
Methodology for Determining Phthalate Residues by Ultrasound–Vortex-Assisted Dispersive Liquid–Liquid Microextraction and GC-IT/MS in Hot Drink Samples by Vending Machines

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Extraction Procedure, Dispersive Liquid–Liquid Microextraction (DLLME)
2.3. Real Sample Analysis
2.4. GC Analysis
2.4.1. GC-FID Analysis
2.4.2. GC-IT/MS Analysis
3. Results and Discussion
3.1. Optimization of PAE Extraction Conditions
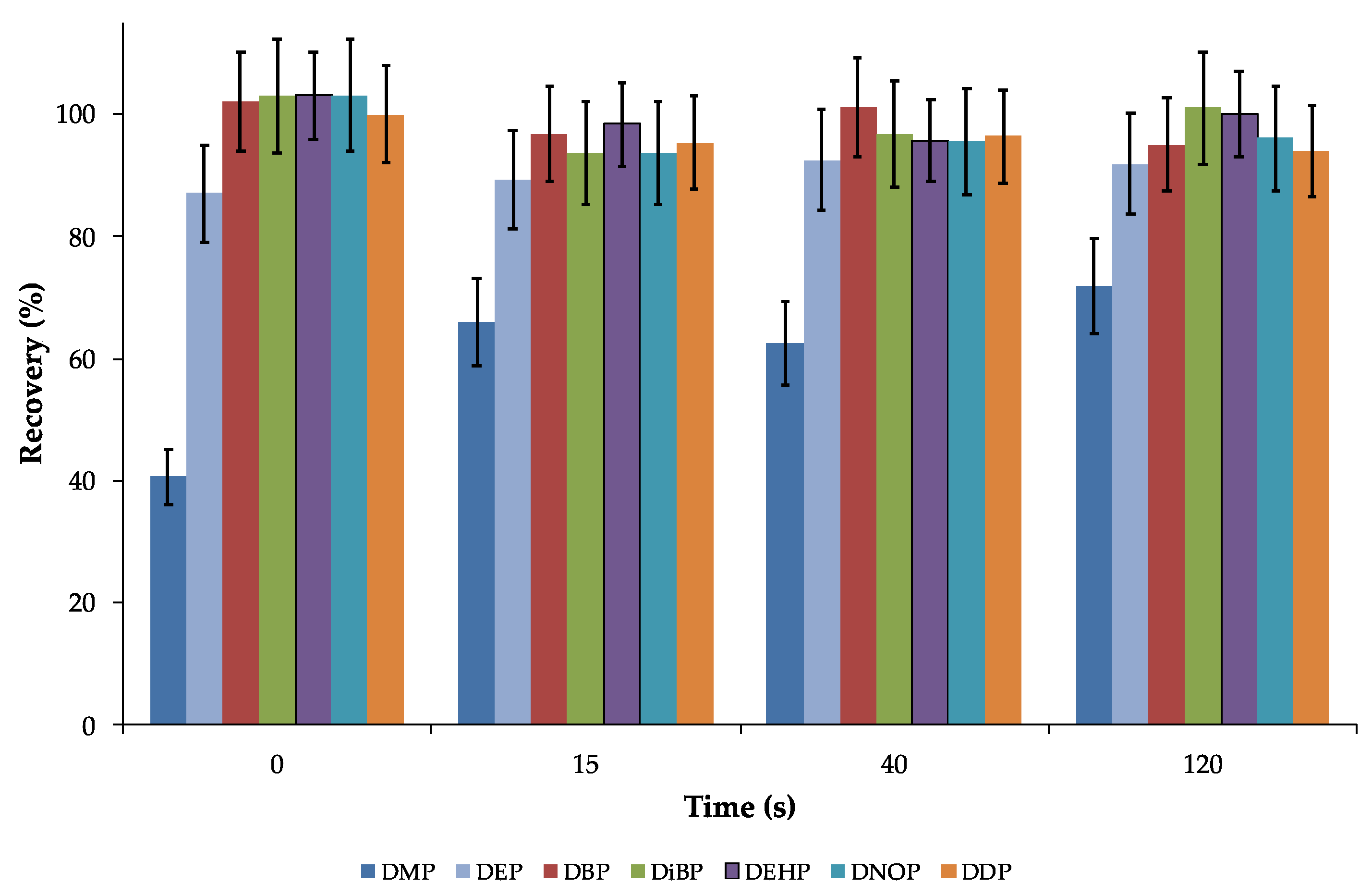
3.2. Influence of Time and Temperature on the PAE Extraction from Hot Drinks
3.3. Matrix Effect
3.4. Analytical Parameters by GC-IT/MS
3.5. LODs and LOQs: A Comparison among Different Procedures
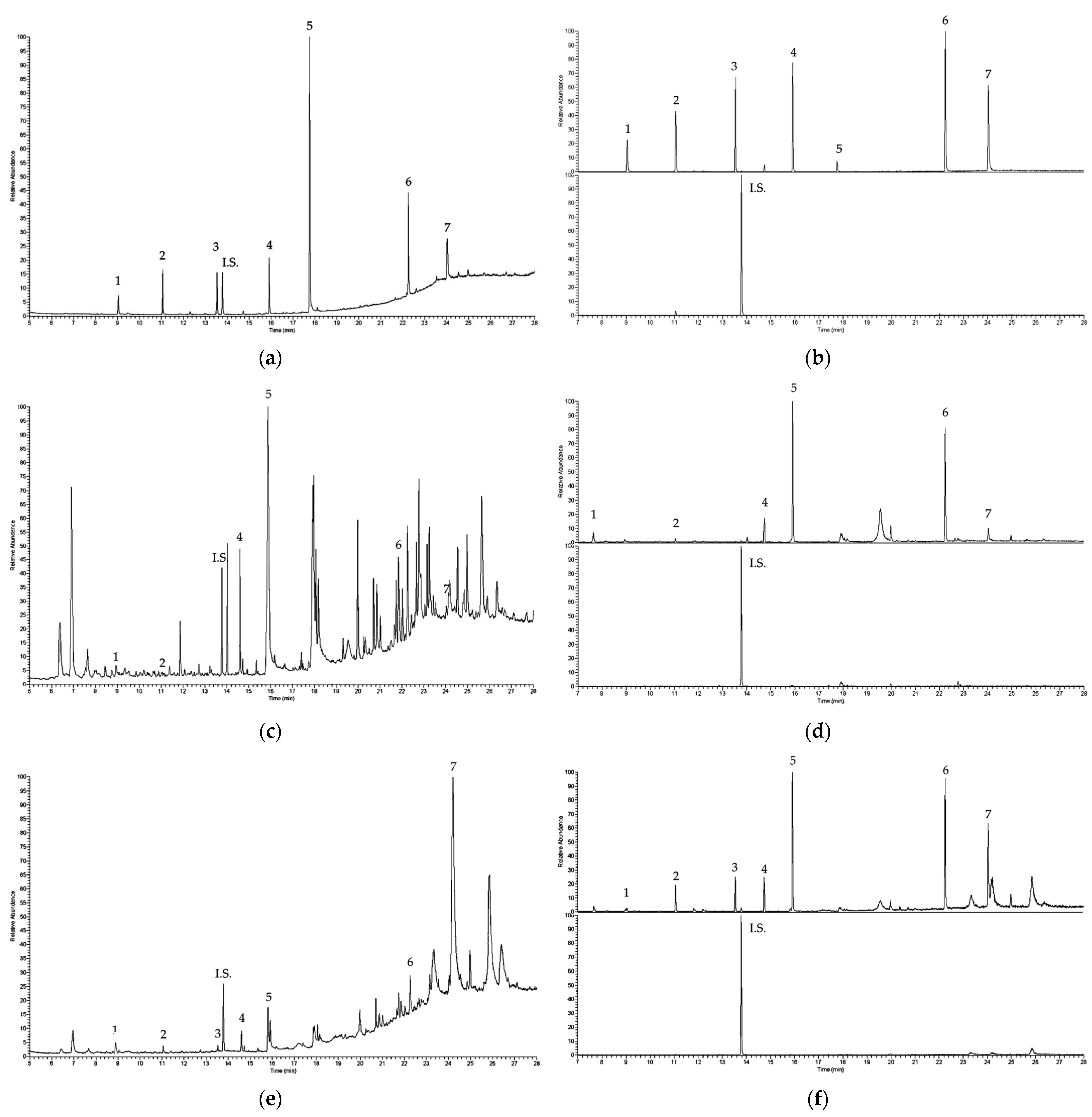
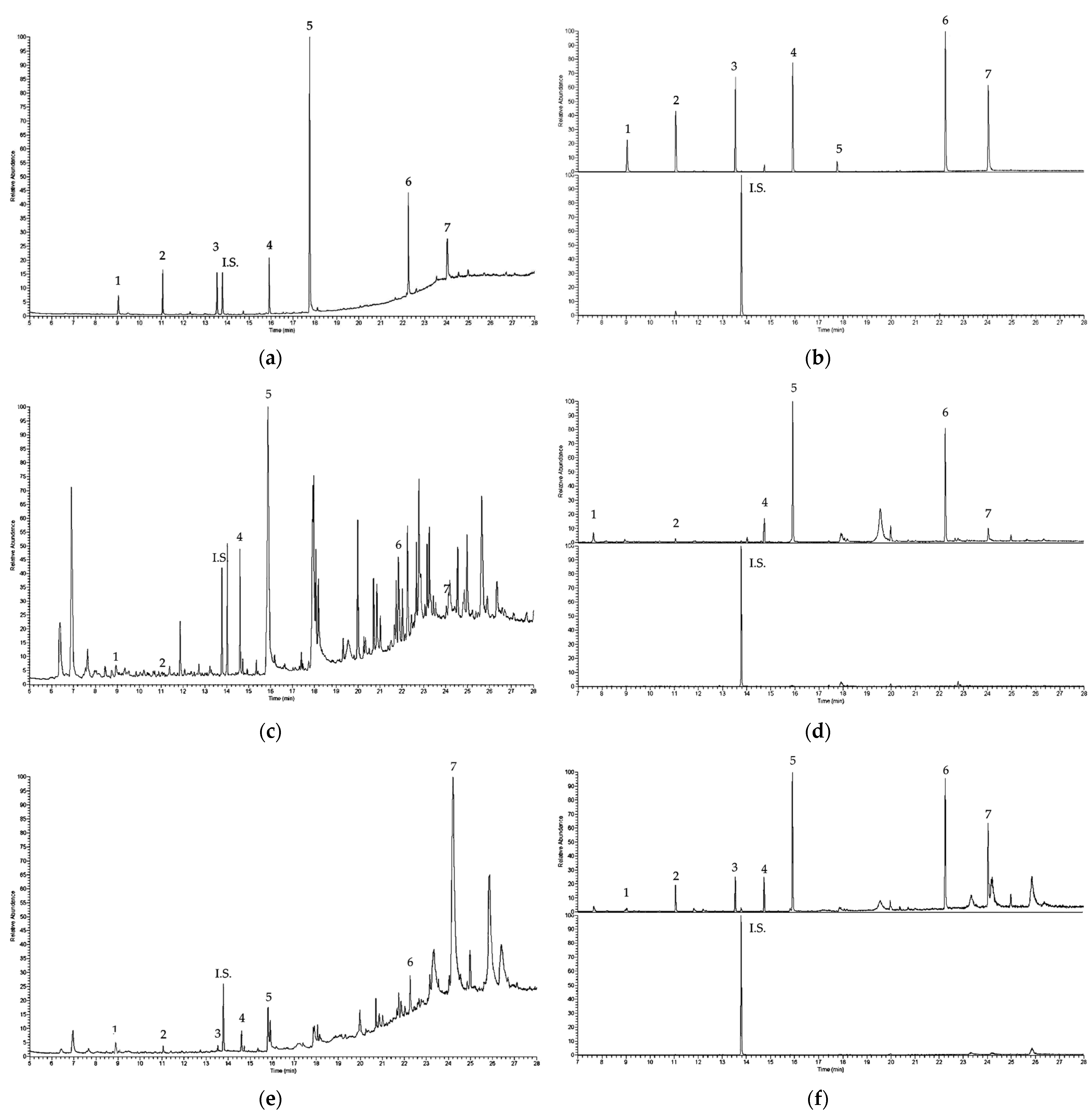
3.6. Application to Real Samples
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martinez-Perez, N.; Arroyo-Izaga, M. Availability, nutritional profile and processing level of food products sold in vending machines in a Spanish public university. Int. J. Environ. Res. Public Health 2021, 18, 6842. [Google Scholar] [CrossRef] [PubMed]
- Moretti, C.; Hamelin, L.; Jakobsen, L.G.; Junginger, M.H.; Steingrimsdottir, M.M.; Høibye, L.; Shen, L. Cradle-to-grave life cycle assessment of single-use cups made from PLA, PP and PET. Resour. Conserv. Recycl. 2021, 169, 105508. [Google Scholar] [CrossRef]
- Fasano, E.; Cirillo, T. Plasticizers and bisphenol as food contaminants: Sources and human risk. Curr. Anal. Chem. 2018, 14, 296–305. [Google Scholar] [CrossRef]
- Wang, W.-R.; Chen, N.-T.; Hsu, N.-Y.; Kuo, I.-Y.; Chang, H.-W.; Wang, J.-Y.; Su, H.-J. Associations among phthalate exposure, DNA methylation of TSLP, and childhood allergy. Clin. Epigen. 2021, 13, 76. [Google Scholar] [CrossRef]
- Prieto-Amador, M.; Caballero, P.; Martínez-Guitarte, J.-L. Analysis of the impact of three phthalates on the freshwater gastropod physella acuta at the transcriptional level. Sci. Rep. 2021, 11, 11411. [Google Scholar] [CrossRef]
- Chang, W.-H.; Herianto, S.; Lee, C.-C.; Hung, H.; Chen, H.-L. The effects of phthalate ester exposure on human health: A review. Sci. Total Environ. 2021, 786, 147371. [Google Scholar] [CrossRef]
- Notardonato, I.; Passarella, S.; Ianiri, G.; Di Fiore, C.; Russo, M.V.; Avino, P. Analytical method development and chemometric approach for evidencing presence of plasticizer residues in nectar honey samples. Int. J. Environ. Res. Public Health 2020, 17, 1692. [Google Scholar] [CrossRef] [Green Version]
- Weizhen, Z.; Xiaowei, Z.; Peng, G.; Ning, W.; Zini, L.; Jian, H.; Zheng, Z. Distribution and risk assessment of phthalates in water and sediment of the pearl river delta. Environ. Sci. Pollut. Res. Int. 2020, 27, 12550–12565. [Google Scholar] [CrossRef]
- Fan, J.-C.; Ren, R.; He, H.-L.; Jin, Q.; Wang, S.-T. Determination of phthalate esters in breast milk before and after frozen storage in milk storage bags. Food Addit. Contam. Part A 2020, 37, 1897–1905. [Google Scholar] [CrossRef]
- Jarošová, A.; Bogdanovičová, S. Phthalates in meat products in dependence on the fat content. Potravinarstvo 2016, 10, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.-M.; Li, C.-Y.; Yang, F.-E.; Zhao, N.; Lv, S.-W.; Liu, J.-C.; Chen, L.-J.; He, Z.; Zhang, Y.; Wang, S. Assessment of migration regularity of phthalates from food packaging materials. Food Sci. Nutr. 2020, 8, 5738–5747. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, W.; Lv, J.; Liu, W.; Sun, S.; Guo, C.; Xu, J. Distribution, source apportionment, and health risk assessment of phthalate esters in indoor dust samples across China. Environ. Sci. Eur. 2021, 33, 19. [Google Scholar] [CrossRef]
- Gkrillas, A.; Dirven, H.; Papadopoulou, E.; Andreassen, M.; Husøy, T. Exposure estimates of phthalates and DINCH from foods and personal care products in comparison with biomonitoring data in 24-hour urine from the Norwegian EuroMix biomonitoring study. Environ. Int. 2021, 155, 106598. [Google Scholar] [CrossRef]
- Dobaradaran, S.; Akhbarizadeh, R.; Mohammadi, M.J.; Izadi, A.; Keshtkar, M.; Tangestani, M.; Moazzen, M.; Shariatifar, N.; Mahmoodi, M. Determination of phthalates in bottled milk by a modified nano adsorbent: Presence, effects of fat and storage time, and implications for human health. Microchem. J. 2020, 159, 105516. [Google Scholar] [CrossRef]
- Sakhi, A.K.; Lillegaard, I.T.L.; Voorspoels, S.; Carlsen, M.H.; Løken, E.B.; Brantsæter, A.L.; Haugen, M.; Meltzer, H.M.; Thomsen, C. Concentrations of phthalates and bisphenol A in Norwegian foods and beverages and estimated dietary exposure in adults. Environ. Int. 2014, 73, 259–269. [Google Scholar] [CrossRef] [PubMed]
- The European Commission. Commission Regulation (EU) No 10/2011 of 14 January 2011 on plastic materials and articles intended to come into contact with food. Off. J. Eur. Union 2011, 54, L 12/1–L 12/89. [Google Scholar]
- Giuliani, A.; Zuccarini, M.; Cichelli, A.; Khan, H.; Reale, M. Critical review on the presence of phthalates in food and evidence of their biological impact. Int. J. Environ. Res. Public Health 2020, 17, 5655. [Google Scholar] [CrossRef]
- Rezaee, M.; Yamini, Y.; Faraji, M. Evolution of dispersive liquid–liquid microextraction method. J. Chromatogr. A 2010, 1217, 2342–2357. [Google Scholar] [CrossRef]
- Hongyuan, Y.; Hui, W. Recent development and applications of dispersive liquid-liquid microextraction. J. Chromatogr. A 2013, 1295, 1–15. [Google Scholar]
- Russo, M.V.; Avino, P.; Perugini, L.; Notardonato, I. Extraction and GC-MS analysis of phthalate esters in food matrices: A review. RSC Adv. 2015, 5, 37023–37043. [Google Scholar] [CrossRef]
- Wypych, A. (Ed.) Phthalates. In Databook of Plasticizers, 2nd ed.; ChemTec Publishing: Toronto, ON, Canada, 2017; pp. 435–560. [Google Scholar]
- Russo, M.V.; Notardonato, I.; Avino, P.; Cinelli, G. Fast determination of phthalate ester residues in soft drinks and light alcoholic beverages by ultrasound/vortex assisted dispersive liquid-liquid microextraction followed by gas chromatography-ion trap mass spectrometry. RSC Adv. 2014, 4, 59655–59663. [Google Scholar] [CrossRef]
- Avino, P.; Notardonato, I.; Perugini, L.; Russo, M.V. New protocol based on high-volume sampling followed by DLLME-GC-IT/MS for determining PAHs at ultra-trace levels in surface water samples. Microchem. J. 2017, 133, 251–257. [Google Scholar] [CrossRef]
- Rosada, A.; Cardone, F.; Avino, P. The astonishing 63Ni radioactivity reduction in radioactive wastes by means of ultrasounds application. SN Appl. Sci. 2019, 1, 1319. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.V.; Notardonato, I.; Avino, P.; Cinelli, G. Determination of phthalate esters at trace levels in light alcoholic drinks and soft drinks by XAD-2 adsorbent and gas chromatography coupled with ion trap-mass spectrometry detection. Anal. Methods 2014, 6, 7030–7037. [Google Scholar] [CrossRef]
- Knoll, J.K. Estimation of the limit of detection in chromatography. J. Chromatogr. Sci. 1985, 23, 422–425. [Google Scholar] [CrossRef]
- Notardonato, I.; Passarella, S.; Iannone, A.; Fiore, C.D.; Russo, M.V.; Protano, C.; Vitali, M.; Avino, P. Comparison of two extraction procedures, SPE and DLLME, for determining plasticizer residues in hot drinks at vending machines. Processes 2021, 9, 1588. [Google Scholar] [CrossRef]
- La Pera, L.; Avellone, G.; Lo Turco, V.; Di Bella, G.; Agozzino, P.; Dugo, G. Influence of roasting and different brewing processes on the ochratoxin A content in coffee determined by high-performance liquid chromatography-fluorescence detection (HPLC-FLD). Food Addit. Contam. A 2008, 25, 1257–1263. [Google Scholar] [CrossRef] [Green Version]
- Pacetti, D.; Boselli, E.; Balzano, M.; Frega, N.G. Authentication of Italian Espresso coffee blends through the GC peak ratio between kahweol and 16-O-methylcafestol. Food Chem. 2012, 135, 1569–1574. [Google Scholar] [CrossRef]
- Caporaso, N.; Genovese, A.; Canela, M.D.; Civitella, A. Neapolitan coffee brew chemical analysis in comparison to espresso, moka and American brews. Food Res. Int. 2014, 61, 152–160. [Google Scholar] [CrossRef]
- Wu, P.-G.; Pan, X.-D.; Ma, B.-J.; Wang, L.-Y.; Zhang, J. Determination of phthalate esters in non-alcoholic beverages by GC–MS and optimization of the extraction conditions. Eur. Food Res. Technol. 2014, 238, 607–612. [Google Scholar] [CrossRef]
- Liu, P.; Chen, H.; Gao, G.; Hao, Z.; Wang, C.; Ma, G.; Chai, Y.; Zhang, L.; Liu, X. Occurrence and residue pattern of phthalate esters in fresh tea leaves and during tea manufacturing and brewing. J. Agric. Food Chem. 2016, 64, 8909–8917. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Rai, S.; Srivastava, A.K.; Panchal, S.; Patel, D.K.; Sharma, V.P.; Jain, S.; Srivastava, L.P. Determination of pesticide and phthalate residues in tea by QuEChERS method and their fate in processing. Environ. Sci. Pollut. Res. 2017, 24, 3074–3083. [Google Scholar] [CrossRef] [PubMed]
- Tashakkori, P.; Erdem, P.; Merdivan, M.; Bozkurt, S.S. Determination of phthalate esters in water and coffee by solid-phase microextraction using vinyl terminated imidazolium based ionic liquid grafted on graphene oxide coatings. ChemistrySelect 2019, 4, 2307–2313. [Google Scholar] [CrossRef]
- Yin, S.; Yang, Y.; Yang, D.; Li, Y.; Jiang, Y.; Wu, L.; Sun, C. Determination of 11 phthalate esters in beverages by magnetic solid-phase extraction combined with high-performance liquid chromatography. J. AOAC Int. 2019, 102, 1624–1631. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, J.R.; Melough, M.M.; Provatas, A.A.; Perkins, C.; Chun, O.K. Evaluation of estrogenic chemicals in capsule and French press coffee using ultra-performance liquid chromatography with tandem mass spectrometry. Toxicol. Rep. 2020, 7, 1020–1024. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Wang, J.-L.; Shu, Y.-Y. Purge-assisted and temperature-controlled headspace solid-phase microextraction combined with gas chromatography–mass spectrometry for determination of six common phthalate esters in aqueous samples. J. Food Meas. Charact. 2020, 14, 1833–1841. [Google Scholar] [CrossRef]
- Song, N.-E.; Lim, M.-C.; Choi, S.-W.; Kim, D.-O.; Nam, T.G. Magnetic solid-phase extraction based on magnetic carbon particles from coffee grounds for determining phthalic acid esters in plastic bottled water. J Food Sci. 2020, 85, 1098–1104. [Google Scholar] [CrossRef]
- Santana-Mayor, Á.; Socas-Rodríguez, B.; Rodríguez-Ramos, R.; Rodríguez-Delgado, M.Á. A Green and simple procedure based on deep eutectic solvents for the extraction of phthalates from beverages. Food Chem. 2020, 312, 125798. [Google Scholar] [CrossRef]
- Ortega-Zamora, C.; Jiménez-Skrzypek, G.; González-Sálamo, J.; Hernández-Borges, J. Extraction of phthalic acid esters from soft drinks and infusions by dispersive liquid-liquid microextraction based on the solidification of the floating organic drop using a menthol-based natural deep eutectic solvent. J. Chromatogr. A 2021, 1646, 462132. [Google Scholar] [CrossRef]
- Santana-Mayor, À.; Herrera-Herrera, A.V.; Rodríguez-Ramos, R.; Socas-Rodríguez, B.; Rodríguez-Delgado, M.A. Development of a green alternative vortex-assisted dispersive liquid−liquid microextraction based on natural hydrophobic deep eutectic solvents for the analysis of phthalate esters in soft drinks. ACS Sustain. Chem. Eng. 2021, 9, 2161–2170. [Google Scholar] [CrossRef]
- Conley, J.M.; Lambright, C.S.; Evans, N.; Cardon, M.; Medlock-Kakaley, E.; Wilson, V.S.; Gray, L.E., Jr. A mixture of 15 phthalates and pesticides below individual chemical no observed adverse effect levels (NOAELs) produces reproductive tract malformations in the male rat. Environ. Inter. 2021, 156, 106615. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Hirosawa, N.; Sakamoto, Y.; Katayama, H.; Moriguchi, T.; Joung, K.E.; Sheen, Y.Y.; Asaoka, K. Phthalate levels in beverages in Japan and Korea. Bull. Environ. Contam. Toxicol. 2002, 68, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Fierens, T.; Servaes, K.; Van Holderbeke, M.; Geerts, L.; De Henauw, S.; Sioen, I.; Vanermen, G. Analysis of phthalates in food products and packaging materials sold on the Belgian market. Food Chem Toxicol. 2012, 50, 2575–2583. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| PAE | Symbol | CAS # | MW | DL50 1 (g kg−1) | ADI 2 (ng kg−1) | Solubility 3 (mg L−1) | Kow 4 (log kow) | Boiling Point 5 (°C) |
|---|---|---|---|---|---|---|---|---|
Dimethyl phthalate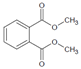 | DMP | 131-11-3 | 194.18 | 8–10 | 79.1 | 4000 | 1.6 | 283.7 |
Diethyl phthalate | DEP | 84-66-2 | 222.24 | 8–10 | 1.4-28.2 | 1080 | 2.47 | 295.0 |
Diisobutyl phthalate | DiBP | 84-69-5 | 278.34 | 8–10 | 105 | 6.2 | 4.11 | 327.0 |
Dibutyl phthalate | DBP | 84-74-2 | 278.35 | 8–10 | 191.8 | 11.2 | 4.5 | 340.0 |
Di-(2-ethylhexyl) phthalate | DEHP | 117-81-7 | 390.56 | 14 | 1458 | 0.27 | 7.6 | 386.9 |
Di-n-octyl phthalate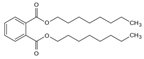 | DNOP | 117-84-0 | 390.56 | 13 | 37 × 106 | 0.022 | 8.1 | 384.0 |
Di-n-decyl phthalate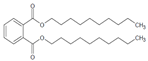 | DDP | 84-77-5 | 446.7 | 17 | N/A | 0.00022 | 9.05 | 268.0 6 |
| PAE 1 | Recovery (%) | ||||
|---|---|---|---|---|---|
| n-Heptane | iso-Octane | Benzene | Xylene | Cyclohexane | |
| DMP | 59.6 ± 5.8 | 33.5 ± 10.1 | 40.5 ± 8.1 | 42.8 ± 6.9 | 11.9 ± 6.2 |
| DEP | 66.7 ± 4.1 | 38.6 ± 7.6 | 60.3 ± 6.6 | 50.3 ± 2.5 | 39.9 ± 2.6 |
| DiBP | 108.9 ± 6.1 | 92.4 ± 4.5 | 109.4 ± 3.1 | 107.4 ± 6.1 | 62.3 ± 3.9 |
| DBP | 104.2 ± 2.6 | 90.4 ± 6.7 | 102.1± 7.7 | 98.9 ± 8.9 | 75.4 ± 7.9 |
| DEHP | 102.4 ± 6.0 | 89.2 ± 5.5 | 100.5 ± 9.2 | 101.2 ± 10.6 | 66.0 ± 5.7 |
| DNOP | 107.0 ± 5.7 | 101.7 ± 10.8 | 95.9 ± 9.1 | 111.0 ± 4.7 | 59.2 ± 8.2 |
| DDP | 100.0 ± 1.0 | 89.8 ± 11.9 | 97.0 ± 12.0 | 103.5 ± 1.6 | 74.4 ± 7.0 |
| PAE 1 | Recovery (%) | |||
|---|---|---|---|---|
| 1 min | 5 min | 7 min | 9 min | |
| DMP | 12.5 ± 1.0 | 22.4 ± 1.6 | 23.7 ± 1.4 | 29.2 ± 1.8 |
| DEP | 20.4 ± 0.8 | 88.6 ± 4.4 | 80.7 ± 4.0 | 83.6 ± 5.0 |
| DiBP | 10.1 ± 0.7 | 95.7 ± 3.7 | 94.1 ± 6.6 | 95.2 ± 5.7 |
| DBP | 12.7 ± 0.6 | 96.2 ± 4.8 | 100.1 ± 4.0 | 101.9 ± 6.1 |
| DEHP | 15.7 ± 0.9 | 99.6 ± 5.0 | 95.9 ± 5.7 | 92.3 ± 3.7 |
| DNOP | 11.1 ± 0.6 | 98.8 ± 4.9 | 97.2 ± 4.7 | 90.9 ± 5.4 |
| PAE 1 | Recovery (%) | |||
|---|---|---|---|---|
| 6 min | 10 min | 14 min | 18 min | |
| DMP | 45.2 ± 3.2 | 41.7 ± 2.9 | 42.9 ± 3.0 | 39.2 ± 2.7 |
| DEP | 78.4 ± 4.7 | 69.6 ± 4.2 | 80.1 ± 4.8 | 81.4 ± 4.9 |
| DiBP | 96.6 ± 3.9 | 99.9 ± 4.0 | 99.2 ± 4.9 | 118.1 ± 4.7 |
| DBP | 97.4 ± 7.8 | 101.7 ± 8.1 | 101.9 ± 8.1 | 116.9 ± 9.3 |
| DEHP | 98.7 ± 6.9 | 102.6 ± 7.2 | 103.3 ± 7.2 | 133.4 ± 9.3 |
| DNOP | 99.5 ± 5.9 | 101.3 ± 6.1 | 102.2 ± 6.1 | 129.8 ± 7.8 |
| PAE 1 | Recovery (%) | ||
|---|---|---|---|
| 10 min | 20 min | 30 min | |
| 3000 rpm | |||
| DEP | 77.9 | 78.4 | 70.3 |
| DiBP | 93.9 | 100.6 | 99.9 |
| DBP | 89.2 | 96.4 | 100.3 |
| DEHP | 92.2 | 95.7 | 97.8 |
| DNOP | 95.9 | 97.5 | 97.6 |
| DDP | 94.6 | 95.9 | 98.5 |
| 4000 rpm | |||
| DEP | 67.3 | 71.2 | 82.1 |
| DiBP | 92.0 | 92.9 | 103.7 |
| DBP | 103.0 | 98.2 | 102.0 |
| DEHP | 92.8 | 98.5 | 99.5 |
| DNOP | 91.7 | 97.4 | 103.6 |
| DDP | 94.7 | 96.4 | 101.2 |
| PAE 1 | y = mx + q | R2 | LOD | LOQ | Recovery | Intra-day | Inter-day |
|---|---|---|---|---|---|---|---|
| DMP | y = 0.0065x − 0.0563 | 0.9782 | 15.4 | 35.8 | 66.7 ± 3.7 | 5.7 | 10.7 |
| DEP | y = 0.0692x − 0.0065 | 0.9993 | 3.8 | 9.0 | 91.7 ± 5.5 | 6.1 | 9.2 |
| DiBP | y = 0.0131x + 0.0035 | 0.9964 | 0.8 | 1.9 | 98.9 ± 3.9 | 4.2 | 11.1 |
| DBP | y = 0.0164x − 0.0053 | 0.9972 | 1.2 | 2.8 | 101.2 ± 4.0 | 3.5 | 10.6 |
| DEHP | y = 0.0192x + 0.0004 | 0.9973 | 0.7 | 1.6 | 99.4 ± 3.9 | 3.8 | 7.3 |
| DNOP | y = 0.0186x + 0.0107 | 0.9971 | 2.2 | 5.0 | 100.7 ± 5.8 | 6.3 | 10.1 |
| DDP | y = 0.0189x + 0.0375 | 0.9972 | 10.7 | 24.9 | 93.1 ± 5.5 | 5.2 | 9.8 |
| PAE 1 | LOD | LOQ | ||
|---|---|---|---|---|
| FID | IT/MS | FID | IT/MS | |
| DMP | 1200 | 15.4 | 2800 | 35.8 |
| DEP | 600 | 3.8 | 2400 | 9.0 |
| DiBP | 600 | 0.8 | 1300 | 1.9 |
| DBP | 400 | 1.2 | 1300 | 2.8 |
| DEHP | 700 | 0.7 | 1200 | 1.6 |
| DNOP | 1600 | 2.2 | 2800 | 5.0 |
| DDP | 500 | 10.7 | 2400 | 24.9 |
| Analytes | Matrix | Extraction Method | LOD/LOQ (ng mL−1) | Recovery (%) | RSD (%) | Ref. |
|---|---|---|---|---|---|---|
| 7 PAEs | coffee, tea | SPE 1 | 3–4/10 | 83–105 | 8–15 | [31] |
| 10 PAEs | tea leaves | LLE | -/1–120 | 85.6–114.1 | <20 | [32] |
| 6 PAEs | tea, infusion | QuEChERS 2 | 9–18/27–58 | 70.1–101.3 | 0.6–1.5 | [33] |
| 6 PAEs | coffee | DI-SPME 3 | 5–30/- | 87.6–100.7 | 3.1–9.2 | [34] |
| 11 PAEs | tea | m-SPE | -/0.03–0.18 | 80–114 | 0–16 | [35] |
| 8 PAEs | coffee | LLE 4 | 0.18–0.67/0.6–2.1 | 80.3–105.1 | 0.7–3.1 | [36] |
| 6 PAEs | coffee | PATC-HS-SPME 5 | 0.04–0.10/- | 75.5–105.3 | 1.8–12.0 | [37] |
| 8 PAEs | coffee | MSPE 6 | 30–200/10–500 | 77.3–119.4 | 0.8–15 | [38] |
| 8 PAEs | iced-tea | VA-DLLME | -/17.2–59.4 | 84–120 | 1–11 | [39] |
| 11 PAEs | tea, infusion | NADES 7 | -/4.3–51.1 | 71–1215 | 1–22 | [40] |
| 14 PAEs | tea, infusion | VA-DLLME | -/25–1250 | 63–124 | 1–19 | [41] |
| 7 PAEs | coffee, ginseng, tea | UVA-DLLME | 0.8–15.4/1.6–35.8 | 66.7–101.2 | 3.5–11.1 | This study |
| Sample | PAE 1 | |||||
|---|---|---|---|---|---|---|
| DMP | DEP | DiBP | DEHP | DNOP | DDP | |
| SML 2 | 60 | 60 | 60 | 1.5 | 60 | 60 |
| Hazard | N/A 3 | 2 4 | 125 5 | 4.8 5 | N/A | N/A |
| Espresso | 0.021 | 0.083 | <LOQ | 0.210 | 0.072 | 0.042 |
| Long espresso | <LOQ | 0.091 | <LOQ | 0.314 | 0.104 | <LOQ |
| Coffee | <LOQ | <LOQ | <LOQ | 0.447 | 0.173 | <LOQ |
| Long coffee | <LOQ | <LOQ | <LOQ | 0.353 | 0.148 | 0.054 |
| Ginseng coffee | <LOQ | <LOQ | <LOQ | 0.536 | 0.122 | 0.049 |
| Tea | <LOQ | <LOQ | <LOQ | 0.731 | 0.237 | <LOQ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ianiri, G.; Di Fiore, C.; Passarella, S.; Notardonato, I.; Iannone, A.; Carriera, F.; Stillittano, V.; De Felice, V.; Russo, M.V.; Avino, P. Methodology for Determining Phthalate Residues by Ultrasound–Vortex-Assisted Dispersive Liquid–Liquid Microextraction and GC-IT/MS in Hot Drink Samples by Vending Machines. Analytica 2022, 3, 213-227. https://doi.org/10.3390/analytica3020015
Ianiri G, Di Fiore C, Passarella S, Notardonato I, Iannone A, Carriera F, Stillittano V, De Felice V, Russo MV, Avino P. Methodology for Determining Phthalate Residues by Ultrasound–Vortex-Assisted Dispersive Liquid–Liquid Microextraction and GC-IT/MS in Hot Drink Samples by Vending Machines. Analytica. 2022; 3(2):213-227. https://doi.org/10.3390/analytica3020015
Chicago/Turabian StyleIaniri, Giuseppe, Cristina Di Fiore, Sergio Passarella, Ivan Notardonato, Alessia Iannone, Fabiana Carriera, Virgilio Stillittano, Vincenzo De Felice, Mario Vincenzo Russo, and Pasquale Avino. 2022. "Methodology for Determining Phthalate Residues by Ultrasound–Vortex-Assisted Dispersive Liquid–Liquid Microextraction and GC-IT/MS in Hot Drink Samples by Vending Machines" Analytica 3, no. 2: 213-227. https://doi.org/10.3390/analytica3020015
APA StyleIaniri, G., Di Fiore, C., Passarella, S., Notardonato, I., Iannone, A., Carriera, F., Stillittano, V., De Felice, V., Russo, M. V., & Avino, P. (2022). Methodology for Determining Phthalate Residues by Ultrasound–Vortex-Assisted Dispersive Liquid–Liquid Microextraction and GC-IT/MS in Hot Drink Samples by Vending Machines. Analytica, 3(2), 213-227. https://doi.org/10.3390/analytica3020015