

Low Hepatic CEACAM1 Tethers Metabolic Dysfunction Steatohepatitis to Atherosclerosis

 , , ,
, , ,  and
and {kind=link}
{kind=link}
Abstract
1. Introduction
2. Pathophysiology of Atherosclerosis: Mechanisms and Cardiovascular Implications
2.1. Atherosclerosis: Prevalence and Clinical Significance
2.2. The Pathophysiological Hallmarks of Atherosclerosis
2.2.1. Endothelial Dysfunction
2.2.2. Lipid Accumulation and Plaque Formation
2.2.3. Vascular Smooth Muscle Cell Migration and Proliferation
2.3. Risk Factors and Their Impact on Atherosclerosis Progression
2.4. Rising Incidence of HFpEF and LV Diastolic Dysfunction in Women Suggests a Potential Link to Hyperinsulinemia
3. Pathophysiology of MASH
3.1. Overview, Prevalence, and Clinical Significance of MASH
3.2. Key Mechanisms in the Development of MASH
3.2.1. Hepatic Steatosis and Lipid Accumulation
3.2.2. Oxidative Stress and Mitochondrial Dysfunction
3.2.3. Hepatic Inflammation and Immune Response
3.2.4. Fibrosis and Progression to Cirrhosis
3.3. Risk Factors and Their Impact on MASH Progression
4. Common Risk Factors and Pathways
5. Loss of CEACAM1 Links Hepatic Fibrosis to Atherosclerosis
5.1. MASLD and Atherosclerosis: Twin Diseases
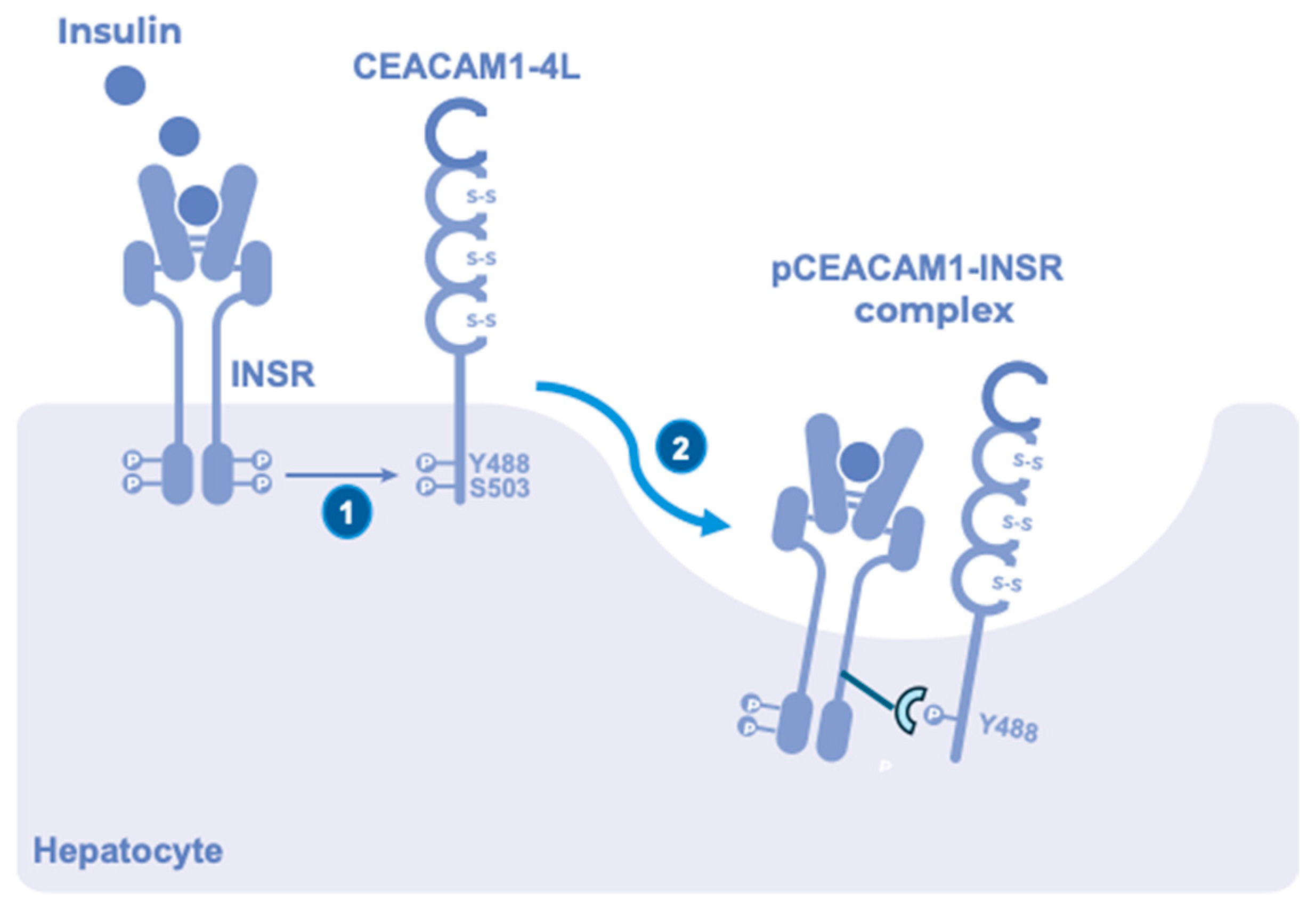
5.2. CEACAM1—Its Loss Links Reduced Insulin Clearance to MASH
5.3. Repositioning CEACAM1 as a Critical Immunometabolic Regulator
5.4. CEACAM1—Its Loss Links MASH to Atherosclerosis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bergheanu, S.C.; Bodde, M.C.; Jukema, J.W. Pathophysiology and treatment of atherosclerosis: Current view and future perspective on lipoprotein modification treatment. Neth. Heart J. 2017, 25, 231–242. [Google Scholar] [CrossRef]
- Timmis, A.; Kazakiewicz, D.; Townsend, N.; Huculeci, R.; Aboyans, V.; Vardas, P. Global epidemiology of acute coronary syndromes. Nat. Rev. Cardiol. 2023, 20, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef]
- Eslam, M.; George, J. Genetic contributions to NAFLD: Leveraging shared genetics to uncover systems biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52. [Google Scholar] [CrossRef]
- Golabi, P.; Paik, J.M.; Arshad, T.; Younossi, Y.; Mishra, A.; Younossi, Z.M. Mortality of NAFLD According to the Body Composition and Presence of Metabolic Abnormalities. Hepatol. Commun. 2020, 4, 1136–1148. [Google Scholar] [CrossRef]
- Mantovani, A.; Csermely, A.; Petracca, G.; Beatrice, G.; Corey, K.E.; Simon, T.G.; Byrne, C.D.; Targher, G. Non-alcoholic fatty liver disease and risk of fatal and non-fatal cardiovascular events: An updated systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2021, 6, 903–913. [Google Scholar] [CrossRef]
- Targher, G.; Day, C.P.; Bonora, E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.M.; Perdomo, G. Hepatic Insulin Clearance: Mechanism and Physiology. Physiology 2019, 34, 198–215. [Google Scholar] [CrossRef]
- Kim, J.A.; Montagnani, M.; Koh, K.K.; Quon, M.J. Reciprocal relationships between insulin resistance and endothelial dysfunction: Molecular and pathophysiological mechanisms. Circulation 2006, 113, 1888–1904. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Ghadieh, H.E.; Abu Helal, R.; Muturi, H.T.; Issa, D.D.; Russo, L.; Abdallah, S.L.; Najjar, J.A.; Benencia, F.; Vazquez, G.; Li, W.; et al. Loss of Hepatic Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 Links Nonalcoholic Steatohepatitis to Atherosclerosis. Hepatol. Commun. 2020, 4, 1591–1609. [Google Scholar] [CrossRef]
- Najjar, S.M.; Ledford, K.J.; Abdallah, S.L.; Paus, A.; Russo, L.; Kaw, M.K.; Ramakrishnan, S.K.; Muturi, H.T.; Raphael, C.K.; Lester, S.G.; et al. Ceacam1 deletion causes vascular alterations in large vessels. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E519–E529. [Google Scholar] [CrossRef]
- Najjar, S.M.; Caprio, S.; Gastaldelli, A. Insulin Clearance in Health and Disease. Annu. Rev. Physiol. 2023, 85, 363–381. [Google Scholar] [CrossRef]
- Heinrich, G.; Muturi, H.T.; Rezaei, K.; Al-Share, Q.Y.; DeAngelis, A.M.; Bowman, T.A.; Ghadieh, H.E.; Ghanem, S.S.; Zhang, D.; Garofalo, R.S.; et al. Reduced Hepatic Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 Level in Obesity. Front. Endocrinol. 2017, 8, 54. [Google Scholar] [CrossRef]
- Lee, W. The CEACAM1 expression is decreased in the liver of severely obese patients with or without diabetes. Diagn. Pathol. 2011, 6, 40. [Google Scholar] [CrossRef]
- DeAngelis, A.M.; Heinrich, G.; Dai, T.; Bowman, T.A.; Patel, P.R.; Lee, S.J.; Hong, E.G.; Jung, D.Y.; Assmann, A.; Kulkarni, R.N.; et al. Carcinoembryonic antigen-related cell adhesion molecule 1: A link between insulin and lipid metabolism. Diabetes 2008, 57, 2296–2303. [Google Scholar] [CrossRef]
- Ghadieh, H.E.; Russo, L.; Muturi, H.T.; Ghanem, S.S.; Manaserh, I.H.; Noh, H.L.; Suk, S.; Kim, J.K.; Hill, J.W.; Najjar, S.M. Hyperinsulinemia drives hepatic insulin resistance in male mice with liver-specific Ceacam1 deletion independently of lipolysis. Metabolism 2019, 93, 33–43. [Google Scholar] [CrossRef]
- Huang, J.; Ledford, K.J.; Pitkin, W.B.; Russo, L.; Najjar, S.M.; Siragy, H.M. Targeted deletion of murine CEACAM 1 activates PI3K-Akt signaling and contributes to the expression of (Pro)renin receptor via CREB family and NF-kappaB transcription factors. Hypertension 2013, 62, 317–323. [Google Scholar] [CrossRef]
- Russo, L.; Muturi, H.T.; Ghadieh, H.E.; Wisniewski, A.M.; Morgan, E.E.; Quadri, S.S.; Landesberg, G.P.; Siragy, H.M.; Vazquez, G.; Scalia, R.; et al. Liver-specific rescuing of CEACAM1 reverses endothelial and cardiovascular abnormalities in male mice with null deletion of Ceacam1 gene. Mol. Metab. 2018, 9, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Imakita, M.; Yutani, C.; Strong, J.P.; Sakurai, I.; Sumiyoshi, A.; Watanabe, T.; Mitsumata, M.; Kusumi, Y.; Katayama, S.; Mano, M.; et al. Second nation-wide study of atherosclerosis in infants, children and young adults in Japan. Atherosclerosis 2001, 155, 487–497. [Google Scholar] [CrossRef]
- Takei, H.; Strong, J.P.; Yutani, C.; Malcom, G.T. Comparison of coronary and aortic atherosclerosis in youth from Japan and the USA. Atherosclerosis 2005, 180, 171–179. [Google Scholar] [CrossRef]
- Hanke, H.; Lenz, C.; Finking, G. The discovery of the pathophysiological aspects of atherosclerosis—A review. Acta Chir. Belg. 2001, 101, 162–169. [Google Scholar] [CrossRef]
- Konstantinov, I.E.; Mejevoi, N.; Anichkov, N.M.; Nikolai, N. Anichkov and his theory of atherosclerosis. Tex. Heart Inst. J. 2006, 33, 417–423. [Google Scholar] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflugers Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.J. Assessment and treatment of endothelial dysfunction in humans. J. Am. Coll. Cardiol. 1999, 34, 631–638. [Google Scholar] [CrossRef]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109 (Suppl. S1), III27–III32. [Google Scholar] [CrossRef] [PubMed]
- Berenji Ardestani, S.; Eftedal, I.; Pedersen, M.; Jeppesen, P.B.; Nørregaard, R.; Matchkov, V.V. Endothelial dysfunction in small arteries and early signs of atherosclerosis in ApoE knockout rats. Sci. Rep. 2020, 10, 15296. [Google Scholar] [CrossRef]
- Mudau, M.; Genis, A.; Lochner, A.; Strijdom, H. Endothelial dysfunction: The early predictor of atherosclerosis. Cardiovasc. J. Afr. 2012, 23, 222–231. [Google Scholar] [CrossRef]
- VanderLaan, P.A.; Reardon, C.A.; Getz, G.S. Site specificity of atherosclerosis: Site-selective responses to atherosclerotic modulators. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 12–22. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef]
- Kang, H.; Cancel, L.M.; Tarbell, J.M. Effect of shear stress on water and LDL transport through cultured endothelial cell monolayers. Atherosclerosis 2014, 233, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Nigro, P.; Abe, J.; Berk, B.C. Flow shear stress and atherosclerosis: A matter of site specificity. Antioxid. Redox Signal 2011, 15, 1405–1414. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.S.; Chien, S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef]
- Souilhol, C.; Serbanovic-Canic, J.; Fragiadaki, M.; Chico, T.J.; Ridger, V.; Roddie, H.; Evans, P.C. Endothelial responses to shear stress in atherosclerosis: A novel role for developmental genes. Nat. Rev. Cardiol. 2020, 17, 52–63. [Google Scholar] [CrossRef]
- Chien, S.; Shyy, J.Y. Effects of hemodynamic forces on gene expression and signal transduction in endothelial cells. Biol. Bull. 1998, 194, 390–391; discussion 392–393. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.J.; Usami, S.; Chien, S. Vascular endothelial responses to altered shear stress: Pathologic implications for atherosclerosis. Ann. Med. 2009, 41, 19–28. [Google Scholar] [CrossRef]
- Shyy, J.Y.; Lin, M.C.; Han, J.; Lu, Y.; Petrime, M.; Chien, S. The cis-acting phorbol ester “12-O-tetradecanoylphorbol 13-acetate”-responsive element is involved in shear stress-induced monocyte chemotactic protein 1 gene expression. Proc. Natl. Acad. Sci. USA 1995, 92, 8069–8073. [Google Scholar] [CrossRef]
- Hsiai, T.K.; Cho, S.K.; Wong, P.K.; Ing, M.; Salazar, A.; Sevanian, A.; Navab, M.; Demer, L.L.; Ho, C.M. Monocyte recruitment to endothelial cells in response to oscillatory shear stress. FASEB J. 2003, 17, 1648–1657. [Google Scholar] [CrossRef]
- Kraiss, L.W.; Geary, R.L.; Mattsson, E.J.; Vergel, S.; Au, Y.P.; Clowes, A.W. Acute reductions in blood flow and shear stress induce platelet-derived growth factor-A expression in baboon prosthetic grafts. Circ. Res. 1996, 79, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, J.N.; Smith, K.M.; Williams, L.T.; Schwartz, S.M.; Gordon, D. Platelet-derived growth factor mRNA detection in human atherosclerotic plaques by in situ hybridization. J. Clin. Investig. 1988, 82, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Hornig, B.; Drexler, H. Endothelial dysfunction in hypercholesterolemia: Mechanisms, pathophysiological importance, and therapeutic interventions. Semin. Thromb. Hemost. 2000, 26, 529–537. [Google Scholar] [CrossRef]
- Chen, J.Y.; Ye, Z.X.; Wang, X.F.; Chang, J.; Yang, M.W.; Zhong, H.H.; Hong, F.F.; Yang, S.L. Nitric oxide bioavailability dysfunction involves in atherosclerosis. Biomed. Pharmacother. 2018, 97, 423–428. [Google Scholar] [CrossRef]
- Carlström, M. Nitric oxide signalling in kidney regulation and cardiometabolic health. Nat. Rev. Nephrol. 2021, 17, 575–590. [Google Scholar] [CrossRef]
- Casino, P.R.; Kilcoyne, C.M.; Quyyumi, A.A.; Hoeg, J.M.; Panza, J.A. The role of nitric oxide in endothelium-dependent vasodilation of hypercholesterolemic patients. Circulation 1993, 88, 2541–2547. [Google Scholar] [CrossRef]
- Panza, J.A.; Casino, P.R.; Kilcoyne, C.M.; Quyyumi, A.A. Role of endothelium-derived nitric oxide in the abnormal endothelium-dependent vascular relaxation of patients with essential hypertension. Circulation 1993, 87, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Esper, R.J.; Nordaby, R.A.; Vilariño, J.O.; Paragano, A.; Cacharrón, J.L.; Machado, R.A. Endothelial dysfunction: A comprehensive appraisal. Cardiovasc. Diabetol. 2006, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Kim, J.; Kwak, S.N.; Lee, K.S.; Lee, D.K.; Ha, K.S.; Won, M.H.; Jeoung, D.; Lee, H.; Kwon, Y.G.; et al. Functional role of NF-kappaB in expression of human endothelial nitric oxide synthase. Biochem. Biophys. Res. Commun. 2014, 448, 101–107. [Google Scholar] [CrossRef]
- Schulz, E.; Gori, T.; Münzel, T. Oxidative stress and endothelial dysfunction in hypertension. Hypertens. Res. 2011, 34, 665–673. [Google Scholar] [CrossRef]
- Alexander, R.W.; Dzau, V.J. Vascular biology: The past 50 years. Circulation 2000, 102 (Suppl. S4), IV112–IV116. [Google Scholar] [CrossRef]
- Chroni, A.; Leondaritis, G.; Karlsson, H. Lipids and lipoproteins in atherosclerosis. J. Lipids 2011, 2011, 160104. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Babitt, J.; Trigatti, B.; Rigotti, A.; Smart, E.J.; Anderson, R.G.; Xu, S.; Krieger, M. Murine SR-BI, a high density lipoprotein receptor that mediates selective lipid uptake, is N-glycosylated and fatty acylated and colocalizes with plasma membrane caveolae. J. Biol. Chem. 1997, 272, 13242–13249. [Google Scholar] [CrossRef] [PubMed]
- Santibanez, J.F.; Blanco, F.J.; Garrido-Martin, E.M.; Sanz-Rodriguez, F.; del Pozo, M.A.; Bernabeu, C. Caveolin-1 interacts and cooperates with the transforming growth factor-beta type I receptor ALK1 in endothelial caveolae. Cardiovasc. Res. 2008, 77, 791–799. [Google Scholar] [CrossRef]
- Wang, D.X.; Pan, Y.Q.; Liu, B.; Dai, L. Cav-1 promotes atherosclerosis by activating JNK-associated signaling. Biochem. Biophys. Res. Commun. 2018, 503, 513–520. [Google Scholar] [CrossRef]
- Takahashi, Y.; Zhu, H.; Yoshimoto, T. Essential roles of lipoxygenases in LDL oxidation and development of atherosclerosis. Antioxid. Redox Signal 2005, 7, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Zheng, X.L.; Tang, C.K. Nuclear Factor-κB Activation as a Pathological Mechanism of Lipid Metabolism and Atherosclerosis. Adv. Clin. Chem. 2015, 70, 1–30. [Google Scholar] [CrossRef]
- Teh, Y.C.; Ding, J.L.; Ng, L.G.; Chong, S.Z. Capturing the Fantastic Voyage of Monocytes Through Time and Space. Front. Immunol. 2019, 10, 834. [Google Scholar] [CrossRef]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, T.; Ley, K. Monocyte trafficking across the vessel wall. Cardiovasc. Res. 2015, 107, 321–330. [Google Scholar] [CrossRef]
- Lin, J.; Kakkar, V.; Lu, X. Impact of MCP-1 in atherosclerosis. Curr. Pharm. Des. 2014, 20, 4580–4588. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- De Paoli, F.; Staels, B.; Chinetti-Gbaguidi, G. Macrophage phenotypes and their modulation in atherosclerosis. Circ. J. 2014, 78, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell Mol. Med. 2016, 20, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Younis, N.; Sharma, R.; Soran, H.; Charlton-Menys, V.; Elseweidy, M.; Durrington, P.N. Glycation as an atherogenic modification of LDL. Curr. Opin. Lipidol. 2008, 19, 378–384. [Google Scholar] [CrossRef]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef]
- Libby, P. Vascular biology of atherosclerosis: Overview and state of the art. Am. J. Cardiol. 2003, 91, 3–6. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Rahmani, M.; Wong, B.W.; Allahverdian, S.; McManus, B.M.; Pickering, J.G.; Chan, T.; Francis, G.A. ATP-binding cassette transporter A1 expression and apolipoprotein A-I binding are impaired in intima-type arterial smooth muscle cells. Circulation 2009, 119, 3223–3231. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.G.; Byrne, H.M.; Macaskill, C.; Myerscough, M.R. A two-phase model of early fibrous cap formation in atherosclerosis. J. Theor. Biol. 2018, 456, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Sharif, H.; Akash, M.S.H.; Rehman, K.; Irshad, K.; Imran, I. Pathophysiology of atherosclerosis: Association of risk factors and treatment strategies using plant-based bioactive compounds. J. Food Biochem. 2020, 44, e13449. [Google Scholar] [CrossRef] [PubMed]
- Cheezum, M.K.; Kim, A.; Bittencourt, M.S.; Kassop, D.; Nissen, A.; Thomas, D.M.; Nguyen, B.; Glynn, R.J.; Shah, N.R.; Villines, T.C. Association of tobacco use and cessation with coronary atherosclerosis. Atherosclerosis 2017, 257, 201–207. [Google Scholar] [CrossRef]
- Elagizi, A.; Kachur, S.; Lavie, C.J.; Carbone, S.; Pandey, A.; Ortega, F.B.; Milani, R.V. An Overview and Update on Obesity and the Obesity Paradox in Cardiovascular Diseases. Prog. Cardiovasc. Dis. 2018, 61, 142–150. [Google Scholar] [CrossRef]
- Kinlen, D.; Cody, D.; O’Shea, D. Complications of obesity. QJM 2018, 111, 437–443. [Google Scholar] [CrossRef]
- Endo, A. A gift from nature: The birth of the statins. Nat. Med. 2008, 14, 1050–1052. [Google Scholar] [CrossRef]
- Fan, J.; Watanabe, T. Atherosclerosis: Known and unknown. Pathol. Int. 2022, 72, 151–160. [Google Scholar] [CrossRef]
- Singh, M.; Kapoor, A.; Bhatnagar, A. Physiological and Pathological Roles of Aldose Reductase. Metabolites 2021, 11, 655. [Google Scholar] [CrossRef]
- Turer, A.T.; Hill, J.A.; Elmquist, J.K.; Scherer, P.E. Adipose tissue biology and cardiomyopathy: Translational implications. Circ. Res. 2012, 111, 1565–1577. [Google Scholar] [CrossRef]
- Liu, J.; Pan, S.; Wang, X.; Liu, Z.; Zhang, Y. Role of advanced glycation end products in diabetic vascular injury: Molecular mechanisms and therapeutic perspectives. Eur. J. Med. Res. 2023, 28, 553. [Google Scholar] [CrossRef]
- Singh, S.; Siva, B.V.; Ravichandiran, V. Advanced Glycation End Products: Key player of the pathogenesis of atherosclerosis. Glycoconj. J. 2022, 39, 547–563. [Google Scholar] [CrossRef]
- D’Souza, A.; Hussain, M.; Howarth, F.C.; Woods, N.M.; Bidasee, K.; Singh, J. Pathogenesis and pathophysiology of accelerated atherosclerosis in the diabetic heart. Mol. Cell Biochem. 2009, 331, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Alpert, M.A.; Omran, J.; Bostick, B.P. Effects of Obesity on Cardiovascular Hemodynamics, Cardiac Morphology, and Ventricular Function. Curr. Obes. Rep. 2016, 5, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Marwick, T.H. Obesity cardiomyopathy: Pathogenesis and pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 436–443. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Cardiomyopathy in obesity, insulin resistance and diabetes. J. Physiol. 2020, 598, 2977–2993. [Google Scholar] [CrossRef] [PubMed]
- Alí, A.; Boutjdir, M.; Aromolaran, A.S. Cardiolipotoxicity, Inflammation, and Arrhythmias: Role for Interleukin-6 Molecular Mechanisms. Front. Physiol. 2018, 9, 1866. [Google Scholar] [CrossRef]
- Cheng, W.; Cui, C.; Liu, G.; Ye, C.; Shao, F.; Bagchi, A.K.; Mehta, J.L.; Wang, X. NF-κB, A Potential Therapeutic Target in Cardiovascular Diseases. Cardiovasc. Drugs Ther. 2023, 37, 571–584. [Google Scholar] [CrossRef]
- Nagueh, S.F.; Smiseth, O.A.; Appleton, C.P.; Byrd, B.F., 3rd; Dokainish, H.; Edvardsen, T.; Flachskampf, F.A.; Gillebert, T.C.; Klein, A.L.; Lancellotti, P.; et al. Recommendations for the Evaluation of Left Ventricular Diastolic Function by Echocardiography: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2016, 29, 277–314. [Google Scholar] [CrossRef]
- Groenewegen, A.; Rutten, F.H.; Mosterd, A.; Hoes, A.W. Epidemiology of heart failure. Eur. J. Heart Fail. 2020, 22, 1342–1356. [Google Scholar] [CrossRef]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2015, 28, 1–39.e14. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, J.H.; Lee, S.Y.; Choi, J.O.; Shin, M.S.; Kim, M.J.; Jung, H.O.; Park, J.R.; Sohn, I.S.; Kim, H.; et al. Normal 2-Dimensional Strain Values of the Left Ventricle: A Substudy of the Normal Echocardiographic Measurements in Korean Population Study. J. Cardiovasc. Ultrasound 2016, 24, 285–293. [Google Scholar] [CrossRef]
- Taylor, R.J.; Moody, W.E.; Umar, F.; Edwards, N.C.; Taylor, T.J.; Stegemann, B.; Townend, J.N.; Hor, K.N.; Steeds, R.P.; Mazur, W.; et al. Myocardial strain measurement with feature-tracking cardiovascular magnetic resonance: Normal values. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 871–881. [Google Scholar] [CrossRef]
- Lieb, W.; Xanthakis, V.; Sullivan, L.M.; Aragam, J.; Pencina, M.J.; Larson, M.G.; Benjamin, E.J.; Vasan, R.S. Longitudinal tracking of left ventricular mass over the adult life course: Clinical correlates of short- and long-term change in the framingham offspring study. Circulation 2009, 119, 3085–3092. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2023 Focused update of the 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. G. Ital. Cardiol. 2024, 25, 202–213. [Google Scholar] [CrossRef] [PubMed]
- van der Ende, M.Y.; Juarez-Orozco, L.E.; Waardenburg, I.; Lipsic, E.; Schurer, R.A.J.; van der Werf, H.W.; Benjamin, E.J.; van Veldhuisen, D.J.; Snieder, H.; van der Harst, P. Sex-Based Differences in Unrecognized Myocardial Infarction. J. Am. Heart Assoc. 2020, 9, e015519. [Google Scholar] [CrossRef] [PubMed]
- Boonman-de Winter, L.J.; Rutten, F.H.; Cramer, M.J.; Landman, M.J.; Zuithoff, N.P.; Liem, A.H.; Hoes, A.W. Efficiently screening heart failure in patients with type 2 diabetes. Eur. J. Heart Fail. 2015, 17, 187–195. [Google Scholar] [CrossRef]
- Garawi, F.; Devries, K.; Thorogood, N.; Uauy, R. Global differences between women and men in the prevalence of obesity: Is there an association with gender inequality? Eur. J. Clin. Nutr. 2014, 68, 1101–1106. [Google Scholar] [CrossRef]
- Abel, E.D. Insulin signaling in the heart. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E130–E145. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Sakaguchi, M.; Kleinridders, A.; Gonzalez-Del Pino, G.; Dreyfuss, J.M.; O’Neill, B.T.; Ramirez, A.K.; Pan, H.; Winnay, J.N.; Boucher, J.; et al. Domain-dependent effects of insulin and IGF-1 receptors on signalling and gene expression. Nat. Commun. 2017, 8, 14892. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Chopra, I.; Li, H.F.; Wang, H.; Webster, K.A. Phosphorylation of the insulin receptor by AMP-activated protein kinase (AMPK) promotes ligand-independent activation of the insulin signalling pathway in rodent muscle. Diabetologia 2012, 55, 783–794. [Google Scholar] [CrossRef]
- Fazio, S.; Mercurio, V.; Fazio, V.; Ruvolo, A.; Affuso, F. Insulin Resistance/Hyperinsulinemia, Neglected Risk Factor for the Development and Worsening of Heart Failure with Preserved Ejection Fraction. Biomedicines 2024, 12, 806. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.; Ditah, I.C.; Saeian, K.; Lalehzari, M.; Aronsohn, A.; Gorospe, E.C.; Charlton, M. Changes in the Prevalence of Hepatitis C Virus Infection, Nonalcoholic Steatohepatitis, and Alcoholic Liver Disease Among Patients With Cirrhosis or Liver Failure on the Waitlist for Liver Transplantation. Gastroenterology 2017, 152, 1090–1099.e1. [Google Scholar] [CrossRef] [PubMed]
- Zafrani, E.S. Non-alcoholic fatty liver disease: An emerging pathological spectrum. Virchows Arch. 2004, 444, 3–12. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J. Toward More Accurate Nomenclature for Fatty Liver Diseases. Gastroenterology 2019, 157, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Sarin, S.K.; Kumar, M.; Eslam, M.; George, J.; Al Mahtab, M.; Akbar, S.M.F.; Jia, J.; Tian, Q.; Aggarwal, R.; Muljono, D.H.; et al. Liver diseases in the Asia-Pacific region: A Lancet Gastroenterology & Hepatology Commission. Lancet Gastroenterol. Hepatol. 2020, 5, 167–228. [Google Scholar] [CrossRef] [PubMed]
- Paik, J.M.; Henry, L.; De Avila, L.; Younossi, E.; Racila, A.; Younossi, Z.M. Mortality Related to Nonalcoholic Fatty Liver Disease Is Increasing in the United States. Hepatol. Commun. 2019, 3, 1459–1471. [Google Scholar] [CrossRef]
- Sayiner, M.; Stepanova, M.; Pham, H.; Noor, B.; Walters, M.; Younossi, Z.M. Assessment of health utilities and quality of life in patients with non-alcoholic fatty liver disease. BMJ Open Gastroenterol. 2016, 3, e000106. [Google Scholar] [CrossRef]
- Lu, Y.C.; Chang, C.C.; Wang, C.P.; Hung, W.C.; Tsai, I.T.; Tang, W.H.; Wu, C.C.; Wei, C.T.; Chung, F.M.; Lee, Y.J.; et al. Circulating fatty acid-binding protein 1 (FABP1) and nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus. Int. J. Med. Sci. 2020, 17, 182–190. [Google Scholar] [CrossRef]
- Wang, G.; Bonkovsky, H.L.; de Lemos, A.; Burczynski, F.J. Recent insights into the biological functions of liver fatty acid binding protein 1. J. Lipid Res. 2015, 56, 2238–2247. [Google Scholar] [CrossRef]
- Koonen, D.P.; Jacobs, R.L.; Febbraio, M.; Young, M.E.; Soltys, C.L.; Ong, H.; Vance, D.E.; Dyck, J.R. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes 2007, 56, 2863–2871. [Google Scholar] [CrossRef]
- Buttet, M.; Poirier, H.; Traynard, V.; Gaire, K.; Tran, T.T.; Sundaresan, S.; Besnard, P.; Abumrad, N.A.; Niot, I. Deregulated Lipid Sensing by Intestinal CD36 in Diet-Induced Hyperinsulinemic Obese Mouse Model. PLoS ONE 2016, 11, e0145626. [Google Scholar] [CrossRef]
- Auinger, A.; Valenti, L.; Pfeuffer, M.; Helwig, U.; Herrmann, J.; Fracanzani, A.L.; Dongiovanni, P.; Fargion, S.; Schrezenmeir, J.; Rubin, D. A promoter polymorphism in the liver-specific fatty acid transport protein 5 is associated with features of the metabolic syndrome and steatosis. Horm. Metab. Res. 2010, 42, 854–859. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Gordon, D.; Stec, D.E.; Hinds, T.D. Bilirubin: A Ligand of the PPARα Nuclear Receptor. In Nuclear Receptors: The Art and Science of Modulator Design and Discovery; Badr, M.Z., Ed.; Springer International Publishing: Cham, Switzerland, 2021; pp. 463–482. [Google Scholar]
- Eberlé, D.; Hegarty, B.; Bossard, P.; Ferré, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Knebel, B.; Haas, J.; Hartwig, S.; Jacob, S.; Kollmer, C.; Nitzgen, U.; Muller-Wieland, D.; Kotzka, J. Liver-specific expression of transcriptionally active SREBP-1c is associated with fatty liver and increased visceral fat mass. PLoS ONE 2012, 7, e31812. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Takao, K.; Yabe, D. ChREBP-Mediated Regulation of Lipid Metabolism: Involvement of the Gut Microbiota, Liver, and Adipose Tissue. Front. Endocrinol. 2020, 11, 587189. [Google Scholar] [CrossRef]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Hosick, P.A.; Chen, S.; Tukey, R.H.; Hankins, M.W.; Nestor-Kalinoski, A.; Stec, D.E. Mice with hyperbilirubinemia due to Gilbert’s syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARalpha. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E244–E252. [Google Scholar] [CrossRef]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin Binding to PPARalpha Inhibits Lipid Accumulation. PLoS ONE 2016, 11, e0153427. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Adeosun, S.O.; Alamodi, A.A.; Stec, D.E. Does bilirubin prevent hepatic steatosis through activation of the PPARalpha nuclear receptor? Med. Hypotheses 2016, 95, 54–57. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; Vanden Heuvel, J.P.; Stec, D.E. Biliverdin Reductase A Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3beta Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) alpha. J. Biol. Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARalpha gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef]
- Hu, X.; Tanaka, N.; Guo, R.; Lu, Y.; Nakajima, T.; Gonzalez, F.J.; Aoyama, T. PPARalpha protects against trans-fatty-acid-containing diet-induced steatohepatitis. J. Nutr. Biochem. 2017, 39, 77–85. [Google Scholar] [CrossRef]
- Ota, T.; Gayet, C.; Ginsberg, H.N. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J. Clin. Investig. 2008, 118, 316–332. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.; Sreekumar, R.; Rasmussen, D.; Lindor, K.; Nair, K.S. Apolipoprotein synthesis in nonalcoholic steatohepatitis. Hepatology 2002, 35, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Sanyal, A.J. Lipotoxicity in NASH. J. Hepatol. 2012, 56, 291–293. [Google Scholar] [CrossRef]
- Sinha, R.A. Autophagy: A Cellular Guardian against Hepatic Lipotoxicity. Genes 2023, 14, 553. [Google Scholar] [CrossRef]
- Rector, R.S.; Thyfault, J.P.; Uptergrove, G.M.; Morris, E.M.; Naples, S.P.; Borengasser, S.J.; Mikus, C.R.; Laye, M.J.; Laughlin, M.H.; Booth, F.W.; et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 2010, 52, 727–736. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Le, B.H.; García-Ruiz, C.; Fernandez-Checa, J.C.; Kaplowitz, N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 2015, 62, 1367–1374. [Google Scholar] [CrossRef]
- Tang, S.P.; Mao, X.L.; Chen, Y.H.; Yan, L.L.; Ye, L.P.; Li, S.W. Reactive Oxygen Species Induce Fatty Liver and Ischemia-Reperfusion Injury by Promoting Inflammation and Cell Death. Front. Immunol. 2022, 13, 870239. [Google Scholar] [CrossRef] [PubMed]
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative Stress in Non-alcoholic Fatty Liver Disease. An Updated Mini Review. Front. Med. 2021, 8, 595371. [Google Scholar] [CrossRef]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306.e20. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Greatorex, S.; Kaur, S.; Xirouchaki, C.E.; Goh, P.K.; Wiede, F.; Genders, A.J.; Tran, M.; Jia, Y.; Raajendiran, A.; Brown, W.A.; et al. Mitochondria- and NOX4-dependent antioxidant defense mitigates progression to nonalcoholic steatohepatitis in obesity. J. Clin. Investig. 2023, 134, e162533. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wu, X.; Liao, R. Mechanism and regulation of mitophagy in nonalcoholic fatty liver disease (NAFLD): A mini-review. Life Sci. 2023, 312, 121162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, Y.; Wang, S.; Li, G.; Xu, K. CREBH alleviates mitochondrial oxidative stress through SIRT3 mediating deacetylation of MnSOD and suppression of Nlrp3 inflammasome in NASH. Free Radic. Biol. Med. 2022, 190, 28–41. [Google Scholar] [CrossRef]
- Tsung, A.; Klune, J.R.; Zhang, X.; Jeyabalan, G.; Cao, Z.; Peng, X.; Stolz, D.B.; Geller, D.A.; Rosengart, M.R.; Billiar, T.R. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 2007, 204, 2913–2923. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Ge, F.; Hu, Y.; Yu, Y.; Guo, K.; Miao, C. Sevoflurane Postconditioning Attenuates Hepatic Ischemia-Reperfusion Injury by Limiting HMGB1/TLR4/NF-kappaB Pathway via Modulating microRNA-142 in vivo and in vitro. Front. Pharmacol. 2021, 12, 646307. [Google Scholar] [CrossRef]
- Schwarzler, J.; Grabherr, F.; Grander, C.; Adolph, T.E.; Tilg, H. The pathophysiology of MASLD: An immunometabolic perspective. Expert. Rev. Clin. Immunol. 2024, 20, 375–386. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallee, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Tirosh, A.; Tuncman, G.; Calay, E.S.; Rathaus, M.; Ron, I.; Tirosh, A.; Yalcin, A.; Lee, Y.G.; Livne, R.; Ron, S.; et al. Intercellular Transmission of Hepatic ER Stress in Obesity Disrupts Systemic Metabolism. Cell Metab. 2021, 33, 319–333.e6. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Kakazu, E.; Mauer, A.S.; Yin, M.; Malhi, H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1alpha-dependent manner. J. Lipid Res. 2016, 57, 233–245. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Yang, G.; Lee, H.E.; Lee, J.Y. A pharmacological inhibitor of NLRP3 inflammasome prevents non-alcoholic fatty liver disease in a mouse model induced by high fat diet. Sci. Rep. 2016, 6, 24399. [Google Scholar] [CrossRef]
- Chen, Y.; Ma, K. NLRC4 inflammasome activation regulated by TNF-alpha promotes inflammatory responses in nonalcoholic fatty liver disease. Biochem. Biophys. Res. Commun. 2019, 511, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, V.; Angeli, P.; Moreau, R.; Jalan, R.; Claria, J.; Trebicka, J.; Fernandez, J.; Gustot, T.; Caraceni, P.; Bernardi, M.; et al. The systemic inflammation hypothesis: Towards a new paradigm of acute decompensation and multiorgan failure in cirrhosis. J. Hepatol. 2021, 74, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Barrow, F.; Khan, S.; Fredrickson, G.; Wang, H.; Dietsche, K.; Parthiban, P.; Robert, S.; Kaiser, T.; Winer, S.; Herman, A.; et al. Microbiota-Driven Activation of Intrahepatic B Cells Aggravates NASH Through Innate and Adaptive Signaling. Hepatology 2021, 74, 704–722. [Google Scholar] [CrossRef]
- Zhang, F.; Jiang, W.W.; Li, X.; Qiu, X.Y.; Wu, Z.; Chi, Y.J.; Cong, X.; Liu, Y.L. Role of intrahepatic B cells in non-alcoholic fatty liver disease by secreting pro-inflammatory cytokines and regulating intrahepatic T cells. J. Dig. Dis. 2016, 17, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.; Cayon, A.; Fernandez-Gil, P.; Hernandez-Guerra, M.; Mayorga, M.; Dominguez-Diez, A.; Fernandez-Escalante, J.C.; Pons-Romero, F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Kamari, Y.; Shaish, A.; Vax, E.; Shemesh, S.; Kandel-Kfir, M.; Arbel, Y.; Olteanu, S.; Barshack, I.; Dotan, S.; Voronov, E.; et al. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J. Hepatol. 2011, 55, 1086–1094. [Google Scholar] [CrossRef]
- Huby, T.; Gautier, E.L. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat. Rev. Immunol. 2022, 22, 429–443. [Google Scholar] [CrossRef]
- Tosello-Trampont, A.C.; Landes, S.G.; Nguyen, V.; Novobrantseva, T.I.; Hahn, Y.S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-alpha production. J. Biol. Chem. 2012, 287, 40161–40172. [Google Scholar] [CrossRef]
- Pan, J.; Ou, Z.; Cai, C.; Li, P.; Gong, J.; Ruan, X.Z.; He, K. Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell Immunol. 2018, 332, 111–120. [Google Scholar] [CrossRef]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T.; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426. [Google Scholar] [CrossRef]
- van der Windt, D.J.; Sud, V.; Zhang, H.; Varley, P.R.; Goswami, J.; Yazdani, H.O.; Tohme, S.; Loughran, P.; O’Doherty, R.M.; Minervini, M.I.; et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 2018, 68, 1347–1360. [Google Scholar] [CrossRef]
- Han, H.; Ge, X.; Komakula, S.S.B.; Desert, R.; Das, S.; Song, Z.; Chen, W.; Athavale, D.; Gaskell, H.; Lantvit, D.; et al. Macrophage-derived Osteopontin (SPP1) Protects From Nonalcoholic Steatohepatitis. Gastroenterology 2023, 165, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Henning, J.R.; Graffeo, C.S.; Rehman, A.; Fallon, N.C.; Zambirinis, C.P.; Ochi, A.; Barilla, R.; Jamal, M.; Deutsch, M.; Greco, S.; et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology 2013, 58, 589–602. [Google Scholar] [CrossRef]
- Deczkowska, A.; David, E.; Ramadori, P.; Pfister, D.; Safran, M.; Li, B.; Giladi, A.; Jaitin, D.A.; Barboy, O.; Cohen, M.; et al. XCR1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat. Med. 2021, 27, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Dudek, M.; Pfister, D.; Donakonda, S.; Filpe, P.; Schneider, A.; Laschinger, M.; Hartmann, D.; Huser, N.; Meiser, P.; Bayerl, F.; et al. Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature 2021, 592, 444–449. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Costantini, S.; Finelli, C.; Capone, F.; Guerriero, E.; La Sala, N.; Gioia, S.; Castello, G. Is serum Interleukin-17 associated with early atherosclerosis in obese patients? J. Transl. Med. 2014, 12, 214. [Google Scholar] [CrossRef]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef]
- Senoo, H.; Yoshikawa, K.; Morii, M.; Miura, M.; Imai, K.; Mezaki, Y. Hepatic stellate cell (vitamin A-storing cell) and its relative—Past, present and future. Cell Biol. Int. 2010, 34, 1247–1272. [Google Scholar] [CrossRef]
- Kamm, D.R.; McCommis, K.S. Hepatic stellate cells in physiology and pathology. J. Physiol. 2022, 600, 1825–1837. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Breitkopf-Heinlein, K.; Meyer, C.; Konig, C.; Gaitantzi, H.; Addante, A.; Thomas, M.; Wiercinska, E.; Cai, C.; Li, Q.; Wan, F.; et al. BMP-9 interferes with liver regeneration and promotes liver fibrosis. Gut 2017, 66, 939–954. [Google Scholar] [CrossRef] [PubMed]
- Vacca, M.; Leslie, J.; Virtue, S.; Lam, B.Y.H.; Govaere, O.; Tiniakos, D.; Snow, S.; Davies, S.; Petkevicius, K.; Tong, Z.; et al. Bone morphogenetic protein 8B promotes the progression of non-alcoholic steatohepatitis. Nat. Metab. 2020, 2, 514–531. [Google Scholar] [CrossRef]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef]
- Hellerbrand, C.; Stefanovic, B.; Giordano, F.; Burchardt, E.R.; Brenner, D.A. The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J. Hepatol. 1999, 30, 77–87. [Google Scholar] [CrossRef]
- Cai, B.; Dongiovanni, P.; Corey, K.E.; Wang, X.; Shmarakov, I.O.; Zheng, Z.; Kasikara, C.; Davra, V.; Meroni, M.; Chung, R.T.; et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2020, 31, 406–421.e7. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef]
- Kagan, P.; Sultan, M.; Tachlytski, I.; Safran, M.; Ben-Ari, Z. Both MAPK and STAT3 signal transduction pathways are necessary for IL-6-dependent hepatic stellate cells activation. PLoS ONE 2017, 12, e0176173. [Google Scholar] [CrossRef]
- Ying, H.Z.; Chen, Q.; Zhang, W.Y.; Zhang, H.H.; Ma, Y.; Zhang, S.Z.; Fang, J.; Yu, C.H. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics (Review). Mol. Med. Rep. 2017, 16, 7879–7889. [Google Scholar] [CrossRef] [PubMed]
- Gieling, R.G.; Wallace, K.; Han, Y.P. Interleukin-1 participates in the progression from liver injury to fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1324–G1331. [Google Scholar] [CrossRef]
- Li, F.; Hao, X.; Chen, Y.; Bai, L.; Gao, X.; Lian, Z.; Wei, H.; Sun, R.; Tian, Z. The microbiota maintain homeostasis of liver-resident gammadeltaT-17 cells in a lipid antigen/CD1d-dependent manner. Nat. Commun. 2017, 7, 13839. [Google Scholar] [CrossRef]
- Fabre, T.; Barron, A.M.S.; Christensen, S.M.; Asano, S.; Bound, K.; Lech, M.P.; Wadsworth, M.H., 2nd; Chen, X.; Wang, C.; Wang, J.; et al. Identification of a broadly fibrogenic macrophage subset induced by type 3 inflammation. Sci. Immunol. 2023, 8, eadd8945. [Google Scholar] [CrossRef] [PubMed]
- Marinovic, S.; Lenartic, M.; Mladenic, K.; Sestan, M.; Kavazovic, I.; Benic, A.; Krapic, M.; Rindlisbacher, L.; Cokaric Brdovcak, M.; Sparano, C.; et al. NKG2D-mediated detection of metabolically stressed hepatocytes by innate-like T cells is essential for initiation of NASH and fibrosis. Sci. Immunol. 2023, 8, eadd1599. [Google Scholar] [CrossRef]
- Ma, H.Y.; Yamamoto, G.; Xu, J.; Liu, X.; Karin, D.; Kim, J.Y.; Alexandrov, L.B.; Koyama, Y.; Nishio, T.; Benner, C.; et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J. Hepatol. 2020, 72, 946–959. [Google Scholar] [CrossRef]
- Najjar, S.M.; Russo, L. CEACAM1 loss links inflammation to insulin resistance in obesity and non-alcoholic steatohepatitis (NASH). Semin. Immunopathol. 2014, 36, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M.; Farpour-Lambert, N.J.; Zhao, L.; Tilg, H. Why we need to curb the emerging worldwide epidemic of nonalcoholic fatty liver disease. Nat. Metab. 2019, 1, 1027–1029. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef]
- Enooku, K.; Kondo, M.; Fujiwara, N.; Sasako, T.; Shibahara, J.; Kado, A.; Okushin, K.; Fujinaga, H.; Tsutsumi, T.; Nakagomi, R.; et al. Hepatic IRS1 and ss-catenin expression is associated with histological progression and overt diabetes emergence in NAFLD patients. J. Gastroenterol. 2018, 53, 1261–1275. [Google Scholar] [CrossRef]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef]
- Mandraffino, G.; Morace, C.; Franzè, M.S.; Nassisi, V.; Sinicropi, D.; Cinquegrani, M.; Saitta, C.; Scoglio, R.; Marino, S.; Belvedere, A.; et al. Fatty Liver as Potential Biomarker of Atherosclerotic Damage in Familial Combined Hyperlipidemia. Biomedicines 2022, 10, 1770. [Google Scholar] [CrossRef]
- Jacobs, K.; Brouha, S.; Bettencourt, R.; Barrett-Connor, E.; Sirlin, C.; Loomba, R. Association of Nonalcoholic Fatty Liver Disease With Visceral Adiposity but Not Coronary Artery Calcification in the Elderly. Clin. Gastroenterol. Hepatol. 2016, 14, 1337–1344.e3. [Google Scholar] [CrossRef]
- Soto, A.; Spongberg, C.; Martinino, A.; Giovinazzo, F. Exploring the Multifaceted Landscape of MASLD: A Comprehensive Synthesis of Recent Studies, from Pathophysiology to Organoids and Beyond. Biomedicines 2024, 12, 397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Q.; Lu, L.G. Nonalcoholic Fatty Liver Disease: Dyslipidemia, Risk for Cardiovascular Complications, and Treatment Strategy. J. Clin. Transl. Hepatol. 2015, 3, 78–84. [Google Scholar] [CrossRef]
- Vos, M.B.; Lavine, J.E. Dietary fructose in nonalcoholic fatty liver disease. Hepatology 2013, 57, 2525–2531. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Chiavaroli, L.; Ayoub-Charette, S.; Khan, T.A.; Zurbau, A.; Au-Yeung, F.; Cheung, A.; Liu, Q.; Qi, X.; Ahmed, A.; et al. Important Food Sources of Fructose-Containing Sugars and Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis of Controlled Trials. Nutrients 2022, 14, 2846. [Google Scholar] [CrossRef]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M.; Nonalcoholic Steatohepatitis Clinical Research Network. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef]
- Andres-Hernando, A.; Orlicky, D.J.; Kuwabara, M.; Ishimoto, T.; Nakagawa, T.; Johnson, R.J.; Lanaspa, M.A. Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab. 2020, 32, 117–127.e3. [Google Scholar] [CrossRef]
- Herman, M.A.; Samuel, V.T. The Sweet Path to Metabolic Demise: Fructose and Lipid Synthesis. Trends Endocrinol. Metab. 2016, 27, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Zelber-Sagi, S.; Henry, L.; Gerber, L.H. Lifestyle interventions in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 708–722. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Davis, T.M.E. Diabetes and metabolic dysfunction-associated fatty liver disease. Metabolism 2021, 123, 154868. [Google Scholar] [CrossRef] [PubMed]
- Ismaiel, A.; Dumitrascu, D.L. Cardiovascular Risk in Fatty Liver Disease: The Liver-Heart Axis-Literature Review. Front. Med. 2019, 6, 202. [Google Scholar] [CrossRef]
- Golabi, P.; Fukui, N.; Paik, J.; Sayiner, M.; Mishra, A.; Younossi, Z.M. Mortality Risk Detected by Atherosclerotic Cardiovascular Disease Score in Patients With Nonalcoholic Fatty Liver Disease. Hepatol. Commun. 2019, 3, 1050–1060. [Google Scholar] [CrossRef]
- Kasper, P.; Martin, A.; Lang, S.; Kutting, F.; Goeser, T.; Demir, M.; Steffen, H.M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2021, 110, 921–937. [Google Scholar] [CrossRef]
- Kim, D.; Kim, W.R.; Kim, H.J.; Therneau, T.M. Association between noninvasive fibrosis markers and mortality among adults with nonalcoholic fatty liver disease in the United States. Hepatology 2013, 57, 1357–1365. [Google Scholar] [CrossRef]
- Tana, C.; Ballestri, S.; Ricci, F.; Di Vincenzo, A.; Ticinesi, A.; Gallina, S.; Giamberardino, M.A.; Cipollone, F.; Sutton, R.; Vettor, R.; et al. Cardiovascular Risk in Non-Alcoholic Fatty Liver Disease: Mechanisms and Therapeutic Implications. Int. J. Environ. Res. Public Health 2019, 16, 3104. [Google Scholar] [CrossRef]
- Haring, R.; Wallaschofski, H.; Nauck, M.; Dorr, M.; Baumeister, S.E.; Volzke, H. Ultrasonographic hepatic steatosis increases prediction of mortality risk from elevated serum gamma-glutamyl transpeptidase levels. Hepatology 2009, 50, 1403–1411. [Google Scholar] [CrossRef]
- Adams, L.A.; Anstee, Q.M.; Tilg, H.; Targher, G. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut 2017, 66, 1138–1153. [Google Scholar] [CrossRef] [PubMed]
- Han, E.; Lee, Y.H.; Kim, Y.D.; Kim, B.K.; Park, J.Y.; Kim, D.Y.; Ahn, S.H.; Lee, B.W.; Kang, E.S.; Cha, B.S.; et al. Nonalcoholic Fatty Liver Disease and Sarcopenia Are Independently Associated With Cardiovascular Risk. Am. J. Gastroenterol. 2020, 115, 584–595. [Google Scholar] [CrossRef]
- Yki-Jarvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Ballestri, S.; Lonardo, A.; Bonapace, S.; Byrne, C.D.; Loria, P.; Targher, G. Risk of cardiovascular, cardiac and arrhythmic complications in patients with non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1724–1745. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Stahl, E.P.; Dhindsa, D.S.; Lee, S.K.; Sandesara, P.B.; Chalasani, N.P.; Sperling, L.S. Nonalcoholic Fatty Liver Disease and the Heart: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 948–963. [Google Scholar] [CrossRef]
- Pasterkamp, G. Methods of accelerated atherosclerosis in diabetic patients. Heart 2013, 99, 743–749. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sarin, S.K.; Wong, V.W.; Fan, J.G.; Kawaguchi, T.; Ahn, S.H.; Zheng, M.H.; Shiha, G.; Yilmaz, Y.; Gani, R.; et al. The Asian Pacific Association for the Study of the Liver clinical practice guidelines for the diagnosis and management of metabolic associated fatty liver disease. Hepatol. Int. 2020, 14, 889–919. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Fargion, S.; Porzio, M.; Fracanzani, A.L. Nonalcoholic fatty liver disease and vascular disease: State-of-the-art. World J. Gastroenterol. 2014, 20, 13306–13324. [Google Scholar] [CrossRef] [PubMed]
- Dogru, T.; Genc, H.; Tapan, S.; Aslan, F.; Ercin, C.N.; Ors, F.; Kara, M.; Sertoglu, E.; Karslioglu, Y.; Bagci, S.; et al. Plasma fetuin-A is associated with endothelial dysfunction and subclinical atherosclerosis in subjects with nonalcoholic fatty liver disease. Clin. Endocrinol. 2013, 78, 712–717. [Google Scholar] [CrossRef]
- Villanova, N.; Moscatiello, S.; Ramilli, S.; Bugianesi, E.; Magalotti, D.; Vanni, E.; Zoli, M.; Marchesini, G. Endothelial dysfunction and cardiovascular risk profile in nonalcoholic fatty liver disease. Hepatology 2005, 42, 473–480. [Google Scholar] [CrossRef]
- Wilcox, J.N.; Subramanian, R.R.; Sundell, C.L.; Tracey, W.R.; Pollock, J.S.; Harrison, D.G.; Marsden, P.A. Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2479–2488. [Google Scholar] [CrossRef]
- Fujimoto, M.; Shimizu, N.; Kunii, K.; Martyn, J.A.; Ueki, K.; Kaneki, M. A role for iNOS in fasting hyperglycemia and impaired insulin signaling in the liver of obese diabetic mice. Diabetes 2005, 54, 1340–1348. [Google Scholar] [CrossRef]
- Chauhan, S.D.; Seggara, G.; Vo, P.A.; Macallister, R.J.; Hobbs, A.J.; Ahluwalia, A. Protection against lipopolysaccharide-induced endothelial dysfunction in resistance and conduit vasculature of iNOS knockout mice. FASEB J. 2003, 17, 773–775. [Google Scholar] [CrossRef]
- Francque, S.; Laleman, W.; Verbeke, L.; Van Steenkiste, C.; Casteleyn, C.; Kwanten, W.; Van Dyck, C.; D’Hondt, M.; Ramon, A.; Vermeulen, W.; et al. Increased intrahepatic resistance in severe steatosis: Endothelial dysfunction, vasoconstrictor overproduction and altered microvascular architecture. Lab. Investig. 2012, 92, 1428–1439. [Google Scholar] [CrossRef]
- Miyao, M.; Kotani, H.; Ishida, T.; Kawai, C.; Manabe, S.; Abiru, H.; Tamaki, K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab. Investig. 2015, 95, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Lafoz, E.; Ruart, M.; Anton, A.; Oncins, A.; Hernández-Gea, V. The Endothelium as a Driver of Liver Fibrosis and Regeneration. Cells 2020, 9, 929. [Google Scholar] [CrossRef]
- Hilscher, M.B.; Sehrawat, T.; Arab, J.P.; Zeng, Z.; Gao, J.; Liu, M.; Kostallari, E.; Gao, Y.; Simonetto, D.A.; Yaqoob, U.; et al. Mechanical Stretch Increases Expression of CXCL1 in Liver Sinusoidal Endothelial Cells to Recruit Neutrophils, Generate Sinusoidal Microthombi, and Promote Portal Hypertension. Gastroenterology 2019, 157, 193–209.e9. [Google Scholar] [CrossRef]
- Ogresta, D.; Mrzljak, A.; Cigrovski Berkovic, M.; Bilic-Curcic, I.; Stojsavljevic-Shapeski, S.; Virovic-Jukic, L. Coagulation and Endothelial Dysfunction Associated with NAFLD: Current Status and Therapeutic Implications. J. Clin. Transl. Hepatol. 2022, 10, 339–355. [Google Scholar] [CrossRef]
- Mazurek, T.; Zhang, L.; Zalewski, A.; Mannion, J.D.; Diehl, J.T.; Arafat, H.; Sarov-Blat, L.; O’Brien, S.; Keiper, E.A.; Johnson, A.G.; et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation 2003, 108, 2460–2466. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Pan, X.; Luo, J.; Xiao, X.; Li, J.; Bestman, P.L.; Luo, M. Association of Inflammatory Cytokines With Non-Alcoholic Fatty Liver Disease. Front. Immunol. 2022, 13, 880298. [Google Scholar] [CrossRef]
- Kathirvel, E.; Chen, P.; Morgan, K.; French, S.W.; Morgan, T.R. Oxidative stress and regulation of anti-oxidant enzymes in cytochrome P4502E1 transgenic mouse model of non-alcoholic fatty liver. J. Gastroenterol. Hepatol. 2010, 25, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Caimi, G.; Lo Presti, R.; Montana, M.; Noto, D.; Canino, B.; Averna, M.R.; Hopps, E. Lipid peroxidation, nitric oxide metabolites, and their ratio in a group of subjects with metabolic syndrome. Oxid. Med. Cell Longev. 2014, 2014, 824756. [Google Scholar] [CrossRef]
- Fernández-Sánchez, A.; Madrigal-Santillán, E.; Bautista, M.; Esquivel-Soto, J.; Morales-González, A.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sánchez-Rivera, G.; Valadez-Vega, C.; Morales-González, J.A. Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef]
- Huang, C.J.; McAllister, M.J.; Slusher, A.L.; Webb, H.E.; Mock, J.T.; Acevedo, E.O. Obesity-Related Oxidative Stress: The Impact of Physical Activity and Diet Manipulation. Sports Med. Open 2015, 1, 32. [Google Scholar] [CrossRef]
- Masenga, S.K.; Kabwe, L.S.; Chakulya, M.; Kirabo, A. Mechanisms of Oxidative Stress in Metabolic Syndrome. Int. J. Mol. Sci. 2023, 24, 7898. [Google Scholar] [CrossRef]
- Potenza, M.A.; Marasciulo, F.L.; Chieppa, D.M.; Brigiani, G.S.; Formoso, G.; Quon, M.J.; Montagnani, M. Insulin resistance in spontaneously hypertensive rats is associated with endothelial dysfunction characterized by imbalance between NO and ET-1 production. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H813–H822. [Google Scholar] [CrossRef]
- Reustle, A.; Torzewski, M. Role of p38 MAPK in Atherosclerosis and Aortic Valve Sclerosis. Int. J. Mol. Sci. 2018, 19, 3761. [Google Scholar] [CrossRef]
- Hong, J.; Zhang, Y.; Lai, S.; Lv, A.; Su, Q.; Dong, Y.; Zhou, Z.; Tang, W.; Zhao, J.; Cui, L.; et al. Effects of metformin versus glipizide on cardiovascular outcomes in patients with type 2 diabetes and coronary artery disease. Diabetes Care 2013, 36, 1304–1311. [Google Scholar] [CrossRef]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Iida, S.; Katsuyama, H. Metabolic-Dysfunction-Associated Steatotic Liver Disease-Its Pathophysiology, Association with Atherosclerosis and Cardiovascular Disease, and Treatments. Int. J. Mol. Sci. 2023, 24, 5473. [Google Scholar] [CrossRef] [PubMed]
- Amor, A.J.; Pinyol, M.; Solà, E.; Catalan, M.; Cofán, M.; Herreras, Z.; Amigó, N.; Gilabert, R.; Sala-Vila, A.; Ros, E.; et al. Relationship between noninvasive scores of nonalcoholic fatty liver disease and nuclear magnetic resonance lipoprotein abnormalities: A focus on atherogenic dyslipidemia. J. Clin. Lipidol. 2017, 11, 551–561.e7. [Google Scholar] [CrossRef]
- DeFilippis, A.P.; Blaha, M.J.; Martin, S.S.; Reed, R.M.; Jones, S.R.; Nasir, K.; Blumenthal, R.S.; Budoff, M.J. Nonalcoholic fatty liver disease and serum lipoproteins: The Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 2013, 227, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Heeren, J.; Scheja, L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Ma, S.; Sun, L.; Gao, J.; Zhao, C. TGF-β1 upregulates the expression of lncRNA-ATB to promote atherosclerosis. Mol. Med. Rep. 2019, 19, 4222–4228. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Franzen, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lu, L.; Dong, Q.; Li, X.; Zhang, N.; Xin, Y.; Xuan, S. Research advances in the relationship between nonalcoholic fatty liver disease and atherosclerosis. Lipids Health Dis. 2015, 14, 158. [Google Scholar] [CrossRef] [PubMed]
- Duell, P.B.; Welty, F.K.; Miller, M.; Chait, A.; Hammond, G.; Ahmad, Z.; Cohen, D.E.; Horton, J.D.; Pressman, G.S.; Toth, P.P.; et al. Nonalcoholic Fatty Liver Disease and Cardiovascular Risk: A Scientific Statement From the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 2022, 42, e168–e185. [Google Scholar] [CrossRef]
- Bieghs, V.; Rensen, P.C.; Hofker, M.H.; Shiri-Sverdlov, R. NASH and atherosclerosis are two aspects of a shared disease: Central role for macrophages. Atherosclerosis 2012, 220, 287–293. [Google Scholar] [CrossRef]
- Sookoian, S.; Gianotti, T.F.; Rosselli, M.S.; Burgueno, A.L.; Castano, G.O.; Pirola, C.J. Liver transcriptional profile of atherosclerosis-related genes in human nonalcoholic fatty liver disease. Atherosclerosis 2011, 218, 378–385. [Google Scholar] [CrossRef]
- Abdallah, L.R.; de Matos, R.C.; YPDM, E.S.; Vieira-Soares, D.; Muller-Machado, G.; Pollo-Flores, P. Non-alcoholic Fatty Liver Disease and Its Links with Inflammation and Atherosclerosis. Curr. Atheroscler. Rep. 2020, 22, 7. [Google Scholar] [CrossRef]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef]
- Zhu, B.; Wu, H.; Li, K.S.; Eisa-Beygi, S.; Singh, B.; Bielenberg, D.R.; Huang, W.; Chen, H. Two sides of the same coin: Non-alcoholic fatty liver disease and atherosclerosis. Vasc. Pharmacol. 2024, 154, 107249. [Google Scholar] [CrossRef]
- Abd El-Kader, S.M.; El-Den Ashmawy, E.M. Non-alcoholic fatty liver disease: The diagnosis and management. World J. Hepatol. 2015, 7, 846–858. [Google Scholar] [CrossRef]
- Pacifico, L.; Nobili, V.; Anania, C.; Verdecchia, P.; Chiesa, C. Pediatric nonalcoholic fatty liver disease, metabolic syndrome and cardiovascular risk. World J. Gastroenterol. 2011, 17, 3082–3091. [Google Scholar] [CrossRef]
- Muzurovic, E.; Peng, C.C.; Belanger, M.J.; Sanoudou, D.; Mikhailidis, D.P.; Mantzoros, C.S. Nonalcoholic Fatty Liver Disease and Cardiovascular Disease: A Review of Shared Cardiometabolic Risk Factors. Hypertension 2022, 79, 1319–1326. [Google Scholar] [CrossRef]
- Hamaguchi, M.; Kojima, T.; Takeda, N.; Nagata, C.; Takeda, J.; Sarui, H.; Kawahito, Y.; Yoshida, N.; Suetsugu, A.; Kato, T.; et al. Nonalcoholic fatty liver disease is a novel predictor of cardiovascular disease. World J. Gastroenterol. 2007, 13, 1579–1584. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Castillo-Nunez, Y.; Almeda-Valdes, P.; Gonzalez-Galvez, G.; Arechavaleta-Granell, M.D.R. Metabolic dysfunction-associated steatotic liver disease and atherosclerosis. Curr. Diab. Rep. 2024, 24, 158–166. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, U.; Beauchemin, N.; Obrink, B. The cytoplasmic domain of CEACAM1-L controls its lateral localization and the organization of desmosomes in polarized epithelial cells. J. Cell Sci. 2004, 117 Pt 7, 1091–1104. [Google Scholar] [CrossRef]
- Sundberg, U.; Obrink, B. CEACAM1 isoforms with different cytoplasmic domains show different localization, organization and adhesive properties in polarized epithelial cells. J. Cell Sci. 2002, 115 Pt 6, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.M.; Yang, Y.; Fernstrom, M.A.; Lee, S.J.; Deangelis, A.M.; Rjaily, G.A.; Al-Share, Q.Y.; Dai, T.; Miller, T.A.; Ratnam, S.; et al. Insulin acutely decreases hepatic fatty acid synthase activity. Cell Metab. 2005, 2, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.M.; Abdolahipour, R.; Ghadieh, H.E.; Jahromi, M.S.; Najjar, J.A.; Abuamreh, B.A.M.; Zaidi, S.; Kumarasamy, S.; Muturi, H.T. Regulation of Insulin Clearance by Non-Esterified Fatty Acids. Biomedicines 2022, 10, 1899. [Google Scholar] [CrossRef]
- Aldroubi, B.G.; Najjar, J.A.; Youssef, T.S.; Rizk, C.E.; Abuamreh, B.A.M.; Aramouni, K.; Ghadieh, H.E.; Najjar, S.M. Cell-specific regulation of insulin action and hepatic fibrosis by CEACAM1. Metab. Target. Organ. Damage 2024, 4, 34. [Google Scholar] [CrossRef]
- Shaheen, M.; Pan, D.; Schrode, K.M.; Kermah, D.; Puri, V.; Zarrinpar, A.; Elisha, D.; Najjar, S.M.; Friedman, T.C. Reassessment of the Hispanic Disparity: Hepatic Steatosis Is More Prevalent in Mexican Americans Than Other Hispanics. Hepatol. Commun. 2021, 5, 2068–2079. [Google Scholar] [CrossRef]
- Bril, F.; Lomonaco, R.; Orsak, B.; Ortiz-Lopez, C.; Webb, A.; Tio, F.; Hecht, J.; Cusi, K. Relationship between disease severity, hyperinsulinemia, and impaired insulin clearance in patients with nonalcoholic steatohepatitis. Hepatology 2014, 59, 2178–2187. [Google Scholar] [CrossRef] [PubMed]
- Watada, H.; Tamura, Y. Impaired insulin clearance as a cause rather than a consequence of insulin resistance. J. Diabetes Investig. 2017, 8, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Patarrao, R.S.; Meneses, M.J.; Ghadieh, H.E.; Herrera, L.; Duarte, S.; Ribeiro, R.T.; Raposo, J.F.; Schmitt, V.; Singer, B.B.; Gastaldelli, A.; et al. Insights into circulating CEACAM1 in insulin clearance and disease progression: Evidence from the Portuguese PREVADIAB2 study. Eur. J. Clin. Investig. 2024, 54 (Suppl. S2), e14344. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; Asalla, S.; Muturi, H.T.; Russo, L.; Abdolahipour, R.; Belew, G.D.; Iglesias, M.B.; Feraudo, M.; Leon, L.; Kuo, E.; et al. Loss of CEACAM1 in hepatocytes causes hepatic fibrosis. Eur. J. Clin. Investig. 2024, 54, e14177. [Google Scholar] [CrossRef]
- Ghadieh, H.E.; Gastaldelli, A.; Najjar, S.M. Role of Insulin Clearance in Insulin Action and Metabolic Diseases. Int. J. Mol. Sci. 2023, 24, 7156. [Google Scholar] [CrossRef]
- Kotronen, A.; Juurinen, L.; Tiikkainen, M.; Vehkavaara, S.; Yki-Jarvinen, H. Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology 2008, 135, 122–130. [Google Scholar] [CrossRef]
- Tiikkainen, M.; Hakkinen, A.M.; Korsheninnikova, E.; Nyman, T.; Makimattila, S.; Yki-Jarvinen, H. Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes 2004, 53, 2169–2176. [Google Scholar] [CrossRef]
- Ghadieh, H.E.; Muturi, H.T.; Russo, L.; Marino, C.C.; Ghanem, S.S.; Khuder, S.S.; Hanna, J.C.; Jash, S.; Puri, V.; Heinrich, G.; et al. Exenatide induces carcinoembryonic antigen-related cell adhesion molecule 1 expression to prevent hepatic steatosis. Hepatol. Commun. 2018, 2, 35–47. [Google Scholar] [CrossRef]
- Muturi, H.T.; Ghadieh, H.E.; Asalla, S.; Lester, S.G.; Belew, G.D.; Zaidi, S.; Abdolahipour, R.; Shrestha, A.P.; Portuphy, A.O.; Stankus, H.L.; et al. Conditional deletion of CEACAM1 in hepatic stellate cells causes their activation. Mol. Metab. 2024, 88, 102010. [Google Scholar] [CrossRef]
- Ghadieh, H.E.; Muturi, H.T.; Najjar, S.M. Exenatide Prevents Diet-induced Hepatocellular Injury in A CEACAM1-Dependent Mechanism. J. Diabetes Treat. 2017, 2017, 10-29011. [Google Scholar] [CrossRef]
- Bakker, L.E.; van Schinkel, L.D.; Guigas, B.; Streefland, T.C.; Jonker, J.T.; van Klinken, J.B.; van der Zon, G.C.; Lamb, H.J.; Smit, J.W.; Pijl, H.; et al. A 5-day high-fat, high-calorie diet impairs insulin sensitivity in healthy, young South Asian men but not in Caucasian men. Diabetes 2014, 63, 248–258. [Google Scholar] [CrossRef]
- Ramakrishnan, S.K.; Khuder, S.S.; Al-Share, Q.Y.; Russo, L.; Abdallah, S.L.; Patel, P.R.; Heinrich, G.; Muturi, H.T.; Mopidevi, B.R.; Oyarce, A.M.; et al. PPARalpha (Peroxisome Proliferator-activated Receptor alpha) Activation Reduces Hepatic CEACAM1 Protein Expression to Regulate Fatty Acid Oxidation during Fasting-refeeding Transition. J. Biol. Chem. 2016, 291, 8121–8129. [Google Scholar] [CrossRef]
- Najjar, S.M.; Shively, J.E. Regulation of lipid storage and inflammation in the liver by CEACAM1. Eur. J. Clin. Investig. 2024, 54 (Suppl. S2), e14338. [Google Scholar] [CrossRef]
- Gandhi, A.K.; Huang, Y.H.; Sun, Z.J.; Kim, W.M.; Kondo, Y.; Hanley, T.; Beauchemin, N.; Blumberg, R.S. Structural aspects of CEACAM1 interactions. Eur. J. Clin. Investig. 2024, 54 (Suppl. S2), e14357. [Google Scholar] [CrossRef]
- Gotz, L.; Rueckschloss, U.; Najjar, S.M.; Ergun, S.; Kleefeldt, F. Carcinoembryonic antigen-related cell adhesion molecule 1 in cancer: Blessing or curse? Eur. J. Clin. Investig. 2024, 54 (Suppl. S2), e14337. [Google Scholar] [CrossRef]
- Dery, K.J.; Yao, S.; Cheng, B.; Kupiec-Weglinski, J.W. New therapeutic concepts against ischemia-reperfusion injury in organ transplantation. Expert. Rev. Clin. Immunol. 2023, 19, 1205–1224. [Google Scholar] [CrossRef]
- Dery, K.J.; Najjar, S.M.; Beauchemin, N.; Shively, J.E.; Kupiec-Weglinski, J.W. Mechanism and function of CEACAM1 splice isoforms. Eur. J. Clin. Investig. 2024, 54 (Suppl. S2), e14350. [Google Scholar] [CrossRef]
- Kim, W.M.; Huang, Y.H.; Gandhi, A.; Blumberg, R.S. CEACAM1 structure and function in immunity and its therapeutic implications. Semin. Immunol. 2019, 42, 101296. [Google Scholar] [CrossRef]
- Yao, S.; Kasargod, A.; Chiu, R.; Torgerson, T.R.; Kupiec-Weglinski, J.W.; Dery, K.J. The Coming Age of Antisense Oligos for the Treatment of Hepatic Ischemia/Reperfusion (IRI) and Other Liver Disorders: Role of Oxidative Stress and Potential Antioxidant Effect. Antioxidants 2024, 13, 678. [Google Scholar] [CrossRef]
- Gotz, L.; Rueckschloss, U.; Ergun, S.; Kleefeldt, F. CEACAM1 in vascular homeostasis and inflammation. Eur. J. Clin. Investig. 2024, 54 (Suppl. S2), e14345. [Google Scholar] [CrossRef]
- Stefan, N.; Haring, H.U.; Cusi, K. Non-alcoholic fatty liver disease: Causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2019, 7, 313–324. [Google Scholar] [CrossRef]
- Alkhouri, N.; Carter-Kent, C.; Elias, M.; Feldstein, A.E. Atherogenic dyslipidemia and cardiovascular risk in children with nonalcoholic fatty liver disease. Clin. Lipidol. 2011, 6, 305–314. [Google Scholar] [CrossRef]
- Di Pino, A.; DeFronzo, R.A. Insulin Resistance and Atherosclerosis: Implications for Insulin-Sensitizing Agents. Endocr. Rev. 2019, 40, 1447–1467. [Google Scholar] [CrossRef]
- Lim, S.; Taskinen, M.R.; Boren, J. Crosstalk between nonalcoholic fatty liver disease and cardiometabolic syndrome. Obes. Rev. 2019, 20, 599–611. [Google Scholar] [CrossRef]
- Muturi, H.T.; Ghadieh, H.E.; Abdolahipour, R.; Stankus, H.L.; Belew, G.D.; Liu, J.K.; Jahromi, M.S.; Lee, A.D.; Singer, B.B.; Angeli-Pahim, I.; et al. Loss of CEACAM1 in endothelial cells causes hepatic fibrosis. Metabolism 2023, 144, 155562. [Google Scholar] [CrossRef]
- Bowman, T.A.; Ramakrishnan, S.K.; Kaw, M.; Lee, S.J.; Patel, P.R.; Golla, V.K.; Bourey, R.E.; Haram, P.M.; Koch, L.G.; Britton, S.L.; et al. Caloric restriction reverses hepatic insulin resistance and steatosis in rats with low aerobic capacity. Endocrinology 2010, 151, 5157–5164. [Google Scholar] [CrossRef]
- Wisloff, U.; Najjar, S.M.; Ellingsen, O.; Haram, P.M.; Swoap, S.; Al-Share, Q.; Fernstrom, M.; Rezaei, K.; Lee, S.J.; Koch, L.G.; et al. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science 2005, 307, 418–420. [Google Scholar] [CrossRef]
- Heinrich, G.; Ghadieh, H.E.; Ghanem, S.S.; Muturi, H.T.; Rezaei, K.; Al-Share, Q.Y.; Bowman, T.A.; Zhang, D.; Garofalo, R.S.; Yin, L.; et al. Loss of Hepatic CEACAM1: A Unifying Mechanism Linking Insulin Resistance to Obesity and Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2017, 8, 8. [Google Scholar] [CrossRef]
- Helal, R.A.; Russo, L.; Ghadieh, H.E.; Muturi, H.T.; Asalla, S.; Lee, A.D.; Gatto-Weis, C.; Najjar, S.M. Regulation of hepatic fibrosis by carcinoembryonic antigen-related cell adhesion molecule 1. Metabolism 2021, 121, 154801. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.; Muturi, H.T.; Ghadieh, H.E.; Ghanem, S.S.; Bowman, T.A.; Noh, H.L.; Dagdeviren, S.; Dogbey, G.Y.; Kim, J.K.; Heinrich, G.; et al. Liver-specific reconstitution of CEACAM1 reverses the metabolic abnormalities caused by its global deletion in male mice. Diabetologia 2017, 60, 2463–2474. [Google Scholar] [CrossRef]
- Abu Helal, R.; Muturi, H.T.; Lee, A.D.; Li, W.; Ghadieh, H.E.; Najjar, S.M. Aortic Fibrosis in Insulin-Sensitive Mice with Endothelial Cell-Specific Deletion of Ceacam1 Gene. Int. J. Mol. Sci. 2022, 23, 4335. [Google Scholar] [CrossRef] [PubMed]
- Muturi, H.T.; Khuder, S.S.; Ghadieh, H.E.; Esakov, E.L.; Noh, H.; Kang, H.; McInerney, M.F.; Kim, J.K.; Lee, A.D.; Najjar, S.M. Insulin Sensitivity Is Retained in Mice with Endothelial Loss of Carcinoembryonic Antigen Cell Adhesion Molecule 1. Cells 2021, 10, 2093. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Khoury, S.; Al Harake, S.N.; Youssef, T.; Risk, C.E.; Helou, N.G.; Doumet, N.M.; Aramouni, K.; Azar, S.; Najjar, S.M.; Ghadieh, H.E. Low Hepatic CEACAM1 Tethers Metabolic Dysfunction Steatohepatitis to Atherosclerosis. Livers 2025, 5, 34. https://doi.org/10.3390/livers5030034
El Khoury S, Al Harake SN, Youssef T, Risk CE, Helou NG, Doumet NM, Aramouni K, Azar S, Najjar SM, Ghadieh HE. Low Hepatic CEACAM1 Tethers Metabolic Dysfunction Steatohepatitis to Atherosclerosis. Livers. 2025; 5(3):34. https://doi.org/10.3390/livers5030034
Chicago/Turabian StyleEl Khoury, Sacha, Sami N. Al Harake, Tya Youssef, Carl E. Risk, Naim G. Helou, Natalie M. Doumet, Karl Aramouni, Sami Azar, Sonia M. Najjar, and Hilda E. Ghadieh. 2025. "Low Hepatic CEACAM1 Tethers Metabolic Dysfunction Steatohepatitis to Atherosclerosis" Livers 5, no. 3: 34. https://doi.org/10.3390/livers5030034
APA StyleEl Khoury, S., Al Harake, S. N., Youssef, T., Risk, C. E., Helou, N. G., Doumet, N. M., Aramouni, K., Azar, S., Najjar, S. M., & Ghadieh, H. E. (2025). Low Hepatic CEACAM1 Tethers Metabolic Dysfunction Steatohepatitis to Atherosclerosis. Livers, 5(3), 34. https://doi.org/10.3390/livers5030034