1. Introduction
The motivation behind the renewed interest in natural product (NP) studies arises from their ability to propose highly diverse and renewable sources of medicinal drugs, cosmetics, dyes, and materials in the broader sense. Dereplication in the context of NP chemistry may be defined as the identification of known chemotypes, so that structure re-elucidation and possibly compound re-isolation can be avoided [
1,
2]. Establishing whether an organic compound is known requires the availability of a collection of identity cards of known compounds, possibly organized as a computer database (DB). The existence, availability, scope, and limitations of the numerous NP DBs has been thoroughly reviewed recently, resulting in the creation of a new DB named COCONUT in which the content of numerous DBs was collected [
3,
4,
5]. An even more recent work led to LOTUS, a comprehensive fully open source open data DB that connects NP molecular structures with the taxonomic classification of the organisms they originate from in an unprecedented way and constitutes a significantly useful source of data for NP chemists [
6,
7]. Moreover, the LOTUS database provides bibliographic links to compound descriptions.
NP chemical studies start from taxonomically well-defined biological resources from which the products of the metabolism, primary and specialized, are extracted. Extraction has considerably evolved during the last decades, involving a wide range of physical and chemical processes adapted to the nature of the starting material and to the desired extraction selectivity [
8]. Crude NP extracts are generally substances made of highly complex compound mixtures. The reward of the subsequent extract complexity reduction by fractionation and purification is a simplification of the identification task. Alternatively, studying complex mixtures results in challenging identification problems, but reduces the investment in separation techniques.
The hyphenation of liquid chromatography (LC) and mass spectrometry (MS) for extract analysis takes advantage of extremely powerful purification devices (UPLC chromatographers) with extremely sensitive detection devices (mass spectrometry, possibly with MS
n capabilities), so that the extract fractionation steps may be as reduced as possible. Compounds are identified from their exact molecular formula, fragmentation pattern, and ionic mobility. Fragmentation pattern analysis has proved to be highly successful and led to initiatives such as Global Natural Products Social molecular networking (GNPS), which results from collaborative efforts among numerous scientists [
9]. LC–MS based methods frequently provide annotations rather than identification, meaning that the collected experimental data may fit with isomer collections. Ideally, identification succeeds when an annotation set can be reduced to a single compound.
The use of LC hyphenated with nuclear magnetic resonance (NMR) spectroscopy is frequently limited by the amount of purified compound that can be analyzed, NMR being far less sensitive than MS. Methods have recently been made available for mixture analysis by NMR with applications to crude natural extracts or to series of extract fractions [
10,
11,
12,
13]. NMR characterizes molecular compounds at the atomic level so that NMR experimental data are less prone than MS data to be compatible with a high number of molecular structures. Ambiguity from NMR arises often from the lack of configuration assignment to chiral structure elements, while planar structures are generally defined to a high level of accuracy [
14]. While NMR spectra offer the possibility of distinguishing between diastereomers, even for compounds in mixtures, enantiomer identification requires either the isolation of pure compounds for their study by chiroptical methods or chemical derivatization by a homochiral reagent and subsequent NMR analysis [
15,
16].
Dereplication relies on the comparison between freshly collected spectroscopic data with those from previous studies and stored in a DB. The extraction of experimental MS and NMR data from publications is a tedious process that may result in copy errors and in the exact copy of erroneous structure or data assignments [
17]. However, the accumulated knowledge gained of the relationships between molecular structures and measurement outcomes has made it possible to design spectroscopic prediction tools that may replace, to some extent, experimental spectral data by predicted ones [
10,
18].
The dereplication of NPs relies on NP DBs containing structural, taxonomic and spectroscopic data [
19]. Merging spectroscopic and biological taxonomy data offers a way to reduce, possibly to one, the number of annotations for a given compound. Restricting the set of candidate structures for dereplication to the chemical entities produced by the organisms that are taxonomically related to the one under study finds its justification in the coevolution of species and of the compounds they produce to establish relationships with their environment. This communication reports a way to create a database related to a given taxon with included data for dereplication through
13C NMR spectroscopy. A similar approach, KnapsackSearch [
19], was reported, resorting to the internet to access the KNApSAcK DB [
20]. The current approach, called CNMR_Predict, relies on the LOTUS DB as a structure provider by the possibility it offers to carry out searches according to taxonomy and to easily export the result of searches [
6]. The use of
13C NMR data for dereplication seems inappropriate in this context, considering the higher sensitivity of
1H NMR. The advantages of
13C NMR lie in the ubiquitous presence of carbon atoms in organic molecules, in the absence of a signal fine structure, so that one carbon atom creates only one spectral peak, in the production of resonances that are narrow (about 1 Hz) by comparison to spectral widths (tens of kHz), resulting in a low probability of peak superimposition, in the low sensitivity of
13C chemical shift to solvent and temperature effects, and in the accurate predictability of these chemical shifts [
10]. These features make of
13C NMR a useful tool for NP dereplication. A general review article about NP mixture analysis has been recently published [
21]; it includes references to the methods that benefit from taxonomy focused NMR databases such as CARAMEL [
10], DerepCrude [
12], or MixONat [
13], but in which the step of
13C NMR chemical shift prediction still constitutes a bottleneck.
3. Results and Discussion
The creation of a taxonomy focused compound library with predicted
13C NMR chemical shift values included is illustrated here for turnip, or
Brassica rapa subsp.
rapa L.
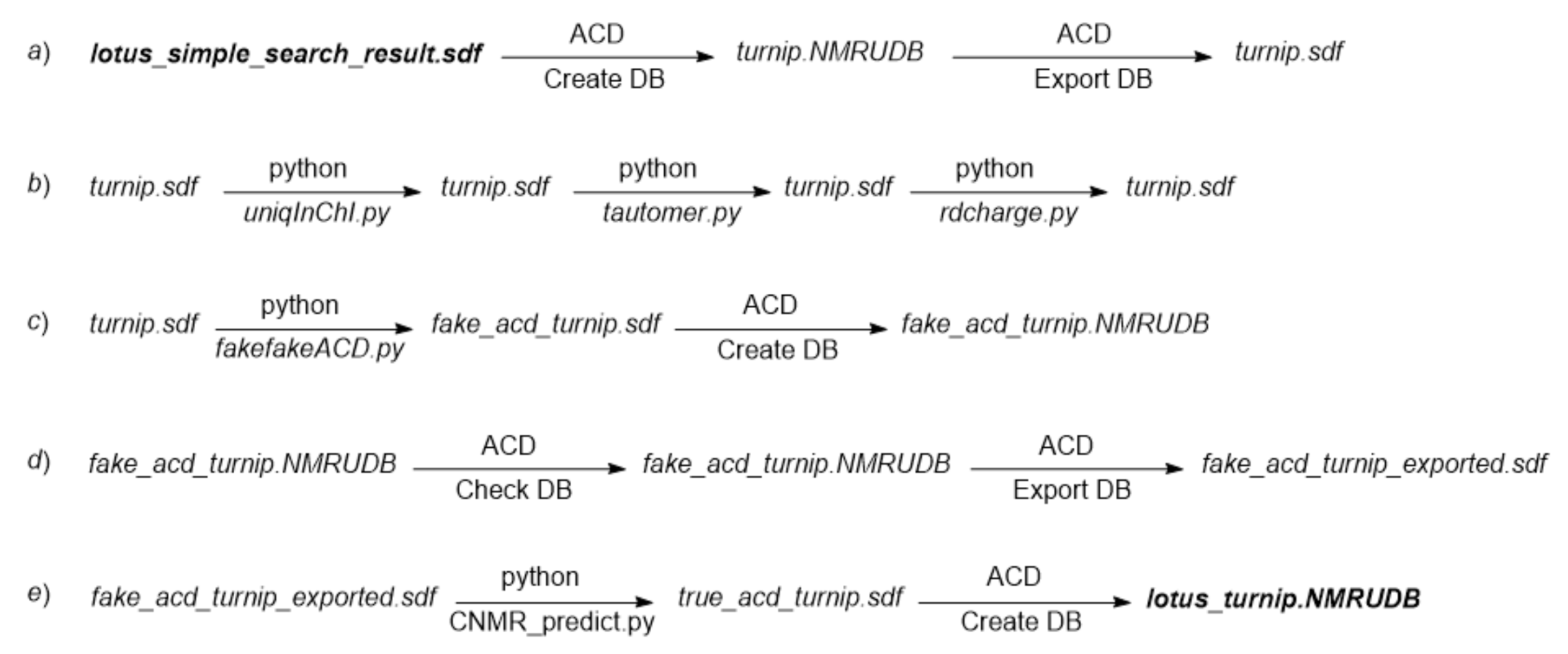
Scheme 1 helps to follow the successive steps of the creation process. The related files are available from the Turnip subdirectory of directory CNMR_Predict [
25]. Submitting species name
Brassica rapa as keyword (thus including subspecies other than turnip) for a simple search in the LOTUS DB resulted in 121 hits. The search result was downloaded as file
lotus_simple_search_result.sdf in V3000 SDF format [
26] and stored in a local computer directory, in which all files related to the turnip project were stored. A new ACD/Labs DB file,
turnip.NMRUDB, was created and filled with data from
lotus_simple_search_result.sdf. Exporting this database in SDF format as file
turnip.sdf converted it to the V2000 SDF format. This conversion (
Scheme 1, step
a) is useful if an SDF file reader that does not decode properly the V3000 format is used for structure viewing.
The
turnip.sdf file then underwent three preparatory operations (
Scheme 1, step
b) before it could be supplemented with chemical shift values. The
turnip.sdf file may have contained identical structures, apparently because different InChI [
27] character strings in LOTUS may have resulted in the production of structures in
lotus_simple_search_result.sdf that were in turn recoded as identical InChI strings by the RDKit library. A python script,
uniqInChI.py, retains only a single occurrence of duplicated structures according to InChI equality and was applied to file
turnip.sdf, in which only one compound out of 121 was removed.
Many structures downloaded from LOTUS were produced by the decoding of InChI strings. This process has the very visible side effect of replacing secondary and primary amide functions by their tautomeric iminol forms. As the central carbon atom in enamine and iminol functional groups have their
13C NMR chemical shift values not identically predicted, it appeared to be necessary to transform aliphatic iminol substructures into their amide tautomer, as achieved by the
tautomer.py script applied to file
turnip.sdf. It should be noticed that the systematic nomenclature [
28] of iminol-containing compounds in LOTUS is determined as if they were really iminols and not amides, resulting, for example, in the difficult identification of peptidic bonds in peptides.
Script
tautomer.py relies on RDKit to write SDF files and makes use of reaction SMARTS [
29]. Electrically charged atoms in structures written by RDKit include a nondefault specification for the nonstandard valence of such atoms (such as four for the nitrogen atom in an ammonium group), in accordance with SDF specification. Such a structure description is not properly interpreted by the ACD/Labs software, thus precluding the prediction of chemical shifts. The
rdcharge.py script resets the valence data piece to the default, nonblocking value and was applied to
turnip.sdf.
The first step toward the automatic calculation of
13C NMR chemical shifts (
Scheme 1, step
c) was to let the ACD/Labs software consider that experimental values were stored in an SDF file it produced, something feasible by supplementing the SDF file with data lines under the purposely created CNMR_SHIFTS SDF tag. These fake data lines include the fake chemical shift value 99.99, one per carbon atom in each molecule. The
fakefakeACD.py script applied to
turnip.sdf transforms it into file
fake_acd_turnip.sdf.
For chemical shift prediction (
Scheme 1, step
d), a new ACD/Labs DB file,
fake_acd_turnip.NMRUDB, was created and filled by importation from the file
fake_acd_turnip.sdf. All carbon atoms appeared with their arbitrarily given 99.99 chemical shift value. The presence of these values allows ACD/Labs DB to check all chemical shift values of all molecules from a single mouse click. Checking the chemical shifts of a DB that does not contain chemical values fails to give a meaningful result, thus justifying the resorting to the
fakefakeACD.py script. Exporting the current DB as file
fake_acd_turnip_exported.sdf first displayed a message that warned that the calculated chemical shifts would not be exported. This is simultaneously true and false. This is true because the calculated values cannot be used for a structure search according to the chemical shift similarity between stored values and a set of targeted values, as required for dereplication. This is also false because the result of the prediction is stored in the resulting file, here
fake_acd_turnip_exported.sdf, under the CNMR_CALC_SHIFTS SDF tag.
The last step toward a DB file usable for dereplication (
Scheme 1, step
e) consists in replacing the 99.99 values under the CNMR_SHIFTS SDF tag that were still present in file
fake_acd_turnip_exported.sdf, with the calculated chemical shift values it contains, written under the CNMR_CALC_SHIFTS SDF tag. This operation was carried out by the script
CNMR_predict.py acting on file
fake_acd_turnip_exported.sdf to produce file
true_acd_turnip.sdf. A new ACD/Labs DB file,
lotus_turnip.NMRUDB was finally created and filled with compounds from file
true_acd_turnip.sdf. The DB
lotus_turnip.NMRUDB was then ready for compound identification according to
13C NMR chemical shift values using the compound search tool included in the ACD/Labs software. The file
true_acd_turnip.sdf also contains SDF tags that make dereplication possible by the MixONat software. The script
ACD_to_DerepCrude.py formatted the predicted chemical shifts for its use with the DerepCrude software. Both DerepCrude [
12] and MixONat [
13] are dedicated to the dereplication by
13C NMR either on crude NP extracts or on extract fractions, as alternatives to the now well established CARAMEL dereplication procedure [
10,
30].
The values that were calculated for the chemical shift checking of an entire DB file, written under the CNMR_CALC_SHIFTS SDF tag in file fake_acd_turnip_exported.sdf, were not exactly those produced by the ACD/Labs CNMR Predictor when run in a compound by compound procedure, but the origin of the difference is difficult to track as no information is available on the details of the underlying algorithms.
The creation of DB
lotus_turnip.NMRUDB is a process that alternates the execution of python scripts from a terminal window and the handling (create/import/predict/export/close) of ACD/Labs DB files. A template text file is proposed with the CNMR_Predict project files so that the actions to perform sequentially can be easily accomplished.
Figure 1 illustrates the content of this template file. CNMR_Predict is a follow up of the KnapsackSearch project that made use of nmrshiftdb2 for the prediction of
13C NMR chemical shift values [
31]. These predicted values may be formatted as experimental values under the CNMR_SHIFTS SDF tag and a template file is also available for this option.
Creating a file such as
lotus_turnip.NMRUDB with the ACD/Labs software from initial file
lotus_simple_search_result.sdf without CNMR_Predict would require a tedious compound by compound operation, lasting about one minute per structure unless some presently undisclosed script is available for calculation process automation [
32]. The prediction involving CNMR_Predict lasts less than one second per structure, making it easy to use for the creation of taxonomically focused collections of natural products. For example, species
Brassica rapa is related to family Brassicaceae and searching for this taxon in LOTUS results in 2271 hits. A ready-to-search database of compounds from Brassicaceae can be thus produced in less than one hour on a standard laptop computer, an hour during which the computer is the only one that performs the repetitive work. The choice of the appropriate taxon type (order, family, genus, species, etc.) is left to the user and can be adapted according to the size of the chemical space to investigate. This taxonomy focused approach is more flexible than the one consisting in precalculating the chemical shifts for all the compounds present in a snapshot of a database such as LOTUS since its content may be steadily updated.
It should be noticed that DBs refer to published data and can propagate errors. For example, glucosinolates constitute an emblematic group of compounds related to the family of Brassicaceae (and more generally to the order of Capparales) that contain an O-sulfated anomeric (
Z)-thiohydroximate function in which the double bond configuration may appear in DBs with the double bond in the (
E) configuration or left undefined [
33]. The library of the compounds from
Brassica rapa, a Brassicaceae species, reported by LOTUS contain such erroneous or incompletely defined structures that would be also found in general purpose databases such as that of the Chemical Abstract Service [
34].



{kind=link}
{kind=link}