016. Available online: https://www.canada.ca/en/health-canada/services/food-nutrition/food-safety/chemical-contaminants/maximum-levels-chemical-contaminants-foods.html (accessed on 11 February 2024).
- Food and Agriculture Organization. Regulations and Monitoring. Marine Biotoxins. 2004. Available online: https://openknowledge.fao.org/server/api/core/bitstreams/cdb8961c-8968-4b17-ae94-0aa735cbbc80/content (accessed on 2 February 2024).
- Marsan, D.W.; Conrad, S.M.; Stutts, W.L.; Parker, C.H.; Deeds, J.R. Evaluation of microcystin contamination in blue-green algal dietary supplements using a protein phosphatase inhibition-based test kit. Heliyon 2018, 4, e00573. [Google Scholar] [CrossRef] [PubMed]
- Cadaillon, A.M.; Mattera, B.; Albizzi, A.; Montoya, N.; Maldonado, S.; Raya Rey, A.; Riccialdelli, L.; Almandoz, G.O.; Schloss, I.R. Multispecies mass mortality in the Beagle Channel associated with paralytic shellfish toxins. Harmful Algae 2024, 132, 102581. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Li, J.; Kuang, J.; Shen, S.; Zhou, X.; Zhao, X.; Huang, B.; Han, B. Okadaic Acid Detection through a Rapid and Sensitive Amplified Luminescent Proximity Homogeneous Assay. Toxins 2023, 15, 501. [Google Scholar] [CrossRef]
- Aboualaalaa, H.; El kbiach, M.L.; Rijal Leblad, B.; Hervé, F.; Hormat-Allah, A.; Baudy, L.; Ennaskhi, I.; Hammi, I.; Ibghi, M.; Elmortaji, H.; et al. Development of harmful algal blooms species responsible for lipophilic and amnesic shellfish poisoning intoxications in southwestern Mediterranean coastal waters. Toxicon 2022, 219, 106916. [Google Scholar] [CrossRef]
- D’Amore, T.; Lo Magro, S.; Vita, V.; Di Taranto, A. Optimization and Validation of a High Throughput UHPLC-MS/MS Method for Determination of the EU Regulated Lipophilic Marine Toxins and Occurrence in Fresh and Processed Shellfish. Mar. Drugs 2022, 20, 173. [Google Scholar] [CrossRef]
- O’Neill, A.; Morrell, N.; Turner, A.D.; Maskrey, B.H. Method performance verification for the combined detection and quantitation of the marine neurotoxins cyclic imines and brevetoxin shellfish metabolites in mussels (Mytilus edulis) and oysters (Crassostrea gigas) by UHPLC-MS/MS. J. Chromatogr. B 2021, 1179, 122864. [Google Scholar] [CrossRef]
- Amzil, Z.; Derrien, A.; Terre Terrillon, A.; Savar, V.; Bertin, T.; Peyrat, M.; Duval, A.; Lhaute, K.; Arnich, N.; Hort, V.; et al. Five Years Monitoring the Emergence of Unregulated Toxins in Shellfish in France (EMERGTOX 2018–2022). Mar. Drugs 2023, 21, 435. [Google Scholar] [CrossRef]
- Merlo, F.; Maraschi, F.; Piparo, D.; Profumo, A.; Speltini, A. Simultaneous pre-concentration and HPLC-MS/MS quantification of phycotoxins and cyanotoxins in Inland and coastal waters. Int. J. Environ. Res. Public Health 2020, 17, 4782. [Google Scholar] [CrossRef] [PubMed]
- Ochi, N.; Suzuki, T. Determination of lipophilic marine biotoxins (azaspiracids, brevetoxins, and okadaic acid group) and domoic acid in mussels by solid-phase extraction and reversed-phase liquid chromatography with tandem mass spectrometry. J. Chromatogr. A 2024, 1720, 464795. [Google Scholar] [CrossRef] [PubMed]
- Quilliam, M. Rapid extraction and cleanup for liquid chromatographic determination of domoic acid in unsalted seafood. J. AOAC Int. 1995, 78, 543–545. [Google Scholar] [CrossRef]
- Sinno-Tellier, S.; Abadie, E.; Guillotin, S.; Bossée, A.; Nicolas, M.; Delcourt, N. Human shellfish poisoning: Implementation of a national surveillance program in France. Front. Mar. Sci. 2023, 9, 1089585. [Google Scholar] [CrossRef]

{kind=link}
| Fungal Toxins (Mycotoxins) | Sample Type and Preparation | HPLC-Based Technique and Chromatographic Conditions | Analytical Parameters | Permissible Limits |
|---|---|---|---|---|
| Aflatoxins B1, B2, G1, and G2 [50] | Peanut butter 12.5 g of locally made peanut butter samples were combined with 2.5 g of NaCl, and 62.5 mL of 70% methanol was blended and then filtered using a No. 1 Whatman filter. Subsequently, 30 mL of water was added to 15 mL of the filtrate. After this, 15 mL were passed through a solid-phase extraction cartridge and rinsed with 10 mL water. Aflatoxins were eluted with 1 mL of ethanol and then 1 mL of water | HPLC-UV Aflatoxins were separated with a C-18 column with a mobile phase of 40:50:10 methanol-water-acetonitrile solution. The UV detection was carried out at 365 nm, with a flow rate of 0.7 mL min−1. The standards for aflatoxins G2, G1, B2, and B1 had 1, 4, 1, and 4 μg mL−1 concentrations, respectively. | Not validated method Total aflatoxin content ranged between 373.6–6741.6 µg kg−1, and aflatoxin B1 content was 54.3–805.8 µg kg−1 [50] | Sum of B1, B2, G1, and G2 in peanuts: 15 μg kg−1 [47] Sum of B1, B2, G1, and G2 in processed peanuts: 4 μg kg−1 [47] |
| Fungal toxins (mycotoxins) | Sample type and preparation | HPLC-based Technique and chromatographic conditions | Analytical parameters | Permissible limits |
| Aflatoxins B1, B2, G1, and G2, and M1 [51] | Rice and flour 5 g of powdered sample was dispersed in 10 mL 70:30 methanol-water and sonicated for 20 min. The supernatant was completed to 45 mL with water. Milk 20 mL samples were centrifuged at 5000 rpm for 15 min, filtered with a 0.22 µm syringe filter, and diluted with 25 mL of deionized water. A MOF-based structure was selected as adsorbent in the extraction and pre-concentration of aflatoxins | HPLC-FD A C18 column (150 × 4.6 mm; 5 µm) was used at 40 °C. A sample size of 25 µL was injected, and the mobile phase consisted of 65% methanol and 35% water, with a flow rate of 1.2 mL min−1. | Validated method LOD: 0.009–0.015 ng mL−1 Linearity: 0.05–8 ng mL−1 | Sum of B1, B2, G1, and G2 for cereals: 4 μg kg−1 [47] Sum of B1, B2, G1, and G2 for mixture of spices: 10 μg kg−1 [47] Aflatoxin M1 in milk: 0.05 μg kg−1 [47] |
| Aflatoxins B1, B2, G1, and G2 [52] | Merkén spice Sample processing not specified. | HPLC-FD A HPLC-FLD with post-column derivation with a Kobra electrochemical cell (R-Biopharm Rhone, Glasgow, UK) was used. | Validated method LOD: 0.3 ng g−1 LOQ: 1.0 ng g−1 Linearity: 0.6–6.2 ng g−1 (B2 and G2), and 1.96–20 ng g−1 | |
| Aflatoxins B1, B2, M1, and M2 [53] | Milk 2.5 mL of sample were defatted by centrifugation at 4000 rpm for 10 min. The sample was then vortex-mixed for 3 min and passed through an in-syringe dispersive micro-solid phase extraction (ISDμSPE) as a sample pretreatment. | HPLC-FD A C18 column Hypersil gold (250 × 4.6 mm; 5 µm) was used at 40°C. The sample size was 25 µL, and the flow rate was 1.2 mL min−1. A gradient was applied using a mobile phase consisting of a mix of 13:74:13 methanol-water-acetonitrile for 6 min, followed by 2 min of a 20:60:20 mix. A 9-min hold followed this, then the initial conditions were restored for 2 min and held until 35 min. The fluorescence detector was set at 360 and 440 nm for excitation and emission wavelengths, respectively. | Validated method LOD: 0.003–0.005 ng mL−1 LOQ: 0.01–0.02 ng mL−1 Linearity: 0.01–1.0 ng mL−1 for Aflatoxins M1, M2, and B2, and 0.02–1.0 ng mL−1 for Aflatoxin B1 | |
| Aflatoxins M1 [54] | Raw milk Samples were filtrated with cellulose nitrate 0.45 µm membrane filters and centrifuged. The supernatant was filtered with 13 mm × 0.2 µm microfilters. | HPLC-FD A C18 column (250 × 4.6 mm; 5 µm) and immunoaffinity columns were used to isolate Aflatoxins M1. A fluorescence detector at 365 and 430 nm was used with a flow rate of 1 mL min−1 at room temperature. The mobile phases used were isocratic: (A) methanol, (B) acetonitrile, and (C) water. | Validated method LOD: 11.99 ng kg−1 and 16.95 ng kg−1 for aflatoxins M1 and M2, respectively. Linearity: 0.6–6.2 ng g−1 (B2 and G2), and 1.96–20 ng g−1 (B1, and G1) | |
| Aflatoxins B1, B2, G1, and G2. Ochratoxin A [55] | Wheat 50 g of sample were extracted with 100 mL of an 8:2 methanol-water mixture and then filtered. Subsequently, 10 mL of filtrate was diluted with 10 mL of phosphate buffer solution (PBS). | HPLC-FD For aflatoxins, an Inertsil ODS-3C C-18 column (250 × 4.6 mm; 5 μm) was utilized at 40 °C. The sample volume was 100 µL, with a flow rate of 1 mL min−1 and a mobile phase consisting of a 6:2:3 water-acetonitrile-acetic acid mix. Post-column derivatization involved using 350 μL of 4M nitric acid and 120 mg of potassium bromide for every 1000 mL mobile phase. Detection occurred via fluorescence at wavelengths 333 and 460 nm. For Ochratoxin A, a mobile phase comprising 47:51:2 acetonitrile-water-acetic acid was employed. | Validated method LOD: 0.014–0.030 μg kg–1 LOQ: 0.045–0.098 μg kg–1 | Sum of B1, B2, G1, and G2 for cereals: 4 μg kg−1 [47] Sum of B1, B2, G1, and G2 in maize: 10 μg kg−1 [47] All foods ready for human consumption: not exceed 10 μg kg−1 of aflatoxin, of which aflatoxin B1 shall not be more than 5 μg kg−1 [56] Ochratoxin A in unprocessed cereals: 5 μg kg−1 [47] |
| Aflatoxins B1, B2, G1, and G2. Ochratoxin A [40] | Processed meat 10 g of minced samples were mixed with 40 mL of a 60:40 acetonitrile-water mix and 0.2 g of NaCl. Then, 4 mL of the mixture were diluted in 44 mL of 2% tween-20-PBS. After that, 0.5 mL of the treated sample was mixed with 0.5 mL of acetonitrile for the cleanup process. | HPLC-FD 100 µL of extract was injected into a C-18 Thermo LC-Si column (250 × 4.6 mm) at 40 °C. The column had a fluorescence detector set at 365 and 435 nm. The mobile phase, consisting of toluene, ethyl acetate, formic acid, and methanol in a 90:5:2.5:2.5, was delivered at a flow rate of 2 mL min−1. | Validated method LOD for aflatoxin B1: 16.5–26.6 µg kg−1 LOD for ochratoxins: 3.8–17 µg kg−1 Linearity: 0.1–20 μg kg−1 | |
| Fungal toxins (mycotoxins) | Sample type and preparation | HPLC-based Technique and chromatographic conditions | Analytical parameters | Permissible limits |
| Aflatoxins B1, B2, G1, and G2 [46] | Wheat, corn, dried fig, or dried coffee beans 5 g of ground sample were mixed with 80% ethanol, filtered, and diluted in phosphate buffer solution (PBS). | HPLC-FD A sample of 50 mL was injected into AflacleanTM immunoaffinity columns at a flow rate of 0.5 mL min−1. The mobile phase consisted of a 60:30:10 water-methanol-acetonitrile solution. Detection was carried out on a UV-Vis at 365 nm. | Validated method LOD: 0.02–0.10 µg kg−1 LOQ: 0.04–0.45 µg kg−1 | Sum of B1, B2, G1, and G2 in corn and dried figs: 10 μg kg−1 [47] Sum of B1, B2, G1, and G2 in peanuts: 15 μg kg−1 [47] Sum of B1, B2, G1, and G2 for cereals: 4 μg kg−1 [47]; Sum of B1, B2, G1, and G2 for processed ground peanuts: 4 μg kg−1 [47] |
| Aflatoxins B1, B2, G1, and G2 [57] | Raw peanuts 25 g of peanuts were blended with 125 mL solution containing 70% methanol, 30% water, and 5.0 g of NaCl. The mixture was then filtered and passed through an Aflatest® IAC column. | HPLC-FD A 40 µL sample was injected into a silica column (LC-Si, 5 µm, 25 cm × 4.6 cm) heated to 30°C at a flow rate of 1.5 mL min−1, using a mobile phase consisting of a 1:1 mixture of solution A (toluene-ethyl acetate-methanol 45:3:2 mL) and solution B (toluene-ethyl acetate-formic acid 45:3:2 mL). | Validated method LOQ: 2.0, 0.5, 1.0, and 0.5 ng g−1 for aflatoxins B1, B2, G1, and G2, respectively | |
| Aflatoxins B1, B2, G1, and G2 [58] | Rice, flattened rice, sorghum, raw and processed peanut, almond, peanut butter, or wheat-based cookies 12.5 g of peanut samples + 12.5 g of water were mixed. 25 g of other samples were extracted with 80% methanol. | HPLC-FD IMMUNOPREP cleanup cartridges and a C18 column (150 × 4.6 mm; 5.0 µm) were utilized, along with a fluorescence detector set at 362 and 455 nm and a flow rate of 1.8 mL min-1. A gradient of solution A (24:52:21 acetonitrile-methanol-water) and solution B (80:20 methanol-water) was employed. The sample size was 1000 µL, and the process was conducted at 30 °C. | Validated method LOQ for peanut, sorghum, rice, and flattened rice: 0.125 ng g−1. LOQ for peanut butter, almond, and wheat-based cookies: 0.5 ng g−1. Linearity: 0.0125–10 ng g−1 | |
| Aflatoxins B1, B2, G1, and G2 [59] | Peanut kernel 50 g of ground sample were mixed with 5 g of NaCl, 200 mL of 80:20 ethanol-water, and 100 mL of n-hexane for 30 min. The mixture was then filtered, and 20 mL of the sample was diluted with 130 mL of deionized water. | HPLC-FD 100 µL of the sample was injected into a reverse-phase C18 column (25 cm × 4.6 mm; 5 μm) at 40°C. The mobile phase consisted of water-methanol-acetonitrile (6:3:2) with 120 µL of potassium bromide and 350 µL of 4 N nitric acid. The flow rate was set at 1 mL min−1 with FARLIB® used as the post-column derivatization cell. A fluorescence detector was employed at 352 nm and 435 nm. | Validated method LOD: 0.06–0.15 µg kg−1 LOQ: 0.20–0.50 µg kg−1 Linearity: 0.4–7.2, 0.08–1.44, 0.4–7.2, and 0.08–1.44 μg L−1 for aflatoxins B1, B2, G1, and G2, respectively | |
| Fungal toxins (mycotoxins) | Sample type and preparation | HPLC-based Technique and chromatographic conditions | Analytical parameters | Permissible limits |
| Aflatoxins B1, B2, G1, and G2 [60] | Rice Samples were treated with a magnetic covalent organic framework. | HPLC-MS and HPLC-MS/MS HPLC method not available. A G1315B diode array detector with 500 nL and 10 mm pathlength coupled to a single quadrupole mass spectrometer with an electrospray ionization source was used. An ESI mode was chosen for instrument performance. The capillary voltage was set at 3000 V, and the analyte ionization voltage was 150 eV. N2 was employed as nebulizing gas at 35 psig with a drying flow of 12 L min−1 at 350 °C. | Validated method LOD: 0.0188–0.1250 μg kg−1 LOQ: 0.037567–0.3750 μg kg−1 Linearity: 0.375–20 μg kg−1 | Sum of B1, B2, G1, and G2 in rice: 10 μg kg−1 [47] Sum of B1, B2, G1, and G2 for cereals: 4 μg kg−1 [47] All foods ready for human consumption should not exceed 10 μg of aflatoxin, of which aflatoxin B1 shall not be more than 5 μg kg−1 [56] Ochratoxin A in unprocessed cereals: 5 μg kg−1 [47] |
| Aflatoxins B1, B2, G1, and G2 [37] | Commercial rice 20 g of ground sample were spiked at 1.43 µg kg−1, and then 100 mL of an 80:20 methanol-water solution was added. The mixture was stirred at 950 rpm for 1 h. After sedimentation, the supernatant was centrifuged at 5000 rpm for 45 min. Next, 7 mL of the treated sample was diluted with 43 mL PBS and filtered through a syringe filter. | HPLC-MS and HPLC-MS/MS A C18 column was used (15 × 0.3 mm; 4 µm). The sample size was 10 µL, with a flow rate of 10 µL min−1. The column was coupled to a diode array and a single quadrupole mass spectrometer. The mobile phase comprised a mixture of formic acid 0.05% and acetonitrile. Diode array and mass spectrometer detectors (cHPLC-DAD/MS) Model 1100 Series, (Agilent Technologies, Madrid, Spain) were used. Also, a G1315B diode array detector (Agilent Technologies, Madrid, Spain) with 500 nL and 10 mm pathlength coupled to a single quadrupole mass spectrometer with an electrospray ionization source (Model 6120 Series, Agilent Technologies) were used. Positive ESI mode was used, the capillary voltage was set at 3000 V, and analyte ionization voltage at 150 eV. N2 was employed as nebulizing gas at 35 psig with a drying flow of 12 Lmin−1 at 350 °C. | Validated method LOD: 0.0967–0.3267 µg kg−1 LOQ: 0.3167–1.0667 µg kg−1 Linearity: 1–25 µg L−1 for all analytes | |
| Aflatoxins B1, B2, G1, and G2 [61] | Cultured fish Fish muscle and liver samples were extracted by an ultrasound-assisted extraction procedure using a 60:40 acetonitrile-0.1 M KH2PO4 aqueous buffer (pH 6.0) mixture. | HPLC-MS and HPLC-MS/MS Sample size: 20 μL, flow rate of 60 μL min−1, mobile phase: 0.1:99.9 formic acid-methanol. A 3200 Q TRAP LC/MS/MS (ABSciex, Concord, Canada) with an electrospray ionization source was used, ions spray voltage (IS), 5500 kV; ion source temperature of 300 °C; N2 as nebulizer gas and curtain gas, 40 psi; N2 as collision gas. | Validated method LOD: 0.2967–0.61 μg kg−1 [61] | |
| Aflatoxins B1, B2, G1, and G2. Ochratoxin A and B. Enniatins A, B, A1, and B1 [62] | Maize Samples were treated with a magnetic covalent organic framework. | HPLC-MS and HPLC-MS/MS A C18 column was used (100 × 3 mm; 1.8 µm) at 40°C. The sample size was 2 µL and detected through a triple-quadrupole mass spectrometer. Mobile phase: gradient mixture of Solvent A (100% acetonitrile) and B (0.1:99.9 formic acid-5 mmol L−1 ammonium formate), flow rate of 0.4 mL min−1. An Agilent 6460 triple-quadrupole mass spectrometer equipped with an electrospray ionization source was used. Mode, positive multiple reaction monitoring (MRM+); drying gas temperature of 300 °C; sheath gas temperature of 250 °C; drying gas flow rate of 5 L min−1; sheath gas flow rate, 10 L min−1; nebulizer pressure of 45 psi; capillary voltage of 3500 V; and nozzle voltage of 0 V. High-purity N2 was used as the drying gas. | Validated method LOD: 0.0267–1.67 µg kg−1 LOQ: 0.07–5.57 µg kg−1 Linearity: 0.05–20 μg kg−1 |
| Bacterial Toxins | Sample Type and Preparation | HPLC-Based Technique and Chromatographic Conditions | Analytical Parameters | Permissible Limits |
|---|---|---|---|---|
| Toxoflavin and fervenulin [81] | Rice bran oil, sweet potato starch, distiller’s yeast, Tremella fuciformis Berk, rice noodles, and fermented corn flour 1.0 g of rice bran oil was extracted twice with 6 mL of methanol. The mixture was then centrifuged at 10,000 rpm for 5 min. The other samples (were weighed and extracted in an ultrasonic bath for 10 min with 10 mL of extraction solvent. For Tremella fuciformis Berk and distiller’s yeast, methanol was used as the extraction solvent, while sweet potato starch, rice noodle, and fermented corn flour were extracted using 0.1:99.9 formic acid–water. These mixtures were then centrifuged at 10,000 rpm for 10 min. Subsequently, all samples were filtered and loaded into SPE cartridges. 6 different SPE cartridges and 6 solvents were tried, with the Oasis HLB having the best recovery, using methanol. | UHPLC-MS/MS A C18 column (150 × 2.1 mm; 5 μm) was used at 35 °C with a mobile phase consisting of 0.1% formic acid (v/v, solvent A) and methanol (solvent B). The flow rate was 0.4 mL/min. Gradient elution was programmed as follows: 0 min, 12% B; 1.00 min, 12% B; 2.50 min, 90% B; 5.00 min, 90% B; 5.01 min, 12% B; 10.00 min, 12% B. The injection volume was 5 μL. An Agilent 6460 triple quadrupole mass spectrometry (Yishun, Singapore) was used. The mass spectrometry was operated in the multiple reaction monitoring (MRM) mode with positive electrospray ionization (ESI+). Ion source conditions were as follows: N2 as drying gas, temperature of 350 °C; drying gas (N2) flow rate of 11 L min−1; nebulizer gas pressure, 3.4 × 105 Pa; capillary voltage, 4000 V. | Validated method LOD: 12–60 μg kg−1 LOQ: 40–200 μg kg−1 Linearity for toxoflavin: 20–1000 μg L−1 Linearity for fervenulin: 40–1000 μg L−1 | Not specified. |
| Bongkrekic and isobongkrekic acids [77] | Rice noodles A 2.0 g sample was mixed with 20 mL of acidic water (pH 4.4) and sonicated for 20 min. The sample solution was then filtered. Subsequently, 81 mg of magnetic nanotubes were added to the filtrate and vortex-mixed for 4.2 min. The magnetic nanotubes were isolated using a strong magnet, and the supernatant was discarded. The magnetic sorbent was eluted thrice with 0.5 mL of acetonitrile containing 1% formic acid. The eluate was collected and dried using a nitrogen stream at 40°C. The residue was diluted to 1.0 mL with a 50% acetonitrile aqueous solution and filtered using a 0.22 μm filter. | HPLC-Orbitrap HRMS technology with magnetic halloysite nanotubes A C18 column (150 × 2.1 mm; 2.6 μm) was used at 35 °C. The mobile phase was used in a gradient elution program consisting of a 0.1% formic acid water solution (A) and acetonitrile (B). The gradient elution program was as follows: 0–3 min, 50% B to 70% B; 3–3.6 min, 70% B to 95% B; 3.6–5.5 min, 95% B; 5.5–5.6 min, 95% B to 50% B; 5.6–7.0 min, 50% B. The flow rate was 0.3 mL min−1, and the injection volume was 5 μL. An HPLC-Orbitrap HRMS (Thermo Fisher Scientific, Germany) was used. Flow rates of sheath gas, auxiliary gas, and sweep gas were set at 45, 8, and 0 arbitrary units, respectively. The spray voltage was set at −3.0 kV. Mass range (m/z) was 100–600. The capillary and auxiliary gas heater temperatures were 320 °C and 350 °C, respectively. | Validated method LOD: 0.3 μg kg−1 LOQ: 1.0 μg kg−1 Linearity: 2–200 μg L−1 | Bonkrekik acid: fatal for humans at 1.0–1.5 mg orally [78] |
| Algal and Cyanobacterial Toxins (Cyanotoxins) | Sample Type and Preparation | HPLC-Based Technique and Chromatographic Conditions | Analytical Parameters | Permissible Limits |
|---|---|---|---|---|
| Cyanobacteria Microcystin-LR and anatoxin-A [85] | Food supplements containing microalgae The samples were extracted three times using 2.5 mL of a 75:25 methanol-water mixture at 60 °C. After extraction, the samples were dried in a Speedvac and reconstituted in 900 mL of methanol. Then, the reconstituted samples were transferred to 2 mL Eppendorf vials equipped with a cellulose-acetate filter (Corning Costar Spin-X centrifuge tube filters) and centrifuged for 5 min at 16,000× g. | HPLC-MS 20 mL of sample were loaded onto a C18 column (150 × 4.6 mm; 5 mm). The mobile phase consisted of water with 0.1% formic acid as eluent A and acetonitrile with 0.1% formic acid as eluent B. The elution program was as follows: 0–2 min at 30% B, a linear increase of B between 2 and 6 min, 6–12 min at 90% B, and a 5-min post-run at 30% B. The flow rate was maintained at 0.5 mL min−1, and the procedure was conducted at 40 °C. A hybrid mass spectrophotometer Agilent Q-TOF 6550, with an ionization source JetStream electrospray + i-Funnel. MS operated in the positive mode and N2 was used as the drying and collision gas. The quadrupole was operated in the unit mode and four spectra/s were recorded. | Not validated method Concentrations of toxins in samples from 0.002 ± 0.0001 µg g−1–0.034 ± 0.002 µg g−1 | Mycrocistin: 1 μg g−1 [93] |
| Cyanobacteria 27 cyanotoxins [86] | Spirulina, Aphanizomenon flos-aquae, Chlorella, and kelp algal dietary supplement samples The samples were extracted twice with 5 mL of methanol + 0.1 M formic acid. The mixture was vortexed for 30 s and then placed in an ultrasonic bath for 15 min. After that, the supernatant was collected following centrifugation at 6000 rpm for 10 min. The supernatants were then treated with 100 mg of graphitized carbon black for clean-up. The resulting mixture was filtered using PES (0.2 μm) and evaporated at 45 °C. Subsequently, the samples were homogenized in 3 mL of water for 5 min in an ultrasonic bath. An aliquot was filtered using PES (0.2 μm). Finally, an oxidation step was performed using 340 μL potassium permanganate 0.4 M, 380 μL sodium periodate 0.35 M, and potassium carbonate 1 M to adjust the pH to 9 [86] | HPLC-MS and HPLC-HRMS 1 mL of sample was applied to a C18 online SPE column (20 × 2.1 mm; 12 μm), and then transferred to a HypersilTM Gold C18 HPLC column (100 × 2.1 mm; 1.9 μm). For individual microcystins: UHPLC-HRMS, Thermo Q-Exactive Orbitrap was used. A heated electrospray ionization interface (HESI-II) operated in positive mode was used for analyte ionization. The ionization spray voltage was set at +3500 V; capillary temperature was set at 350 °C; the vaporizer temperature was set at 250 °C; sheath gas and auxiliary gas flow were set at 60 and 15 arbitrary units, respectively. For total microcystins: UHPLC-MS/MS, Thermo TSQ Quantiva was used. An SPE column Hypersyl Gold aQ C18 (20 × 2.1 mm; 12 μm) was used, and the analytical column Hypersil Gold C18 (50 mm × 2.1 mm, 1.9 μm particle size | Validated method LOD: 0.1–35 ng g−1 LOQ: 0.3–105 ng g−1 Linearity: 10–1000 ng g−1 | |
| Algae Paralytic shellfish toxins [94] | Stomach content and liver samples from dead kelp gulls, Magellanic penguins, Papua penguins, and imperial cormorants, along with zooplankton, squat lobsters, Fuegian sprat, and seabirds Samples from DGLTM ** were extracted with cold 0.04 M acetic acid and sonicated 1 min at 4 s intervals. Samples were pre-treated with C18 SPE columns activated with methanol. [94] | HPLC-UV A C18 column (25 cm × 4.6 mm; 5–10 µm), at 40 °C, rate flow of 1.0–1.5 mL min−1. The mobile phase was a mixture of 100:899:1 acetonitrile-water- trifluoroacetic acid. | Not validated method LOD: 400 μg Kg−1 LOQ: 3–8 pmol mL −1 | For paralytic shellfish toxins: 80 mg STX eq/100 g [92] |
| Algal and cyanobacterial toxins (cyanotoxins) | Sample type and preparation | HPLC-based Technique and chromatographic conditions | Analytical parameters | Permissible limits |
| Algae Okadaic acid [95] | Shellfish 0.9 mL of methanol was added to the samples, vortex-mixed, and sonicated. The resulting supernatants were centrifuged at 8000 rpm for 5 min. The extracts were then adjusted to a volume of 2 mL. Next, 2.5 M sodium hydroxide was added, and the mixture was incubated at 70 °C for 1 h. After cooling, an equivalent amount of hydrochloric acid was added, and the solution was filtered using a 0.22 µm filter. Finally, 6 mL of methanol and 6 mL of water were added. | HPLC-UV A C18 column with a UV-vis detector was used at 200 nm, with a flow rate of 2 mL min-1, an injection volume of 20 µL, and a mobile phase 35:65 (water-acetonitrile). | Validated method LOD: 4.55 × 10−3 ng mL−1 [95] | Okadaic acid, dinophysis toxins, and pecteno-toxins together: 160 mg OA equivalents kg−1 [92] |
| Algae Domoic acid [96] | Mollusks 4 g of shredded and homogenized samples were mixed with 16 mL of solvent extraction (1:1 methanol-water solution), homogenized for 3 min at 10,000 rpm, then centrifuged for 10 min at 4000 rpm. | HPLC-UV A C18 column (250 × 4.6 mm; 5 μm) was used with a flow rate of 1 mL min−1, a detection wavelength of 242 nm, an injection volume of 20 μL, and an oven temperature for the column of 40 °C. | Validated method LOD: 0.05 mg kg−1 | Domoic acid: 20 µg g−1 of tissue [91] |
| Algae Okadaic acid, yessotoxin, pectenotoxin, and azaspiracid [97] | Processed food Deionized water was added (30% of the weighted sample), and 5 mL of the sample was diluted with 5 mL of ultrapure water. | HPLC-MS and UHPLC-MS The sample was loaded into an SPE cartridge and eluted with 2 mL of methanol and 3% ammonium hydroxide. Later, it was passed through a C18 column (100 × 2.1 mm; 1.7 µm) with detection by MS/MS at 40 °C. The injection volume was 5 µL with a flow rate of 0.2 mL min−1. Different gradients of solution A (water) and B (acetonitrile), both with 0.04% v/v of ammonium hydroxide, were used, starting at B 20%, then B 85%, B 98%, and finally B 20%. A triple quadrupole mass spectrometer TSQ-Endura (Thermo Fisher Scientific, Waltham, MA, USA), equipped with a heated electrospray source (H-ESI II) operating in both positive (ESI+) and negative mode (ESI−). N2 was used as sheath and auxiliary gas, while Argon was used as collision gas. The optimized parameters were: capillary voltage (3500 V in ESI+ and 2700 V in ESI−), sheath gas flow rate (30 arbitrary units), auxiliary gas flow rate (10 arbitrary units), ion transfer tube temperature (270 °C), vaporizer temperature (240 °C) and collision gas pressure (2.5 mTorr). | Validated method LOQ: 3–8 µg kg−1 | Okadaic acid, dinophysis toxins and pecteno-toxins together: 160 mg Okadaic Acid (OA) equivalents kg−1 [92] Yessotoxins: 1 mg YTX equivalents kg−1 [92] Azaspiracid: 160 µg kg−1 [92] |
| Algal and cyanobacterial toxins (cyanotoxins) | Sample type and preparation | HPLC-based Technique and chromatographic conditions | Analytical parameters | Permissible limits |
| Algae Cyclic imine analogues spirolide, gymnodimine, and pinnatoxin groups [98] | Mussel and oysters 1 g samples were extracted using 9 mL of methanol, vortex-mixed for 3 min, and centrifuged at 3400× g for 8 min at 20 °C. The resulting supernatants were then filtered through a 0.2 µm nylon filter. | HPLC-MS and UHPLC-MS A C18 column (50 × 2.1 mm; 1.7 µm) was used at 30 °C with an injection volume of 2 µL and flow rates of 0.4–0.6 mL min−1. Various mobile phases were used, and 1 mM ammonium fluoride in methanol had the best signal intensity. A Xevo-TQ-S triple quadrupole mass spectrometer (MS/MS) with electrospray ionization was used, with a 150 °C source temperature, 600 °C desolvation temperature, 1100 L/Hr desolvation gas flow, 150 L/Hr cone gas flow and a 1.0 kV capillary voltage. | Validated method LOD: 0.01–16.5 µg kg−1 LOQ: 0.03–55 µg kg−1 Linearity: 0.1–80 µg kg−1, 0.1–40 µg kg−1, 0.4–40 µg kg−1, 0.1–20 µg kg−1, and 1–320 µg kg−1 for PnTx-E and PnTx-F, PnTx-G and 20-Me-SPX-G, GYM-A and 13-desMe-SPX-C, 13,19-didesMe-SPX C, and 12-Me-GYM toxins, respectively | Not regulated [99] |
| Algae: Phycotoxins and cyanotoxins [100] | Fresh and salt waters: 500 mL samples were acidified to pH 3 with formic acid. | HPLC-MS and UHPLC-MS: Samples were loaded onto an SPE cartridge at a flow rate of 5 mL min−1. The analytes were eluted using methanol with 1% formic acid. Subsequently, a C18 column (150 × 2.1 mm; 3.5 µm) preceding a pre-column C18, both maintained at 30 °C, was employed. A gradient was performed using solvent A (water) and solvent B (acetonitrile), containing 0.5% formic acid. The gradient involved a change from 95% A to 2% A with corresponding changes in B, at a flow rate of 0.2 mL min−1 and an injection volume of 10 µL. | LOD: 0.3–29 ng L−1 LOQ: 1–88 ng L−1 Linearity: 2.5–250 µg L−1 | For phycotoxins (domoic and okadaic acids): Okadaic acid, dinophysis toxins and pecteno-toxins together: 160 mg OA equivalents kg−1 [92] Domoic acid: 20 µg g−1 of tissue [91] For cyanotoxins (microcystin): 1 μg g−1 [93] |
| Algae: Azaspiracids, brevetoxins, okadaic acid group, and domoic acid [101] | Mussels: Homogenized samples were extracted with different acetic acid and methanol concentrations. Then, the extracts were loaded in C18 SPE cartridges. | HPLC-MS and UHPLC-MS: An ODS-3 C18 column (150 × 2.1 mm; 3 µm) was used at 40 °C, with a 2 µL injection volume and a flow rate of 0.2 mL min−1. The mobile phase comprised 0.1% aqueous formic acid (A) and acetonitrile (B). A linear gradient of 5–60% B, 60–70% B, and 90% B was used to separate lipophilic biotoxins. A 10% solution of B was utilized to analyze domoic acid. | LOD: 0.002–0.017 mg kg−1 LOQ: 0.007–0.058 mg kg−1 Detection range: 0.008–0.2 mg Kg−1 to 0.06–6 mg kg−1 | For azaspiracid: 160 µg kg−1 [92] Brevetoxins: not regulated [99] Okadaic acid, dinophysis toxins and pecteno-toxins together: 160 mg OA equivalents kg−1 [92] Domoic acid: 20 µg g−1 of tissue [91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quintanilla-Villanueva, G.E.; Sánchez-Álvarez, A.; Núñez-Salas, R.E.; Rodríguez-Delgado, M.M.; Luna-Moreno, D.; Villarreal-Chiu, J.F. Recent Advances in Monitoring Microbial Toxins in Food Samples by HPLC-Based Techniques: A Review. Analytica 2024, 5, 512-537. https://doi.org/10.3390/analytica5040035
Quintanilla-Villanueva GE, Sánchez-Álvarez A, Núñez-Salas RE, Rodríguez-Delgado MM, Luna-Moreno D, Villarreal-Chiu JF. Recent Advances in Monitoring Microbial Toxins in Food Samples by HPLC-Based Techniques: A Review. Analytica. 2024; 5(4):512-537. https://doi.org/10.3390/analytica5040035
Chicago/Turabian StyleQuintanilla-Villanueva, Gabriela Elizabeth, Araceli Sánchez-Álvarez, Raisa Estefanía Núñez-Salas, Melissa Marlene Rodríguez-Delgado, Donato Luna-Moreno, and Juan Francisco Villarreal-Chiu. 2024. "Recent Advances in Monitoring Microbial Toxins in Food Samples by HPLC-Based Techniques: A Review" Analytica 5, no. 4: 512-537. https://doi.org/10.3390/analytica5040035
APA StyleQuintanilla-Villanueva, G. E., Sánchez-Álvarez, A., Núñez-Salas, R. E., Rodríguez-Delgado, M. M., Luna-Moreno, D., & Villarreal-Chiu, J. F. (2024). Recent Advances in Monitoring Microbial Toxins in Food Samples by HPLC-Based Techniques: A Review. Analytica, 5(4), 512-537. https://doi.org/10.3390/analytica5040035